一种改进的甲基丙烯酸二烷氨基乙酯的制备方法与流程
本发明涉及一种改进的甲基丙烯酸二烷氨基乙酯的制备方法,具体地说是一种以正烷烃为携甲醇溶剂,采用分水器从反应体系中不断地分出甲醇的酯交换方法,属于精细有机合成领域。
背景技术:
甲基丙烯酸二烷氨基乙酯是一类重要的阳离子单体,可用于制备水处理剂、抗静电剂、助留剂、施胶剂、粘合剂、医用缓释剂等许多精细化学品[罗娅君,张新申,王照丽.甲基丙烯酸二甲氨基乙酯的生产和应用前景.精细石油化工,2004,21(2):58-60;魏晓奕,常刚,李积华,等.纤维素-g-聚甲基丙烯酸-n,n-二甲氨基乙酯对so42-的吸附性能研究.离子交换与吸附,2014,30(5):468-474;王西锋,张光华,牛恒.阳离子型无皂苯丙乳液纸张表面施胶剂的研制.中国胶粘剂,2011,20(7):28-31]和功能性聚合物[杜凯迪,唐二军,袁淼,等.[amim]cl离子液体中纤维素接枝pdeaema分子刷的均相可控聚合.化工学报,2015,66(10):4275-4280;唐新颖,刘文涛,杨阳,等.聚乳酸-聚甲基丙烯酸二甲氨基乙酯两亲性聚合物的结晶性能.高分子材料科学与工程,2015,31(6):50-54;justinra,suryakm.novelatomtransferradicalpolymerizationmethodtoyieldcopper-freeblockcopolymericbiomaterials.macromol.chem.phys.2013,214,1321-1325;sudhinag,bishnupb,victorm,etal.multi-stimuli-responsivepolymericmaterials.chemeurj,2015,21,13164–13174]等,也可作为纤维的改性单体,以改善纤维对颜料和染料的亲合性[wangyazhen,shenxinyuan,dushuping,etal.fabricationandcharacterizationofaciddyeablepolyacrylo-nitrileblendfiber.jmacromol,scipartb,2007,46(2):231-237;wangyazhen,shenxinyuan,gaoyue,etal.synthesisofaciddyeableacrylonitrilecopolymer.jmacromol,sciparta,2006,43(11):1773-1778;徐静,季春晓.酸性可染腈纶聚合工艺研究.合成纤维工业,2011,34(3):34-38]。这类单体的合成有直接酯化法[罗娅君,张新申,王照丽.甲基丙烯酸二甲氨基乙酯的生产和应用前景.精细石油化工,2004,21(2):58-60]、甲基丙烯酰氯酯化法[贺灵芝,曾庆友,许瑞安.甲基丙烯酸二乙氨基乙酯的合成.精细化工中间体,2009,39(6):69-70]和酯交换法[冯大春,鲁红,尹家贵.n,n-二甲氨基丙烯酸乙酯的制备.合成化学,2001,9(4):362-364;吴殿义,丁伟,刘永建.钛酸四丁酯催化合成甲基丙烯酸二乙氨基乙酯.大庆石油学院学报,2003,27(3):34,236;张光旭,徐鑫,龙艳,夏涛.钛酸异丙酯催化酯交换合成甲基丙烯酸二甲氨基乙酯.石油化工,2008,37(11):1160-1163;刘祥臣,王波,杨东辉等.二乙基氨基甲基丙烯酸乙酯的研制.化学世界,2002,43(1):138-39;曲慧,毕玉遂,胡婉莹.二苯基二氯化锡催化合成甲基丙烯酸二甲氨基乙酯.精细石油化工,2007,24(5):53-57]等。直接酯化法由于甲基丙烯酸呈酸性,氨基醇呈碱性,用碱或酸作催化剂都避免不了反应物和催化剂发生反应,因而造成催化剂用量大,并易生成副产物。另外,氨基醇空间位阻较大,反应活性较低,酯化较困难。甲基丙烯酰氯酯化法具有收率高、反应时间短、后处理简单等优点,但原料容易腐蚀设备,酰氯类化合物的性质活泼,遇水分解,使用过程需保证无水,因而操作麻烦。另外,付产的大量hcl难以处理。酯交换法具有催化剂用量少、原料对设备的腐蚀性小、成本低、步骤简单等优点而成为目前最常用的一种方法,但酯交换法也存在如下问题:酯交换为平衡反应,为了使反应完全,必须不断蒸出副产物-甲醇,但由于甲醇和甲基丙烯酸甲酯易共沸,并形成恒沸物,因此,必须采用精馏装置,以致酯化设备复杂,甲基丙烯酸甲酯用量较多;用于酯交换法的催化剂很多,如钛酸酯、有机锡等,但均存在催化活性低,反应温度较高,一般在100℃左右,在该温度条件下,所用原料甲基丙烯酸甲酯和产品易聚合,控制不当而造成废物。
技术实现要素:
为了解决酯交换法存在的问题,本发明的发明者对酯交换法进行了深入的研究,发明了一种新的酯交换工艺。该工艺利用正烷烃与甲醇不互溶的特性,以正烷烃为携甲醇剂,采用分水器不断地从反应体系中分出甲醇而使反应完全。
一种甲基丙烯酸二烷氨基乙酯的合成方法,其工艺步骤如下:
(1)合成:将甲基丙烯酸甲酯、二烷氨基乙醇、氢氧化锂、正烷烃和少量的阻聚剂加入带搅拌、温度计和分水器的反应釜中,加热至65℃~73℃,通过回流不断地分出甲醇,反应至无甲醇分出时停止反应(约5h)。
(2)提纯:冷却至室温,加去离子水洗涤两次。油相先逐步减压(0~100mmhg)于60℃~100℃蒸出正己烷、未反应的甲基丙烯酸甲酯和二烷氨基乙醇,再于5~10mmhg和80℃~100℃下减压蒸出甲基丙烯酸二烷氨基乙酯。
进一步地,所述的二烷氨基乙醇主要为二甲氨基乙醇和二乙氨基乙醇。
所述的二烷氨基乙醇与甲基丙烯酸甲酯的质量比为0.25~0.60:1,其中二乙氨基乙醇与甲基丙烯酸甲酯的质量比优选为0.45~0.55:1,二甲氨基乙醇与甲基丙烯酸甲酯的质量比优选为0.35~0.45:1。
所述的氢氧化锂与甲基丙烯酸甲酯的质量比为0.015~0.05:1,优选为0.03:1。
所述的正烷烃为正己烷和正庚烷,优先为正己烷,正己烷与甲基丙烯酸甲酯的质量比为0.80~1.0:1,
所述的阻聚剂主要为对苯二酚、酚噻嗪等,阻聚剂与甲基丙烯酸甲酯的质量比为0.001~0.003:1,优选为0.002:1。
本发明的优点在于:以正烷烃为携甲醇溶剂,采用分水器从反应体系中不断地分出甲醇,和常规精馏分甲醇相比,酯化设备简单,反应温度明显降低,因而可有效地防止甲基丙烯酸甲酯和产品在反应过程中发生聚合。
附图说明
图1为本发明实施例1得到的甲基丙烯酸二乙氨基乙酯的红外光谱;
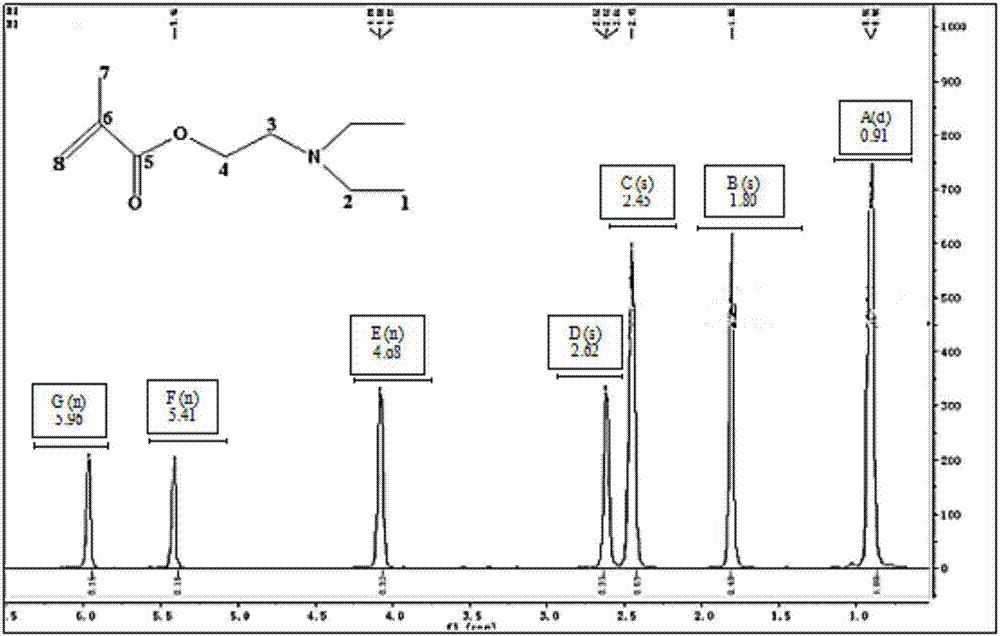
图2为本发明实施例1得到的甲基丙烯酸二乙氨基乙酯的核磁共振氢谱;
图3为本发明实施例1得到的甲基丙烯酸二乙氨基乙酯的核磁共振碳谱。
图4为本发明所采用的实验室反应装置。
具体实施方式
以下对本发明的优选实施例进行说明,应当理解,此处所描述的优选实施例仅用于说明和解释本发明,并不用于限定本发明。
除非另有说明,本发明中所采用的百分数均为质量百分数。
原料、产品及甲醇的含量均采用气相色谱分析,所用仪器为岛津gc-14c气相色谱仪,分析条件如下:柱型:ac1.10,进样温度:80℃,检测温度:280℃,采用程序升温,每分钟10℃。色谱数据采用浙大智能n2000数据工作站处理,含量采用面积归一法计算。
实施例1
一种以二乙氨基乙醇和甲基丙烯酸甲酯为原料,正己烷为携甲醇剂,采用分水器从反应体系中不断地分出甲醇,合成甲基丙烯酸二乙氨基乙酯的方法,工艺步骤如下:
(1)合成:将240g(2.4mol)甲基丙烯酸甲酯、117g(1.0mol)二乙氨基乙醇、200g正己烷、8g氢氧化锂和0.5g酚噻嗪加入带搅拌、温度计和分水装置的1000ml三口烧瓶中,加热至65℃~73℃,不断回流分出甲醇,回流至无甲醇分出时停止反应(约5h)。分出甲醇34.5g,气相色谱分析表明,其中甲醇含量为88.6%,甲基丙烯酸甲酯含量为11.2%。
(2)提纯:冷却至室温,加300ml×2的去离子水洗涤两次。油相先逐步减压(0~100mmhg)于60℃~100℃蒸出正己烷、未反应的甲基丙烯酸甲酯和二乙氨基乙醇,再于5~10mmhg和90℃~100℃下减压蒸出甲基丙烯酸二乙氨基乙酯。所得产品重量为175.7g(理论产量185.26g),产率94.1%。气相色谱分析表明,甲基丙烯酸二乙氨基乙酯的含量为99.2%。
本发明通过红外光谱和核磁共振谱测定对本实施例得到的产物结构进行了表征。图1为本发明实施例1得到的甲基丙烯酸二乙氨基乙酯的红外光谱图;图2为本发明实施例1得到的甲基丙烯酸二乙氨基乙酯的1hnmr(500mhz,cdcl3);图3为本发明实施例1得到的甲基丙烯酸二乙氨基乙酯的13cnmr(126mhz,cdcl3)。
图1中,2970.61cm-1为甲基的伸缩振动吸收峰,2933.25cm-1为亚甲基的伸缩振动吸收峰,1721cm-1为酯基伸缩振动吸收峰,1638.58cm-1为-c=c-的伸缩振动吸收峰,1453.71cm-1和1380.94cm-1为c-h键的弯曲振动吸收峰,1164.11cm-1为c-n的伸缩振动吸收峰。以上分析说明图1中的红外光谱符合甲基丙烯酸二乙氨基乙酯的结构特征。
图2中,0.91(d,3h)为1号位甲基的氢质子峰,1.80(s,3h)为7号位甲基的氢质子峰,2.45(s,2h)为2号位亚甲基的氢质子峰,2.62(s,2h)为3号位亚甲基的氢质子峰,4.08(m,2h)为4号位亚甲基的氢质子峰,5.41(s,1h)和5.96(s,1h)为8号位亚甲基的氢质子峰。以上分析说明图2中氢核的化学位移符合甲基丙烯酸二乙氨基乙酯的结构特征。
图3中,12.03(d)为1号位甲基的碳核峰,18.16(d)为7号位甲基的碳核峰,47.74(s)为2号位亚甲基的碳核峰,50.89(s)为3号位亚甲基的碳核峰,63.11(s)为4号位亚甲基的碳核峰,,77.29(m)为溶剂cdcl3的碳核峰,125.12(s)为8号位亚甲基的碳核峰,136.28(s)为6号位的碳核峰,167.12(s)为5号位的碳核峰。以上分析说明图3中碳核的化学位移符合甲基丙烯酸二乙氨基乙酯的结构特征。
以上分析表明本实施例合成的产物为甲基丙烯酸二乙氨基乙酯。
实施例2
一种以二乙氨基乙醇和甲基丙烯酸甲酯为原料,正己烷为携甲醇剂,采用分水器从反应体系中不断地分出甲醇,合成甲基丙烯酸二乙氨基乙酯的方法,工艺步骤如下:
(1)合成:将240g(2.4mol)甲基丙烯酸甲酯、117g(1.0mol)二乙氨基乙醇、9g氢氧化锂和0.5g酚噻嗪加入带搅拌、温度计和分水装置的1000ml三口烧瓶中,加热至65℃~73℃,不断回流分出甲醇,回流至无甲醇分出时停止反应(约5h)。分出甲醇34.7g,气相色谱分析表明,其中甲醇含量为90.2%,甲基丙烯酸甲酯含量为9.3%。
(2)提纯:冷却至室温,加300ml×2的去离子水洗涤两次。油相先逐步减压(0~100mmhg)于60℃~100℃蒸出正己烷、未反应的甲基丙烯酸甲酯和二乙氨基乙醇,再于5~10mmhg和90℃~100℃下减压蒸出甲基丙烯酸二乙氨基乙酯。所得产品重量为176.1g(理论产量185.26g),产率94.1%。气相色谱分析表明,甲基丙烯酸二乙氨基乙酯的含量为99.0%。
按照实施例1中的表征方式对本实施例的产品进行检测,证明本实施例得到的产物为目标产物。
实施例3
一种以二乙氨基乙醇和甲基丙烯酸甲酯为原料,正己烷为携甲醇剂,采用分水器从反应体系中不断地分出甲醇,合成甲基丙烯酸二乙氨基乙酯的方法,工艺步骤如下:
(1)合成:将240g(2.4mol)甲基丙烯酸甲酯、117g(1.0mol)二乙氨基乙醇、9g氢氧化锂和0.5g对苯二酚加入带搅拌、温度计和分水装置的1000ml三口烧瓶中,加热至65℃~73℃,不断回流分出甲醇,回流至无甲醇分出时停止反应(约5h)。分出甲醇34.1g,气相色谱分析表明,其中甲醇含量为88.4%,甲基丙烯酸甲酯含量为11.0%。
(2)提纯:冷却至室温,加300ml×2的去离子水洗涤两次。油相先逐步减压(0~100mmhg)于60℃~100℃蒸出正己烷、未反应的甲基丙烯酸甲酯和二乙氨基乙醇,再于5~10mmhg和90℃~100℃下减压蒸出甲基丙烯酸二乙氨基乙酯。所得产品重量为173.2g(理论产量185.26g),产率92.5%。气相色谱分析表明,甲基丙烯酸二乙氨基乙酯的含量为98.9%。
按照实施例1中的表征方式对本实施例的产品进行检测,证明本实施例得到的产物为目标产物。
实施例4
一种以二甲氨基乙醇和甲基丙烯酸甲酯为原料,正己烷为携甲醇剂,采用分水器从反应体系中不断地分出甲醇,合成甲基丙烯酸二甲氨基乙酯的方法,工艺步骤如下:
(1)合成:将240g(2.4mol)甲基丙烯酸甲酯、89g(1.0mol)二甲氨基乙醇、8g氢氧化锂和0.5g对苯二酚加入带搅拌、温度计和分水装置的1000ml三口烧瓶中,加热至65℃~73℃,不断回流分出甲醇,回流至无甲醇分出时停止反应(约5h)。分出甲醇33.4g,气相色谱分析表明,其中甲醇含量为91.0%,甲基丙烯酸甲酯含量为8.7%。
(2)提纯:冷却至室温,加300ml×2的去离子水洗涤两次。油相先逐步减压(0~100mmhg)于60℃~100℃蒸出正己烷、未反应的甲基丙烯酸甲酯和二甲氨基乙醇,再于5~10mmhg和80℃~90℃下减压蒸出甲基丙烯酸二甲氨基乙酯。所得产品重量为146.7g(理论产量157.21g),产率92.8%。气相色谱分析表明,甲基丙烯酸二甲氨基乙酯的含量为99.5%。
按照实施例1中的表征方式对本实施例的产品进行检测,证明本实施例得到的产物为目标产物。
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!