肝癌微球体细胞表面特异性靶向多肽及其制备方法和应用与流程
本发明涉及肝癌微球体细胞表面特异性靶向多肽及其制备方法和应用,具体属于生物技术领域。
背景技术:
肝癌合并癌栓(cancerembolus)在临床的发生率很高,而且往往预后不良,诊断和治疗非常棘手。研究认为,肝癌癌栓是有肝癌干细胞(hepaticcanerstemcell,hcsc)在门静脉或胆管微环境下诱导产生,同时保留了一些干细胞的原始分化特性。肝癌微球体(hepatomamicrospheres)是研究肝癌干细胞的理想模型,同时也表现出癌栓的特性,成为研究癌栓的理想模型。以癌栓作为靶点进行诊断和治疗,是目前肝癌合并癌栓临床努力的方向,寻找靶向癌栓的分子,则是其中的关键环节。
在肿瘤靶向治疗研究中,特异性的多肽类小分子受到了越来越多的关注。抗体类作为诊断和治疗癌症的靶向配体,难以穿透被致密基质包裹的肿瘤细胞且成本高、分子量大、结构复杂,有免疫原性等问题。与之相比,多肽类分子具有亲和力高、特异性高、相对分子质量小、易于合成加工、毒性低、生物相容性好等优点,具有良好的应用前景。在筛选与靶标分子或细胞特异性结合多肽的众多技术中,噬菌体展示技术(phagedisplaytechnology)是目前运用较广且相当高效的一项筛选技术。噬菌体展示技术是由smith等在1985年建立,其核心原理是具有在噬菌体衣壳蛋白表面展示多肽的作用。多肽在表面展示的原理是通过基因工程技术将一定长度的随机肽序列基因整合到噬菌体衣壳蛋白基因中,当基因表达后对应多肽序列会和衣壳蛋白形成融合蛋白并将其展示于衣壳蛋白表面,从而使表型和基因型之间建立了联系。至今,其作为一种简便、高效筛选工具,已广泛应用于抗肿瘤药物、肿瘤诊断标志物等的筛选和研发。综上所述,筛选出一种肝癌微球体细胞表面特异性靶向多肽,显得尤为必要。
技术实现要素:
为解决现有技术的不足,本发明的目的在于提供一种肝癌微球体细胞表面特异性靶向多肽及其制备方法和应用,所得多肽可以与肝癌微球体细胞特异性结合,其制备方法操作简单,条件易控制,准确度高;所得多肽可用于制备肝癌癌栓诊断试剂或肝癌靶向治疗药物中。
为了实现上述目标,本发明采用如下的技术方案:
肝癌微球体细胞表面特异性靶向多肽,所述多肽为七肽,其氨基酸序列为asn-thr-gly-ser-pro-tyr-glu。
优选地,前述肝癌微球体细胞表面特异性靶向多肽,为环状的环七肽,其结构式为:cyclo-(asn-thr-gly-ser-pro-tyr-glu)。
一种肝癌微球体细胞表面特异性靶向多肽的制备方法,包括以下步骤:(1)贴壁细胞及肝癌微球体的培养;(2)以hcclm3细胞悬浮培养的肝癌微球体为靶细胞,贴壁培养的hcclm3细胞和lo2细胞为吸附细胞,采用噬菌体展示技术进行差减筛选;(3)经dna测序确定目标多肽。
前述肝癌微球体细胞表面特异性靶向多肽的制备方法中,步骤(1)中,贴壁细胞培养具体为:人肝癌细胞株hcclm3用含15%fbs的mem培养基培养,人正常肝细胞lo2用含10%fbs的高糖dmem培养基培养;取对数生长期的hcclm3细胞及lo2细胞,常规消化后按1×106/皿接种于6cm培养皿中,置于37℃、含体积浓度为5%的co2条件下培养至细胞贴壁长满皿底,备用。采用本发明中贴壁细胞培养方法,能长期传代,保持活性,便于监控检测结构功能和生命活动。不同代次细胞可长期保存,既可开展同代次不同条件和方法研究,又可观察不同代次的动态变化。该方法耗资少、经济、成本低、可大量培养,利于生物制品的生产。
前述肝癌微球体细胞表面特异性靶向多肽的制备方法中,所述步骤(1)中,肝癌微球体培养具体为:取5×105个处于对数生长期的hcclm3细胞于超低吸附培养瓶中进行悬浮培养,每7ml培养体系中含有:6776μl的dmem-f12培养基、140μl的b27为、70μl的肝素、终浓度为100μg/ml的hegf和终浓度为50μg/ml的bfgf,细胞置于37℃、体积浓度为5%的co2条件下的培养箱中培养48h,得到肝癌微球体,备用。本发明中肝癌微球体培养体系,即肿瘤干细胞无血清培养体系,提供给肝癌细胞类似于肿瘤干细胞“龛”的生长环境,非肿瘤干细胞在这样的环境中则不能生存。相对于从癌细胞中分选出肿瘤干细胞(微球体),肿瘤干细胞培养体系能保证培养细胞的完整性以及减少被污染的概率。
前述肝癌微球体细胞表面特异性靶向多肽的制备方法中,所述步骤(2)以hcclm3细胞悬浮培养的肝癌微球体为靶细胞,贴壁培养的hcclm3细胞和lo2细胞为吸附细胞,采用噬菌体展示技术进行差减筛选,具体为:
(2.1)取贴壁长满皿底的lo2细胞,pbs洗3次,加入bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用pbst缓冲液洗lo2细胞3次;吸取噬菌体原始肽库溶液并与pbs混匀得到混合液;将该混合液加入贴壁生长的lo2细胞中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;转出上清液至一个新的1.5mlepp管中,备用;
(2.2)取贴壁长满皿底的hccml3细胞,pbs洗3次,加入bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用pbst缓冲液洗贴壁生长的hcclm3细胞3次,加入步骤
(2.1)保留的上清液,继续在37℃、50rpm条件下,恒温培养振荡器中孵育1h,转出上清液至一个新的1.5mlepp管中,备用;
(2.3)将步骤(1)中肝癌微球体培养物吸至10ml离心管中,离心,吸弃陈旧培养基;微球体用pbs洗3次,离心,吸弃pbs;加入bsa,小心吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中封闭30min;随后将混悬液吸至一个新的10ml离心管中,离心,吸弃上清液;用pbst缓冲液洗微球体3次,离心,弃上清液;加入步骤(2.2)保留的上清液,轻轻吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;随后将混悬液吸至一个新的5ml离心管中,离心,吸弃上清液;再用pbst缓冲液洗微球体6次,离心,吸弃上清液;随后在细胞沉淀中加入ph=2.2的甘氨酸-盐酸缓冲液,轻轻吹打混匀后,置于37℃、50rpm条件下,恒温培养振荡器中洗脱10min;随后离心,将上清液转入一个新的1.5mlepp管内,加入ph=9.1的tris-hcl缓冲液进行中和,测定洗脱液中所含噬菌体滴度,随后取该洗脱液进行扩增,扩增后噬菌体通过离心以及peg/nacl沉淀的方法进行纯化,并测定扩增、纯化后噬菌体滴度,完成第一轮筛选;
(2.4)重复以上步骤数次,筛选结束。
进一步地,肝癌微球体细胞表面特异性靶向多肽的制备方法中,步骤(2)以hcclm3细胞悬浮培养的肝癌微球体为靶细胞,贴壁培养的hcclm3细胞和lo2细胞为吸附细胞,采用噬菌体展示技术进行差减筛选,具体为:
(2.1)取贴壁长满皿底的lo2细胞,pbs洗3次,加入1.5ml质量浓度为1%的bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用0.1%~0.5%pbst缓冲液洗lo2细胞3次;吸取噬菌体原始肽库溶液,该溶液滴度为2×1011pfu,并与1.5mlpbs混匀得到混合液;将该混合液加入贴壁生长的lo2细胞中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;转出上清液至一个新的1.5mlepp管中,备用;
(2.2)取贴壁长满皿底的hcclm3细胞,pbs洗3次,加入1.5ml质量浓度为1%的bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用0.1%~0.5%pbst缓冲液洗贴壁生长的hcclm3细胞3次,加入步骤(2.1)保留的上清液,继续在37℃、50rpm条件下,恒温培养振荡器中孵育1h,转出上清液至一个新的1.5mlepp管中,备用;
(2.3)将步骤(1)中肝癌微球体培养物吸至10ml离心管中,1000rpm条件下离心3min,吸弃陈旧培养基;微球体用pbs洗3次,1000rpm条件下离心3min,吸弃pbs;加入1.5ml质量浓度为1%的bsa,小心吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中封闭30min;随后将混悬液吸至一个新的10ml离心管中,1000rpm条件下离心3min,吸弃上清液;用0.1%~0.5%pbst缓冲液洗微球体3次,1000rpm条件下离心3min,弃上清液;加入步骤(2.2)保留的上清液,轻轻吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;随后将混悬液吸至一个新的5ml离心管中,1000rpm条件下离心3min,吸弃上清液;再用0.1%~0.5%pbst缓冲液洗微球体6次,1000rpm条件下离心3min,吸弃上清液;随后在细胞沉淀中加入1ml0.2mol/l的ph=2.2的甘氨酸-盐酸缓冲液,轻轻吹打混匀后,置于37℃、50rpm条件下,恒温培养振荡器中洗脱10min;再在2000rpm条件下离心10min,将上清液转入一个新的1.5mlepp管内,加入150μl1mol/l的ph=9.1的tris-hcl缓冲液进行中和;测定洗脱液中所含噬菌体滴度,随后取该洗脱液100μl感染宿主菌e.colier2738进行扩增,扩增后噬菌体通过离心以及peg/nacl沉淀的方法进行纯化,并测定扩增、纯化后噬菌体滴度,完成第一轮筛选;其中,所用0.1%~0.5%pbst缓冲液是指含tween-20体积浓度为0.1%~0.5%的pbs缓冲液;
(2.4)重复以上步骤,共完成3~5轮筛选,筛选结束。
优选地,肝癌微球体细胞表面特异性靶向多肽的制备方法中,步骤(2)中共完成4轮筛选,其中,第一轮筛选中所用pbst缓冲液为为0.1%pbst缓冲液,即含tween-20体积浓度为0.1%的pbs缓冲液;第二轮筛选中所用pbst缓冲液为0.2%pbst缓冲液,即含tween-20体积浓度为0.2%的pbs缓冲液;第三轮筛选中所用pbst缓冲液为0.3%pbst缓冲液,即含tween-20体积浓度为0.3%的pbs缓冲液;第四轮筛选中所用pbst缓冲液为0.5%pbst缓冲液,即含tween-20体积浓度为0.5%的pbs缓冲液;第四轮筛选时洗脱液进行滴度测定后不再进行扩增纯化。
前述肝癌微球体细胞表面特异性靶向多肽的制备方法中,步骤(3)经dna测序确定目标多肽,具体为:取最后一轮筛选后的噬菌体洗脱液铺于x-gal/lb平板上检测滴度,待噬菌斑长出后,随机挑取若干个噬菌斑进行扩增及纯化,将噬菌体单克隆分别与肝癌微球体、hcclm3细胞和lo2细胞结合,计算噬菌体单克隆的特异性,选取与肝癌微球体特异性结合最高的噬菌体,随后进行测序,其所展示的多肽即为目标多肽。
肝癌微球体细胞表面特异性靶向多肽在制备肝癌癌栓诊断试剂或肝癌靶向治疗药物中的应用。
为确保本发明技术方案的科学、有效,发明人进行了一系列的实验。以在环七肽库中筛选为例,具体如下所示。
1、材料与方法
1.1、材料
1.1.1主要仪器及设备
激光共聚焦扫描显微镜:德国zeiss公司,型号是lsm710;co2恒温培养箱:美国thermo公司,型号是model310;生物安全柜:新加坡esco公司,型号是ac2-4s1;多功能酶标仪:美国biotech,型号是synergy;细胞培养瓶:美国corning公司;恒温水浴箱:杭州蓝天化验仪器厂,型号是zd-420;微量可调加样器:德国eppendorf公司;超纯水机:四川沃特尔水处理设备有限公司,型号是sc082628;恒温培养振荡器:上海智城分析有限公司,型号是zhwy-103d;手提式压力蒸气灭菌器:上海博迅实业有限公司医疗设备厂;台式高速冷冻离心机:美国beckman公司,型号是allegra64rcentrifuge;全自动制冰机:常熟市雪科电器有限公司,型号是ims-40;超低温冰箱:中国haier公司,型号是dw-86l626;电子天平:德国satourious公司,型号是bs-124s;磁力搅拌器:德国ika公司,型号是rh-basic-1;漩涡混合器:德国ika公司,型号是ms-3b-s25;倒置显微镜:德国leica公司,型号是dm1lled;ph计:德国satourious公司,型号是pb-10。
1.1.2、微生物试验所用试剂配方:
含15%fbs的mem培养基:按照体积分数计,含有15%胎牛血清(购自gibco公司)和1%双抗(链霉素+青霉素,购自哈药集团),余量为dmem培养基混合液(购自hyclone公司)。用于培养人肝癌细胞株hcclm3。
含10%fbs的高糖dmem培养基:按照体积分数计,含有10%胎牛血清(购自gibco公司)和1%双抗(链霉素+青霉素,购自哈药集团),余量为dmem培养基混合液(购自hyclone公司)。用于培养人正常肝细胞lo2。
lb培养基:每升含:10gbacto-tryptone(胰蛋白胨),5gyeastextract(酵母提取物),5gnacl。高压灭菌,室温贮存。
iptg/xgal配方:将1.25giptg(isopropylβ–d-thiogalactoside)和1gxgal(5-bromo-4-chloro-3-indolyl-β-d-galactoside)溶于25ml二甲基甲酰胺中。溶液-20℃避光贮存。
lb/iptg/xgal平板:lb培养基+15g/l琼脂粉。高压灭菌,冷却至低于70℃时,加入1mliptg/xgal,混匀倒平板。平板4℃避光贮存。
顶层琼脂:每升含:10gbacto-tryptone,5gyeastextract,5gnacl,1gmgcl2·6h2o和7g琼脂粉。高压灭菌,分成50ml等份。固体培养基室温贮存,用时微波炉融化。
四环素贮液:以20mg/ml的浓度溶于乙醇中。-20℃避光贮存。用前摇匀。
lb-tet平板:lb培养基+15g/l琼脂粉。高压灭菌,冷却至低于70℃时,加入1ml四环素贮液,混匀倒平板。平板4℃避光贮存。
tbs:溶液中含有50mmol/ltris-hcl(ph=7.5)和150mmol/lnacl。高压灭菌,室温贮存。
peg/nacl:溶液中含有20%(w/v)peg-8000和2.5mol/lnacl。高压灭菌,室温贮存。
1.1.3、细胞试验所用试剂配方:
pbs溶液:1l规格的pbs粉剂,溶于1l超纯水中,溶解完全后,0.22μm微孔滤膜过滤除菌和杂质,移至高温灭菌过的广口瓶中,高压灭菌30min,4℃保存。
质量浓度为0.25%的胰酶消化液:称取0.5g的胰酶溶解于200ml的pbs溶液中(在冰浴中进行),搅拌溶解完全后,0.22μm的微孔滤膜过滤除菌,分装后,-20℃保存。
细胞冻存液:取dmem/mem培养基170ml、dmso10ml和gibco的胎牛血清20ml,混匀完全后分装,-20℃保存。
hegf:称取10μghegf粉末溶解于100μl无菌超纯水中,配成浓度为100mg/l的hegf溶液,0.22μm的微孔滤膜过滤后,分装,-20℃保存。
bfgf:称取50μgbfgf粉末溶解于200μl无菌超纯水中,配成浓度为250mg/l的bfgf溶液,0.22μm的微孔滤膜过滤后,分装,-20℃保存。
2、实验方法
2.1、贴壁细胞及微球体的培养
2.1.1、贴壁细胞的传代、培养、冻存和复苏
(a)传代和培养:人肝癌细胞株hcclm3用含15%fbs的mem培养基培养,人正常肝细胞lo2用含10%fbs的dmem(高糖)培养基培养,取对数生长期的hcclm3细胞及lo2细胞,常规消化后按1×106/皿接种于6cm培养皿中,置于37℃、含体积浓度为5%的co2条件下培养至细胞贴壁长满皿底,备用。
(b)细胞冻存:取对数生长期的细胞,弃瓶内培养基,pbs洗瓶3遍,向瓶中加入1ml质量浓度为0.25%的胰酶消化液于显微镜下观察:当细胞胞质回缩、细胞间隙增大、细胞变圆变亮时,加入2ml含血清的完全培养基终止消化,小心将细胞从壁上吹打下来并转移至新鲜10ml离心管中,1000rpm条件下离心5min,之后弃上清,加入冻存液轻轻吹打细胞使其重悬,将重悬液移入冻存管中。将冻存管放入4℃冰箱中放置30min,-20℃放置1h,之后放入-80℃冰箱24h,最后放于液氮中长期保存。
(c)细胞的复苏:从液氮中取出细胞冻存管,快速将细胞冻存管浸入37℃的水浴箱中,摇动使其融化;细胞冻存液融化后,快速从37℃水浴箱中取出细胞冻存管,用75%的酒精喷拭;在超净台内打开细胞冻存管盖子,用移液器吸出细胞悬液,加到10ml的无菌离心管中;1000rpm条件下离心5min;弃上清,加入新鲜含血清培养基重悬细胞,轻轻吹打混匀,将细胞混悬液分装移入25cm2培养瓶(带气孔)中,每瓶补加含血清培养基,使总体积为5ml,继续在37℃、体积浓度为5%的co2条件下的培养箱中进行培养;培养6h后换液。
2.1.2、微球体的培养和换液
(a)微球体的培养:取5×105个处于对数生长期的hcclm3细胞于超低吸附培养瓶中进行悬浮培养,培养体系如下:dmem-f12培养基为6776μl、b27为140μl、肝素为70μl、hegf为7μl(终浓度:100μg/ml)和bfgf为7μl(终浓度:50μg/ml),细胞置于37℃、体积浓度为5%的co2条件下的培养箱中进行培养48h,得到肝癌微球体,并观察和记录微球体生长情况。
(b)微球体的换液:将超低吸附培养瓶竖立放置并以瓶高的棱角为接触面在桌面呈45°角倾斜。待绝大多数细胞下沉后,用移液器小心吸弃超低吸附培养瓶中的部分陈旧培养基,随后再加入步骤(a)中所用培养体系,使总体积保持在7ml,继续在37℃、体积浓度为5%的co2条件下培养箱中进行培养。
2.2、噬菌体随机肽库体外快速差减筛选
2.2.1、第1轮筛选:
(a)取贴壁长满皿底的lo2细胞,pbs洗3次,加入1.5ml质量浓度为1%的bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min;0.1%pbst缓冲液洗lo2细胞3次,吸取噬菌体环七肽库溶液,该溶液滴度为2×1011pfu,并与1.5mlpbs混匀,得到混合液;将该混合液加入贴壁生长的lo2细胞中在37℃、50rpm条件下,恒温培养振荡器中孵育1h;转出上清液至一个新的1.5mlepp管中,待下一步骤使用;
(b)取贴壁长满皿底的hccml3细胞,pbs洗3次,加入1.5ml质量浓度为1%的bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min;随后用0.1%pbst缓冲液洗贴壁生长的hcclm3细胞3次,加入上一步骤保留的上清液;继续在37℃、50rpm条件下,恒温培养振荡器中孵育1h;转出上清液至一个新的1.5mlepp管中,保留至下一步骤使用;
(c)将肝癌微球体培养物吸至10ml离心管中,1000rpm条件下离心3min;吸弃陈旧培养基,微球体用pbs洗3次,1000rpm条件下离心3min;吸弃pbs,加入1.5ml质量浓度为1%的bsa,小心吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中封闭30min;将混悬液吸至一个新的10ml离心管中,1000rpm条件下离心3min;吸弃上清液,0.1%pbst缓冲液洗微球体3次,1000rpm条件下离心3min;弃上清液,加入上一步骤保留的上清液,轻轻吹打混匀后,将混悬液吸至一个新的6cm培养皿中;在37℃、50rpm条件下,恒温培养振荡器中孵育1h;将混悬液吸至一个新的5ml离心管中,1000rpm条件下离心3min;吸弃上清液,0.1%pbst缓冲液洗微球体6次,1000rpm条件下离心3min;吸弃上清液,细胞沉淀中加入1ml0.2mol/l的甘氨酸-盐酸缓冲液(ph=2.2),轻轻吹打混匀后,置于37℃、50rpm条件下,恒温培养振荡器中洗脱10min;2000rpm条件下离心10min,将上清液转入一个新的1.5mlepp管内,加入150μl1mol/l的tris-hcl(ph=9.1)进行中和;取10μl洗脱液进行滴度测定,剩余洗脱液取100μl扩增并测滴度,同时准备下一轮筛选所需细胞。
2.2.2、第2-4轮筛选:
方法同第1轮,每一轮噬菌体洗脱液扩增后,均取吸取噬菌体环七肽库溶液,溶液滴度为2×1011pfu,作为下一轮筛选的初始投入。2-4轮所用的pbst缓冲液按0.2%、0.3%、0.5%的梯度浓度逐轮递增。第4轮筛选完成后,收集到的噬菌体洗脱液不再扩增,测滴度后,随机挑选噬菌体单克隆进行测序。所用0.1%~0.5%pbst缓冲液缓冲液是指含tween-20体积浓度为0.1%~0.5%的pbs缓冲液。
2.2.3、噬菌体的扩增和纯化:
取50μler2738大肠杆菌接种至盛有30mllb培养基的250ml锥形瓶中,在200rpm、37℃条件下摇至对数期前期(od600=0.2)。加入待扩增的噬菌体样品(噬菌体洗脱液或x-gal/iptglb平板上挑取的噬菌体单克隆),在200rpm、37℃条件下培养5h。将混合培养物转移至50ml离心管中,12000rpm条件下离心15min,吸取80%上清转移至50ml无菌离心管中并加入1/5体积的peg/nacl溶液,放置于4℃预沉淀。将预沉淀物按12000rpm于4℃离心10min,弃上清,用1mltbs溶液(4℃)重悬沉淀并转移至1.5ml离心管中,加入200μlpeg/nacl后置于4℃沉淀1h。将沉淀物以12000rpm于4℃离心10min,弃上清用200μltbs(4℃)重悬沉淀,10000rpm于4℃离心5min,吸取上清至1.5ml无菌离心管中,加入等体积甘油混匀于-20℃存放。
2.2.4、噬菌体滴度测定:
取10μler2738大肠杆菌接种至盛有10mllb培养基的100ml锥形瓶中,在200rpm、37℃条件下摇至对数期中期(od600=0.5-0.6之间),保留至下一步骤使用。将噬菌体样品制备成10倍系列稀释溶液(洗脱液稀释倍数范围:102-104,纯化液稀释倍数范围:108-1010)。取10μl噬菌体样品稀释液加入盛有200μl大肠杆菌(对数中期)1.5ml离心管中,混匀于室温孵育2min后,迅速将上述混合物加入盛有3ml顶层琼脂(45℃)10ml离心管中并混匀,将混合溶液迅速倒至x-gal/iptglb平板上,琼脂冷却凝固后,将平板置于37℃培养箱倒置避光培养过夜。隔天观察平板对噬菌体克隆进行计数。
2.2.5、第4轮噬菌体洗脱液挑单菌落测序
(a)第4轮筛选的噬菌体洗脱液铺于x-gal/lb平板上检测滴度,待噬菌斑长出后,随机挑取36个噬菌斑进行扩增及纯化。纯化后的噬菌体样品按挑斑顺序编为p1-p36号,委托江苏金唯智生物技术有限公司提取噬菌体基因组,以-96giii测序引物5’-hoccctcaagttagcgtaacg-3’对噬菌体基因组采用sanger双脱氧链终止法(sangerchainterminationmethod)进行测序。
(b)得到测序结果后,查找knpi(ggtacc)和eagi(cggccg)两处酶切位点,噬菌体随机肽库的外源嵌入基因片段就位于两个酶切位点之间,即5’-(knpi)ggtacctttctattctcactctgctcgaaca(nnm)7acgccacctccatcggccg(eagi)-3’,加粗字体序列(cgaaca(nnm)7acgccacctcca)即为外源嵌入的基因片段,该片段中(nnm)7即为编码环七肽的碱基序列,(nnm)7两端aca和acg是编码环七肽两侧相互连接成环半胱氨酸的密码子。
2.3、cell-elisa检测阳性噬菌体克隆
(a)取对数期hcclm3细胞,按1×105/孔的密度铺于96孔酶标板,使用微球体培养体系在37℃培养24h,取hcclm3和lo2细胞按相同密度使用贴壁细胞培养体系在37℃培养24h;
(b)吸弃培养基,使用预热的pbs洗涤5min×3次,加入4%多聚甲醛固定30min(室温),弃甲醛,用pbs洗涤5min×3次;
(c)每孔加入100μl3%过氧化氢溶液后置入恒温培养箱,37℃孵育30min,pbs洗3次,每次5min;
(d)用质量浓度为1%的bsa封闭30min(室温);
(e)吸弃封闭液,每孔加入1×109pfu噬菌体溶液,37℃孵育1h;
(f)0.1%pbst缓冲液洗3次,每次5min,每孔加入50μl稀释的hrp标记的抗m13单克隆抗体溶液(稀释比例为1:5000),37℃孵育1h;
(g)吸弃抗体溶液,0.1%pbst缓冲液洗3次,每次5min;每孔加入50μltmb显色液在室温显色30min(避光);
(h)每孔加入50μl2mol/lh2so4终止反应,用酶标仪测定490nm波长下的od值,计算各组细胞od490nm的均值和噬菌体克隆与细胞结合特异性(selectivity),如公式(1.1)所示。
其中,ods1代表微球体组的吸光度值,ods2代表贴壁细胞hcclm3组的吸光度值,ods3代表贴壁细胞lo2组的吸光度值,odc1代表微球体阴性对照组的吸光度值;odc2代表hcclm3阴性对照组的吸光度;odc3代表lo2阴性对照组的吸光度。
2.4、多肽合成
根据elisa试验结果,选取特异性最高的噬菌体克隆,依照测序结果读取该噬菌体克隆编码环七肽的基序,将编码环七肽的基序翻译成氨基酸序列并在两末端各加上一个半胱氨酸残基,按照翻译出来的氨基酸残基序列委托吉尔生化(上海)有限公司进行多肽合成,两端的半胱氨酸以二硫键连接形成环七肽,以七肽前三个氨基酸残基对其命名为xxx肽。如序列为ntgspye的多肽,两端各加一个半胱氨酸残基后进行合成并使两端的半胱氨酸残基以二硫键连接,最终合成环七肽,命名为ntg肽。
2.5、竞争抑制试验
(a)取对数期hcclm3细胞,按1×105/孔的密度铺于96孔酶标板,使用微球体培养体系在37℃、体积浓度为5%co2条件下培养48h,获得hcclm3微球体;
(b)取对数生长期hcclm3和lo2细胞,按1×105/孔的密度铺于96孔酶标板,使用贴壁细胞培养体系在37℃培养、体积浓度为5%co2条件下培养至细胞贴壁长满孔底;
(c)吸弃96孔酶标板中培养基,使用预热的pbs洗3次,每次5min,加入4%多聚甲醛固定30min(室温),弃甲醛,用pbs洗3次,每次5min;
(d)每孔加入100μl3%过氧化氢溶液后置入恒温培养箱,37℃孵育30min,pbs洗3次,每次5min;
(e)用质量浓度为1%的bsa封闭30min(室温);弃封闭液,pbs洗3遍;
(f)分别以0.1μmol/l、1μmol/l、10μmol/l、100μmol/l、1000μmol/l不同浓度的多肽溶液加入各个孔中,37℃孵育1h;
(g)0.1%pbst缓冲液洗3次,每次5min,每个孔分别加入与阳性多肽对应的噬菌体肽109pfu,37℃孵育1h;
(h)0.1%pbst缓冲液洗3次,每次5min,每孔加入50μl稀释的hrp标记的抗m13单克隆抗体溶液(稀释比例为1:5000),37℃孵育1h;
(i)吸弃抗体溶液,0.1%pbst缓冲液洗3次,每次5min;每孔加入50μltmb显色液在室温显色30min(避光);
(j)每孔加入50μl2mol/lh2so4终止反应,用酶标仪测定450nm波长下的od值。
(k)记录各组细胞的od450nm值并按照下列公式(公式(1.2))计算多肽对噬菌体肽与细胞结合的抑制率(inhibitionrate),
其中,odc代表未加多肽的所测的od值,odp代表加入多肽的各组所测的od值。
2.6、细胞免疫荧光试验
2.6.1、贴壁细胞步骤:
(a)包被:多聚赖氨酸包被激光共聚焦专用玻底皿,放于培养箱中过夜;
(b)包被好的玻底皿用pbs洗2次,每次2min,之后用培养液浸洗皿洗2次,每次2min;
(c)分别取对数生长期的hcclm3细胞、lo2细胞、人肝癌细胞系(hcclm6细胞、mhcc97h细胞、mhcc97l细胞)、人乳腺癌细胞系mda-mb-468细胞、小鼠乳腺癌细胞系4t1,常规消化后按密度为3×105/ml接种于玻底皿中,放于37℃、体积浓度为5%co2的培养箱中,培养至细胞密度为70%左右;
(d)预热的pbs洗2遍,加入1ml体积浓度为4%的多聚甲醛(室温),室温固定1h,pbs洗3遍;
(e)加入1ml质量浓度为1%的bsa于37℃封闭30min,pbs洗3遍;
(f)加入浓度为5μmol/l的fitc基团标记多肽500μl,37℃结合1h;
(g)用含tritonx-100体积浓度为0.2%的pbs洗3次,每次5min;
(h)加入100μldapi染核,室温孵育30min(避光);
(i)回收dapi,pbs洗3遍;
(j)加入1mlpbs,在激光共聚焦显微镜下检测fitc和dapi信号。
2.6.2、悬浮细胞步骤:
(a)包被:将鼠尾胶用1%的冰醋酸稀释至10%浓度,每个玻底皿加入100ul鼠尾胶,将皿盖打开放入培养箱中一小时待液体蒸发;
(b)包被好的玻底皿用pbs洗2次,每次2min,之后用培养液浸洗皿洗2次,每次2min;
(c)取对数生长期的hccml3细胞、hcclm6细胞、mhcc97h细胞、4t1细胞按微球体培养体系培养成相应的微球体;
(d)用巴氏滴管小心吸取1ml左右微球体悬液加入玻底皿中,静置2min,使微球体粘附于鼠尾胶上,用200ul枪头小心吸去部分上层液体,加入1ml体积浓度为4%的多聚甲醛(室温),室温固定1h。
(e)离心:将玻底皿放入六孔板中,离心(1500r/min,15min),pbs洗3遍;
(f)加入1ml质量浓度为1%的bsa于37℃封闭30min,pbs洗3遍;
(g)加入浓度为5μmol/l的fitc基团标记多肽500μl,37℃结合1h;
(h)0.1%pbst缓冲液洗3次,每次5min;
(i)加入100μldapi染核,室温孵育30min(避光);
(j)回收dapi,pbs洗3遍;
(k)加入1mlpbs,在激光共聚焦显微镜下检测fitc和dapi信号。
2.6.3、细胞增殖试验
(a)取对数期hcclm3细胞,按1×105/孔的密度铺于超低吸附96孔酶标板,使用微球体培养体系进行培养,取hcclm3和lo2细胞进行贴壁培养,以1×105/孔的密度铺于96孔酶标板,以上三种细胞均在37℃、体积浓度为5%的co2条件下培养;
(b)向微球体体系和贴壁细胞中分别加入1mmol/l、10mmol/l、100mmol/l不同浓度的ntg肽溶液50μl后,进行连续培养;
(c)当微球体培养物和贴壁细胞培养至0、12、24、48h时,将wst-1溶液分别加入各个时间段的微球体培养体系和贴壁细胞中,以无多肽溶液的微球体培养物和贴壁细胞为阴性对照,将培养基与wst-1试剂的混合溶液作为空白对照;
(d)在37℃、体积浓度为5%的co2条件下孵育1h后,使用酶标仪检测450nm处每孔的od值;
(e)按以下公式(公式(1.3))计算各组微球体细胞和贴壁细胞的存活率(survivalrate):
其中,od1代表实验组的od值,od2代表阴性对照组的od值,od3代表空白对照组的od值。
3、结果
3.1、无血清悬浮培养使hcclm3形成微球体
取处于对数生长期的hcclm3细胞于超低吸附培养瓶中进行悬浮培养,干细胞培养体系如下:dmem-f12培养基为6776μl、b27为140μl、肝素为70μl、hegf为7μl(终浓度:100μg/ml)和bfgf为7μl(终浓度:50μg/ml)。观察hcclm3细胞在球体形成试验的生长情况发现:无血清干细胞悬浮培养体系能促进hcclm3细胞在体外形成微球体(见图1)。
3.2、噬菌体随机肽库差减筛选在肝癌微球体上富集率
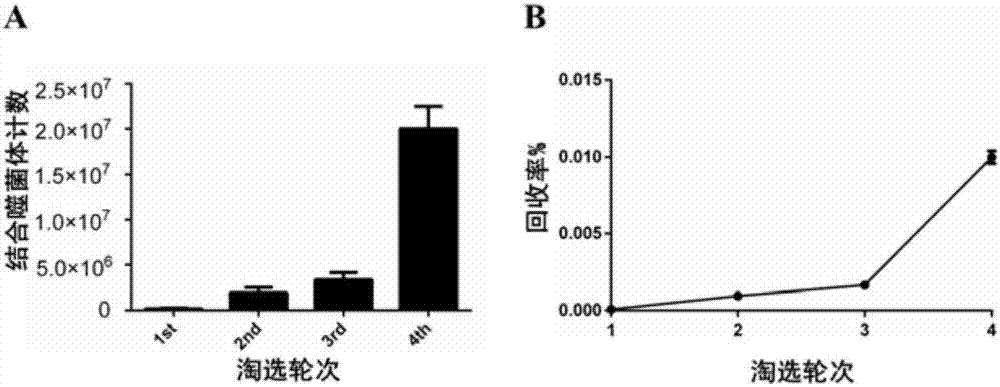
以hcclm3细胞悬浮培养的肝癌微球体为靶标细胞,贴壁培养的hcclm3和lo2作为吸附细胞,在噬菌体环七肽库中进行4轮差减筛选,记录肝癌微球体噬菌体结合量以及富集率。如表1所示,其中噬菌体克隆回收量从第一轮的1.67×105±3.30×104(pfu)升至第四轮的2.0×107±8.16×105(pfu)(p<0.001),回收量提高明显;如图2-a所示,回收率从第一轮的8.50×10-5±1.70×10-5(%)明显提高至第四轮的1.0×10-2±4.08×10-4(%)(p<0.001)(见图2-b),说明了噬菌体肽库中能与肝癌微球体结合的噬菌体克隆数量在第4轮时呈现明显的富集现象,提高了获取高亲和力噬菌体的几率。
表1体外筛选噬菌体克隆的回收量和富集率
3.3、第4轮洗脱液的噬菌体克隆测序
从第4轮筛选后的噬菌体洗脱中,随机挑取36个单克隆送至金唯智(江苏)生物有限公司进行测序,根据测序峰值图在knpi和eagi位点之间查找编码环七肽的基序,即5’-(knpi)ggtacctttctattctcactctgctgcttgt(nnk)7tgcggtggaggtccatcggccg(eagi)-3’(见图3),该序列中(nnk)7即为编码环七肽的基序,该基序两侧的tgt、tgc密码子是编码环七肽成环的半胱氨酸。按噬菌体遗传密码表对编码多肽基序(nnk)7进行翻译(见图4),翻译结果如表2所示。
表2所筛选噬菌体克隆展示肽的序列
3.4、cell-elisa鉴定阳性噬菌体克隆
将第4轮差减筛选的噬菌体洗脱液铺于x-gal/lb平板上,待噬菌斑长出后,随机从平板上挑取36个单克隆扩增并纯化,将36个噬菌体单克隆分别与肝癌微球体、hcclm3和lo2细胞结合,洗去未结合的噬菌体后,加入抗m13抗体(标记hrp),最后加入显色底物后在酶标仪下检测od值,以od值大小来衡量各个单克隆噬菌体分别与上述三种细胞的结合能力。如图5所示,用公式1.1分别计算噬菌体单克隆的特异性。数据表明,p26号噬菌体在所有挑选的单克隆中特异性是最高的。如图6所示。因此,分析p26号噬菌体dna编码展示多肽的基序,将其翻译成氨基酸序列并进行下一步验证。
3.5、多肽的合成
委托吉尔生化(上海)生物公司进行阳性肽ntg和乱序对照肽qmv的合成。选择p26号阳性噬菌体,在其展示肽序列(ntgspye)两端各连接一个半胱氨酸,之后再将两端的半胱氨酸残基进行连接,形成环七肽(见图7a)。合成对照肽qmv(序列为:qmvqdfr)并按上述方法进行环化(见图7b)。多肽经纯化后,纯度为95%以上(见图8、9)。此外,用fitc分别对ntg肽和qmv肽进行标记,标记荧光的多肽纯度均大于95%(见图10、11)。
3.6、竞争抑制试验初步证明合成多肽与靶细胞结合的独立性
运用竞争抑制试验来检测ntg肽对p26噬菌体与靶细胞结合的抑制作用。预先将ntg肽以不同的浓度分别与肝癌微球体细胞结合,洗去未结合多肽后,加入p26噬菌体竞争结合,洗去未结合的噬菌体,用抗m13抗体(标记hrp)检测各组细胞中噬菌体结合的量并计算多肽对噬菌体的抑制率。结果显示:ntg肽对p26噬菌体与微球体的结合有抑制作用并随浓度的升高抑制率逐渐增高,而对照肽qmv则无此效应。(**p<0.01或***p<0.001)(见图12)该试验说明了ntg肽在脱离噬菌体载体后,仍具有与肝癌干细胞特异性结合的能力。
3.7、细胞免疫荧光试验检测多肽的亲和力和特异性
运用激光共聚焦显微镜观察ntg肽与细胞结合的情况,进一步验证ntg肽与肝癌微球体结合的亲和力和特异性。用fitc绿色荧光基团分别标记阳性多肽ntg及阴性对照肽qmv(记作ntg-fitc和qmv-fitc),单独使用fitc染料与细胞结合作为阴性对照并以此作为背影荧光强度(非特异性结合)。使用细胞核染料dapi(蓝色荧光)对待检测的细胞核进行染核,运用激光共聚焦显微镜观察ntg肽与靶细胞(hcclm3微球体)结合的亲和力,并同时检测ntg阳性多肽与吸附细胞(贴壁培养的hcclm3细胞、lo2细胞)、贴壁培养的人肝癌细胞系(hcclm6细胞、mhcc97h细胞、mhcc97l细胞)、贴壁培养的人乳腺癌细胞系mda-mb-468细胞和贴壁培养的小鼠乳腺癌细胞系4t1细胞,以及检测ntg阳性多肽与各种悬浮培养的微球体(hcclm6spheres、mhcc97hspheres、4t1spheres)的结合的情况,以此来进一步验证ntg肽与肝癌微球体结合的亲和力和特异性。
检测ntg肽与靶细胞(hcclm3微球体)结合的情况,结果表明:单独使用fitc荧光染料与靶细胞结合,结果检测到有微弱的fitc荧光信号,以此作为背景荧光信号(见图13)。标记fitc的ntg肽与hcclm3微球体结合时,检测到高强度的fitc荧光信号,结果呈阳性(见图13、14、15-a);用fitc标记的对照肽qmv与靶细胞结合中检测到的fitc荧光信号强度同背景荧光强度,结果呈阴性,(见图13);说明ntg肽具有与hcclm3微球体结合的能力。
检测ntg肽与吸附细胞(贴壁培养的hcclm3细胞、lo2细胞)结合的情况,结果显示:fitc-ntg肽分别与贴壁培养的hcclm3细胞及lo2细胞结合时,检测到微弱fitc荧光信号,结果呈阴性(图14),表明了ntg肽不与贴壁培养的hcclm3细胞及lo2细胞结合。为了进一步验证ntg肽的特异性,观察ntg肽与贴壁培养的人肝癌细胞系hcclm6细胞、mhcc97h细胞、mhcc97l细胞、人乳腺癌细胞系mda-mb-468细胞及小鼠乳腺癌细胞系4t1细胞的结合情况,结果显示:用fitc标记的ntg肽分别与贴壁培养的hcclm6细胞、mhcc97h细胞、mhcc97l细胞、mda-mb-468细胞及4t1细胞结合时,检测到微弱绿色荧光信号,结果呈阴性(图15-b、15-c、15-d、15-e、15-f),说明ntg肽与无关细胞不结合。
此外,检测ntg与其它微球体结合的结果表明:在与hcclm6微球体的结合也显示为阳性(见图16),但与mhcc97h微球体和4t1微球体结合呈阴性(见图16)。表明:ntg肽除了能与hcclm3微球体结合外,还具有与hcclm6微球体结合的能力,与mhcc97h微球体和4t1微球体不结合。hcclm6和hcclm3均为具有相对较高转移能力的肝癌细胞系,体外培养的hcclm6微球体及hcclm3微球体具有肝癌干细胞及肝癌癌栓的特征,表明ntg肽可能具有与高转移肝癌细胞形成的癌栓特异性结合的能力。
计算ntg肽与细胞结合的相对荧光强度(fitc/dapi)的结果见图17。
细胞免疫荧光试验检测的结果表明ntg肽与肝癌微球体的结合具有高亲和力和高特异性。
3.8、检测ntg肽对细胞生长的影响
分别向hcclm3微球体细胞、hcclm3细胞和lo2细胞中,加入不同浓度的ntg肽(1μmol/l、10μmol/l、100μmol/l),之后在0h、12h、24h和48h时,每孔分别加入等量wst-1试剂,反应一段时间后在酶标仪下读取每组的od值,以不加多肽的细胞作为对照组,分别计算各个时段的各组细胞的存活率。结果显示:在三种细胞中,不同浓度给药组在各个时段的存活率均无明显差异(p>0.05)(见图18)。初步表明了ntg肽对细胞的生长无影响,是潜在的良好的靶向药物载体
3.9、统计学处理
采用统计软件对数据进行处理,计量资料用均数±标准差(x±s表示)。若方差齐性时,整体比较采用one-wayanova,多组之间的两两比较采用lsd检验;若方差不齐,整体比较用welch方法,以为p<0.05差异有统计学意义。
3.10、讨论
本发明为了筛选到肝癌微球体细胞表面特异性靶向多肽,将肝癌hcclm3细胞系悬浮培养获得的肝癌微球体作为靶细胞,以hcclm3和lo2为吸附细胞,采用噬菌体展示技术进行差减筛选。发明人以在环七肽库中筛选为例,使用噬菌体环七肽库进行了四轮淘选后,观察结果发现第四轮微球体结合的噬菌体回收率升高明显,之后在第四轮噬菌体洗脱液中随机挑选36个单克隆进行测序。根据噬菌体测序的结果,去除空载体后,运用elisa对各个噬菌体单克隆与靶细胞和吸附细胞结合的亲和力进行初步的检测,同时根据各个噬菌体克隆的亲和力来计算噬菌体克隆与肝癌微球体结合的特异性。结果显示p26号噬菌体具有最高的特异性,将其作为候选噬菌体。为了验证p26号噬菌体与肝癌微球体的结合是依靠展示在噬菌体衣壳蛋白的多肽序列的特异性,而与噬菌体载体无关,发明人进行了接下来的试验:分析p26号噬菌体编码环七肽的基序,翻译并在体外合成并纯化,以前三个氨基酸残基的缩写将其命名为ntg肽,运用竞争抑制试验证明了ntg肽在脱离了噬菌体载体的情况下,依然能够和肝癌微球体进行结合。在细胞免疫荧光进一步检测ntg肽与肝癌微球体结合的特异性的实验中,结果显示:ntg肽只与肝癌微球体的结合具有较高的特异性,不与吸附细胞及其它无关细胞结合。细胞增值率试验检测ntg肽对于肝癌微球体及吸附细胞的生长影响,结果显示:ntg肽对细胞的生长无影响。
运用噬菌体展示技术,通过差减筛选并经elisa验证得到的阳性噬菌体克隆以及ntg肽,与靶细胞肝癌微球体具有高特异性,该方法是获取与选定靶细胞高特异性结合多肽的高效且简便的方法。
综上所述,通过噬菌体展示筛选得到ntg肽,与靶细胞肝癌微球体的结合具有高亲和力和高特异性,初步实验表明ntg肽无生物学活性,在作为诊断试剂和靶向药物的载体的应用上具有良好前景。
4、结论
hcclm3肝癌细胞系经干细胞悬浮体系培养可快速且稳定地形成肝癌微球体。筛选到的p26号噬菌体与肝癌微球体的结合具有最高特异性,其展示的环七肽序列为ntgspye。合成的ntg肽具有与肝癌微球体靶向结合的能力,且对细胞的生长无影响,是良好的载体。因此,ntg肽作为介导与肝癌干细胞特异性结合的靶向载体,具有高潜力的应用前景。
本发明的有益之处在于:本发明提供的一种肝癌微球体细胞表面特异性靶向多肽,能够与肝癌微球体细胞进行特异性结合,与肝癌微球体靶向结合的能力强,具有高亲和力和高特异性,且对细胞的生长无影响。本发明的制备方法是以hcclm3细胞悬浮培养的肝癌微球体为靶细胞,贴壁培养的hcclm3细胞和lo2细胞为吸附细胞,采用噬菌体展示技术进行差减筛选,并最终得到一种与肝癌微球体细胞结合且具有高度特异性的多肽。本发明制备方法中库容量大,筛选成功率高,操作简单,成本低。本发明所筛选到的多肽可作为研究转移性肝癌及其癌栓的生物试剂原料,可用于制备肝癌癌栓诊断试剂或肝癌靶向治疗药物中,可以为肝癌的癌栓诊断和靶向治疗提供新的有效手段。
附图说明
图1是本发明的无血清悬浮培养体系促进lm3细胞形成微球体结果图(×400);
图2是体外筛选噬菌体克隆的回收量和富集率结果图;
图3是噬菌体测序峰值图;
图4是噬菌体的遗传密码图;
图5是各个噬菌体克隆与不同细胞的亲和力测试结果图;
图6是噬菌体克隆与微球体结合测试结果图;
图7是多肽的结构图;
图8是合成多肽的高效液相色谱图;
图9是合成多肽的质谱检测图;
图10是标记fitc荧光基团的多肽高效液相色谱图;
图11是标记fitc荧光基团的质谱检测图;
图12是ntg肽对p26噬菌体与肝癌微球体结合的竞争抑制效应图;
图13是激光共聚焦显微镜检测标记fitc的ntg肽与肝癌微球体结合的亲和力和特异性结果图;
图14是ntg肽与吸附细胞结合情况的激光共聚焦显微镜图;
图15是ntg肽与无关细胞结合情况的激光共聚焦显微镜图;
图16是ntg与其它微球体结合情况的激光共聚焦显微镜图;
图17是ntg肽与细胞结合的相对荧光强度结果图;
图18是ntg肽对细胞存活率的影响图;
图中附图标记的含义:图2:a-噬菌体富集率;b-噬菌体回收量;图5:a-微球体,b-hcclm3细胞,c-lo2细胞;图7:a-ntg结构;b-qmv结构;图12:1-ntg肽,2-对照肽qmv;图17:a-ntg-fitc,b-fitc;图18:a-ntg肽对微球体存活率的影响,b-ntg肽对hcclm3细胞存活率的影响,c-ntg肽对lo2细胞存活率的影响,1-对照组,2-1μmol/l、3-10μmol/l、4-100μmol/l。
具体实施方式
以下结合具体实施例对本发明作进一步的介绍。
实施例1肝癌微球体细胞表面特异性靶向多肽,所述多肽为七肽,其氨基酸序列为asn-thr-gly-ser-pro-tyr-glu。
实施例2肝癌微球体细胞表面特异性靶向多肽,为环状的环七肽,其结构式为:cyclo-(asn-thr-gly-ser-pro-tyr-glu)。
实施例3一种肝癌微球体细胞表面特异性靶向多肽的制备方法,包括以下步骤:
(1)贴壁细胞及肝癌微球体的培养:按照常规方法进行肝癌微球体、hcclm3细胞和lo2细胞培养;
(2)以hcclm3细胞悬浮培养的肝癌微球体为靶细胞,贴壁培养的hcclm3细胞和lo2细胞为吸附细胞,采用噬菌体展示技术进行差减筛选,具体为:(2.1)取贴壁长满皿底的lo2细胞,pbs洗3次,加入bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用pbst缓冲液洗lo2细胞3次;吸取噬菌体原始肽库溶液,并与pbs混匀得到混合液;将该混合液加入贴壁生长的lo2细胞中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;转出上清液至一个新的1.5mlepp管中,备用;(2.2)取贴壁长满皿底的hcclm3细胞,pbs洗3次,加入bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用pbst缓冲液洗贴壁生长的hcclm3细胞3次,加入步骤(2.1)保留的上清液,继续在37℃、50rpm条件下,恒温培养振荡器中孵育1h,转出上清液至一个新的1.5mlepp管中,备用;(2.3)将步骤(1)中肝癌微球体培养物吸至10ml离心管中,离心,吸弃陈旧培养基;微球体用pbs洗3次,离心,吸弃pbs;加入bsa,小心吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中封闭30min;随后将混悬液吸至一个新的10ml离心管中,离心,吸弃上清液;用pbst缓冲液洗微球体3次,离心,弃上清液;加入步骤(2.2)保留的上清液,轻轻吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;随后将混悬液吸至一个新的5ml离心管中,离心,吸弃上清液;再用pbst缓冲液洗微球体6次,离心,吸弃上清液;随后在细胞沉淀中加入ph=2.2的甘氨酸-盐酸缓冲液,轻轻吹打混匀后,置于37℃、50rpm条件下,恒温培养振荡器中洗脱10min;随后离心,将上清液转入一个新的1.5mlepp管内,加入ph=9.1的tris-hcl缓冲液进行中和,测定洗脱液中所含噬菌体滴度,随后取该洗脱液进行扩增,扩增后噬菌体通过离心以及peg/nacl沉淀的方法进行纯化,并测定扩增、纯化后噬菌体滴度,完成第一轮筛选:所述pbst缓冲液为含tween-20的pbs缓冲液;(2.4)重复以上步骤数次,筛选结束;
(3)经dna测序确定目标多肽,具体为:取最后一轮筛选后的噬菌体洗脱液铺于x-gal/lb平板上检测滴度,待噬菌斑长出后,随机挑取若干个噬菌斑进行扩增及纯化,将噬菌体单克隆分别与肝癌微球体、hcclm3细胞和lo2细胞结合,计算噬菌体单克隆的特异性,选取与肝癌微球体特异性结合最高的噬菌体,随后进行测序,其所展示的多肽即为目标多肽。
实施例4一种肝癌微球体细胞表面特异性靶向多肽的制备方法,包括以下步骤:
(1)贴壁细胞及肝癌微球体的培养:按照常规方法进行肝癌微球体;贴壁细胞培养具体为:取对数生长期的hcclm3细胞及lo2细胞,常规消化后按1×106/皿接种于6cm培养皿中,置于37℃、含体积浓度为5%的co2条件下培养至细胞贴壁长满皿底,备用;
(2)以hcclm3细胞悬浮培养的肝癌微球体为靶细胞,贴壁培养的hcclm3细胞和lo2细胞为吸附细胞,采用噬菌体展示技术进行差减筛选,具体为:(2.1)取贴壁长满皿底的lo2细胞,pbs洗3次,加入1.5ml质量浓度为1%的bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用0.1%pbst缓冲液洗lo2细胞3次;吸取噬菌体七肽库溶液,该溶液滴度为2×1011pfu,并与1.5mlpbs混匀得到混合液;将该混合液加入贴壁生长的lo2细胞中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;转出上清液至一个新的1.5mlepp管中,备用;(2.2)取贴壁长满皿底的hcclm3细胞,pbs洗3次,加入1.5ml质量浓度为1%的bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用0.1%pbst缓冲液洗贴壁生长的hcclm3细胞3次,加入步骤(2.1)保留的上清液,继续在37℃、50rpm条件下,恒温培养振荡器中孵育1h,转出上清液至一个新的1.5mlepp管中,备用;(2.3)将步骤(1)中肝癌微球体培养物吸至10ml离心管中,1000rpm条件下离心3min,吸弃陈旧培养基;微球体用pbs洗3次,1000rpm条件下离心3min,吸弃pbs;加入1.5ml质量浓度为1%的bsa,小心吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中封闭30min;随后将混悬液吸至一个新的10ml离心管中,1000rpm条件下离心3min,吸弃上清液;用0.1%pbst缓冲液洗微球体3次,1000rpm条件下离心3min,弃上清液;加入步骤(2.2)保留的上清液,轻轻吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;随后将混悬液吸至一个新的5ml离心管中,1000rpm条件下离心3min,吸弃上清液;再用0.1%pbst缓冲液洗微球体6次,1000rpm条件下离心3min,吸弃上清液;随后在细胞沉淀中加入1ml0.2mol/l的ph=2.2的甘氨酸-盐酸缓冲液,轻轻吹打混匀后,置于37℃、50rpm条件下,恒温培养振荡器中洗脱10min;再在2000rpm条件下离心10min,将上清液转入一个新的1.5mlepp管内,加入150μl1mol/l的ph=9.1的tris-hcl缓冲液进行中和;测定洗脱液中所含噬菌体滴度,随后取该洗脱液100μl感染宿主菌e.colier2738进行扩增,扩增后噬菌体通过离心以及peg/nacl沉淀的方法进行纯化,并测定扩增、纯化后噬菌体滴度,完成第一轮筛选;其中,所用0.1%pbst缓冲液是指含tween-20体积浓度为0.1%的pbs缓冲液;(2.4)重复以上步骤,共完成5轮筛选,筛选结束。
(3)经dna测序确定目标多肽,具体为:取最后一轮筛选后的噬菌体洗脱液铺于x-gal/lb平板上检测滴度,待噬菌斑长出后,随机挑取若干个噬菌斑进行扩增及纯化,将噬菌体单克隆分别与肝癌微球体、hcclm3细胞和lo2细胞结合,计算噬菌体单克隆的特异性,选取与肝癌微球体特异性结合最高的噬菌体,随后进行测序,其所展示的多肽即为目标多肽。
实施例5一种肝癌微球体细胞表面特异性靶向多肽的制备方法,包括以下步骤:
(1)贴壁细胞及肝癌微球体的培养:贴壁细胞培养具体为:人肝癌细胞株hcclm3用含15%fbs的mem培养基培养,人正常肝细胞lo2用含10%fbs的高糖dmem培养基培养,取对数生长期的hcclm3细胞及lo2细胞,常规消化后按1×106/皿接种于6cm培养皿中,置于37℃、含体积浓度为5%的co2条件下培养至细胞贴壁长满皿底,备用;
肝癌微球体培养具体为:取5×105个处于对数生长期的hcclm3细胞于超低吸附培养瓶中进行悬浮培养,每7ml培养体系中含有:6776μl的dmem-f12培养基、140μl的b27为、70μl的肝素、终浓度为100μg/ml的hegf和终浓度为50μg/ml的bfgf,细胞置于37℃、体积浓度为5%的co2条件下的培养箱中培养48h,得到肝癌微球体,备用;
(2)以hcclm3细胞悬浮培养的肝癌微球体为靶细胞,贴壁培养的hcclm3细胞和lo2细胞为吸附细胞,采用噬菌体展示技术进行差减筛选,具体为:(2.1)取贴壁长满皿底的lo2细胞,pbs洗3次,加入1.5ml质量浓度为1%的bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用0.5%pbst缓冲液洗lo2细胞3次;吸取噬菌体环七肽库溶液,该溶液滴度为2×1011pfu,并与1.5mlpbs混匀得到混合液;将该混合液加入贴壁生长的lo2细胞中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;转出上清液至一个新的1.5mlepp管中,备用;(2.2)取贴壁长满皿底的hcclm3细胞,pbs洗3次,加入1.5ml质量浓度为1%的bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用0.5%pbst缓冲液洗贴壁生长的hcclm3细胞3次,加入步骤(2.1)保留的上清液,继续在37℃、50rpm条件下,恒温培养振荡器中孵育1h,转出上清液至一个新的1.5mlepp管中,备用;(2.3)将步骤(1)中肝癌微球体培养物吸至10ml离心管中,1000rpm条件下离心3min,吸弃陈旧培养基;微球体用pbs洗3次,1000rpm条件下离心3min,吸弃pbs;加入1.5ml质量浓度为1%的bsa,小心吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中封闭30min;随后将混悬液吸至一个新的10ml离心管中,1000rpm条件下离心3min,吸弃上清液;用0.5%pbst缓冲液洗微球体3次,1000rpm条件下离心3min,弃上清液;加入步骤(2.2)保留的上清液,轻轻吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;随后将混悬液吸至一个新的5ml离心管中,1000rpm条件下离心3min,吸弃上清液;再用0.5%pbst缓冲液洗微球体6次,1000rpm条件下离心3min,吸弃上清液;随后在细胞沉淀中加入1ml0.2mol/l的ph=2.2的甘氨酸-盐酸缓冲液,轻轻吹打混匀后,置于37℃、50rpm条件下,恒温培养振荡器中洗脱10min;再在2000rpm条件下离心10min,将上清液转入一个新的1.5mlepp管内,加入150μl1mol/l的ph=9.1的tris-hcl缓冲液进行中和;测定洗脱液中所含噬菌体滴度,随后取该洗脱液100μl感染宿主菌e.colier2738进行扩增,扩增后噬菌体通过离心以及peg/nacl沉淀的方法进行纯化,并测定扩增、纯化后噬菌体滴度,完成第一轮筛选;其中,所用0.5%pbst缓冲液是指含tween-20体积浓度为0.5%的pbs缓冲液;(2.4)重复以上步骤,共完成3轮筛选,筛选结束。
(3)经dna测序确定目标多肽,具体为:取最后一轮筛选后的噬菌体洗脱液铺于x-gal/lb平板上检测滴度,待噬菌斑长出后,随机挑取若干个噬菌斑进行扩增及纯化,将噬菌体单克隆分别与肝癌微球体、hcclm3细胞和lo2细胞结合,计算噬菌体单克隆的特异性,选取与肝癌微球体特异性结合最高的噬菌体,随后进行测序,其所展示的多肽即为目标多肽。
实施例6一种肝癌微球体细胞表面特异性靶向多肽的制备方法,包括以下步骤:
(1)贴壁细胞及肝癌微球体的培养:按照常规方法进行hcclm3细胞和lo2细胞培养;肝癌微球体培养具体为:取5×105个处于对数生长期的hcclm3细胞于超低吸附培养瓶中进行悬浮培养,每7ml培养体系中含有:6776μl的dmem-f12培养基、140μl的b27为、70μl的肝素、终浓度为100μg/ml的hegf和终浓度为50μg/ml的bfgf,细胞置于37℃、体积浓度为5%的co2条件下的培养箱中培养48h,得到肝癌微球体,备用;
(2)以hcclm3细胞悬浮培养的肝癌微球体为靶细胞,贴壁培养的hcclm3细胞和lo2细胞为吸附细胞,采用噬菌体展示技术进行差减筛选,具体为:(2.1)取贴壁长满皿底的lo2细胞,pbs洗3次,加入1.5ml质量浓度为1%的bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用0.3%pbst缓冲液洗lo2细胞3次;吸取噬菌体环七肽库溶液,该溶液滴度为2×1011pfu,并与1.5mlpbs混匀得到混合液;将该混合液加入贴壁生长的lo2细胞中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;转出上清液至一个新的1.5mlepp管中,备用;(2.2)取贴壁长满皿底的hcclm3细胞,pbs洗3次,加入1.5ml质量浓度为1%的bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用0.3%pbst缓冲液洗贴壁生长的hcclm3细胞3次,加入步骤(2.1)保留的上清液,继续在37℃、50rpm条件下,恒温培养振荡器中孵育1h,转出上清液至一个新的1.5mlepp管中,备用;(2.3)将步骤(1)中肝癌微球体培养物吸至10ml离心管中,1000rpm条件下离心3min,吸弃陈旧培养基;微球体用pbs洗3次,1000rpm条件下离心3min,吸弃pbs;加入1.5ml质量浓度为1%的bsa,小心吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中封闭30min;随后将混悬液吸至一个新的10ml离心管中,1000rpm条件下离心3min,吸弃上清液;用0.3%pbst缓冲液洗微球体3次,1000rpm条件下离心3min,弃上清液;加入步骤(2.2)保留的上清液,轻轻吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;随后将混悬液吸至一个新的5ml离心管中,1000rpm条件下离心3min,吸弃上清液;再用0.3%pbst缓冲液洗微球体6次,1000rpm条件下离心3min,吸弃上清液;随后在细胞沉淀中加入1ml0.2mol/l的ph=2.2的甘氨酸-盐酸缓冲液,轻轻吹打混匀后,置于37℃、50rpm条件下,恒温培养振荡器中洗脱10min;再在2000rpm条件下离心10min,将上清液转入一个新的1.5mlepp管内,加入150μl1mol/l的ph=9.1的tris-hcl缓冲液进行中和;测定洗脱液中所含噬菌体滴度,随后取该洗脱液100μl感染宿主菌e.colier2738进行扩增,扩增后噬菌体通过离心以及peg/nacl沉淀的方法进行纯化,并测定扩增、纯化后噬菌体滴度,完成第一轮筛选;其中,所用0.3%pbst缓冲液是指含tween-20体积浓度为0.3%的pbs缓冲液;(2.4)重复以上步骤,共完成4轮筛选,筛选结束。
(3)经dna测序确定目标多肽,具体为:取最后一轮筛选后的噬菌体洗脱液铺于x-gal/lb平板上检测滴度,待噬菌斑长出后,随机挑取若干个噬菌斑进行扩增及纯化,将噬菌体单克隆分别与肝癌微球体、hcclm3细胞和lo2细胞结合,计算噬菌体单克隆的特异性,选取与肝癌微球体特异性结合最高的噬菌体,随后进行测序,其所展示的多肽即为目标多肽。
实施例7一种肝癌微球体细胞表面特异性靶向多肽的制备方法,包括以下步骤:
(1)贴壁细胞及肝癌微球体的培养:贴壁细胞培养具体为:人肝癌细胞株hcclm3用含15%fbs的mem培养基培养,人正常肝细胞lo2用含10%fbs的高糖dmem培养基培养,取对数生长期的hcclm3细胞及lo2细胞,常规消化后按1×106/皿接种于6cm培养皿中,置于37℃、含体积浓度为5%的co2条件下培养至细胞贴壁长满皿底,备用;
肝癌微球体培养具体为:取5×105个处于对数生长期的hcclm3细胞于超低吸附培养瓶中进行悬浮培养,每7ml培养体系中含有:6776μl的dmem-f12培养基、140μl的b27为、70μl的肝素、终浓度为100μg/ml的hegf和终浓度为50μg/ml的bfgf,细胞置于37℃、体积浓度为5%的co2条件下的培养箱中进行培养培养48h,得到肝癌微球体,备用;
(2)以hcclm3细胞悬浮培养的肝癌微球体为靶细胞,贴壁培养的hcclm3细胞和lo2细胞为吸附细胞,采用噬菌体展示技术进行差减筛选,具体为:(2.1)取贴壁长满皿底的lo2细胞,pbs洗3次,加入1.5ml质量浓度为1%的bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用pbst缓冲液洗lo2细胞3次;吸取噬菌体环七肽库溶液,该溶液滴度为2×1011pfu,并与1.5mlpbs混匀得到混合液;将该混合液加入贴壁生长的lo2细胞中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;转出上清液至一个新的1.5mlepp管中,备用;(2.2)取贴壁长满皿底的hcclm3细胞,pbs洗3次,加入1.5ml质量浓度为1%的bsa,在37℃、50rpm条件下,恒温培养振荡器中封闭30min,随后用pbst缓冲液洗贴壁生长的hcclm3细胞3次,加入步骤(2.1)保留的上清液,继续在37℃、50rpm条件下,恒温培养振荡器中孵育1h,转出上清液至一个新的1.5mlepp管中,备用;(2.3)将步骤(1)中肝癌微球体培养物吸至10ml离心管中,1000rpm条件下离心3min,吸弃陈旧培养基;微球体用pbs洗3次,1000rpm条件下离心3min,吸弃pbs;加入1.5ml质量浓度为1%的bsa,小心吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中封闭30min;随后将混悬液吸至一个新的10ml离心管中,1000rpm条件下离心3min,吸弃上清液;用pbst缓冲液洗微球体3次,1000rpm条件下离心3min,弃上清液;加入步骤(2.2)保留的上清液,轻轻吹打混匀后,将混悬液吸至一个新的6cm培养皿中,在37℃、50rpm条件下,恒温培养振荡器中孵育1h;随后将混悬液吸至一个新的5ml离心管中,1000rpm条件下离心3min,吸弃上清液;再用pbst缓冲液洗微球体6次,1000rpm条件下离心3min,吸弃上清液;随后在细胞沉淀中加入1ml0.2mol/l的ph=2.2的甘氨酸-盐酸缓冲液,轻轻吹打混匀后,置于37℃、50rpm条件下,恒温培养振荡器中洗脱10min;再在2000rpm条件下离心10min,将上清液转入一个新的1.5mlepp管内,加入150μl1mol/l的ph=9.1的tris-hcl缓冲液进行中和;测定洗脱液中所含噬菌体滴度,随后取该洗脱液100μl感染宿主菌e.colier2738进行扩增,扩增后噬菌体通过离心以及peg/nacl沉淀的方法进行纯化,并测定扩增、纯化后噬菌体滴度,完成第一轮筛选;(2.4)重复以上步骤,共完成4轮筛选,筛选结束,其中,第一轮筛选中所用pbst缓冲液为0.1%pbst缓冲液,即为含tween-20体积浓度为0.1%的pbs缓冲液;第二轮筛选中所用pbst缓冲液为0.2%pbst缓冲液,即含tween-20体积浓度为0.2%的pbs缓冲液;第三轮筛选中所用pbst缓冲液为0.3%pbst缓冲液,即含tween-20体积浓度为0.3%的pbs缓冲液;第四轮筛选中所用pbst缓冲液为0.5%pbst缓冲液,即含tween-20体积浓度为0.5%的pbs缓冲液;第四轮筛选时洗脱液进行滴度测定后不再进行扩增纯化。
(3)经dna测序确定目标多肽,具体为:取最后一轮筛选后的噬菌体洗脱液铺于x-gal/lb平板上检测滴度,待噬菌斑长出后,随机挑取若干个噬菌斑进行扩增及纯化,将噬菌体单克隆分别与肝癌微球体、hcclm3细胞和lo2细胞结合,计算噬菌体单克隆的特异性,选取与肝癌微球体特异性结合最高的噬菌体,随后进行测序,其所展示的多肽即为目标多肽。
实施例8一种肝癌微球体细胞表面特异性靶向多肽的制备方法,与实施例7的区别在于:选用的筛选肽库为七肽库。
实施例9肝癌微球体细胞表面特异性靶向多肽,其为七肽,氨基酸序列为asn-thr-gly-ser-pro-tyr-glu,能够在制备肝癌癌栓诊断试剂或肝癌靶向治疗药物中的应用。
- 还没有人留言评论。精彩留言会获得点赞!