一种制备培哚普利叔丁胺盐的绿色合成新工艺的制作方法
本发明属于医药技术领域,涉及一种培哚普利叔丁胺盐的绿色制备方法。
发明背景
培哚普利(perindopril)是一种重要的非巯基的血管紧张素ⅰ转换酶(angiotensinconvertingenzyme,ace)抑制剂的前药,通过在体内迅速代谢为活性二酸培哚普利拉而发挥作用。口服给药,吸收迅速、完全,生物利用度高于70%,且不受食物影响。培哚普利代谢所得的培哚普利拉与ace结合产生的酶复合物离解缓慢,药物半衰期为25~30h,因此显示出给药后降压作用强,持续时间久特征。临床上主要是通过抑制血管紧张素ⅱ生成及减少缓激肽降解,达到较好的降低血管阻力,增加大动脉顺应性的降压效果。同时发现该药对高血压患者的并发症及一些伴发疾病也具有良好影响,主要是延缓糖尿病性肾病进展、减轻肾小球硬化、减轻左心室肥厚、改善左心收缩功能、降低心梗后并发症及死亡率;同时还具有阻断醛固酮对利尿流失na+产生的对抗作用。除此之外,该药也用于充血性心衰治疗,通过逆转充血性心衰的心肌结构改变,减轻心脏肥厚,使心内膜下胶原含量和心肌生化指标正常化,改变左室功能参数,使受损心脏收缩和舒张功能正常化及冠脉血流动力学正常化,并明显地阻止电生理异常,恢复充血性心衰正常的神经内分泌,降低交感活性,降低心钠素分泌而达到缓解充血性心衰作用。正是由于这种原因,有关培哚普利的制备新工艺一直是人们关注焦点。
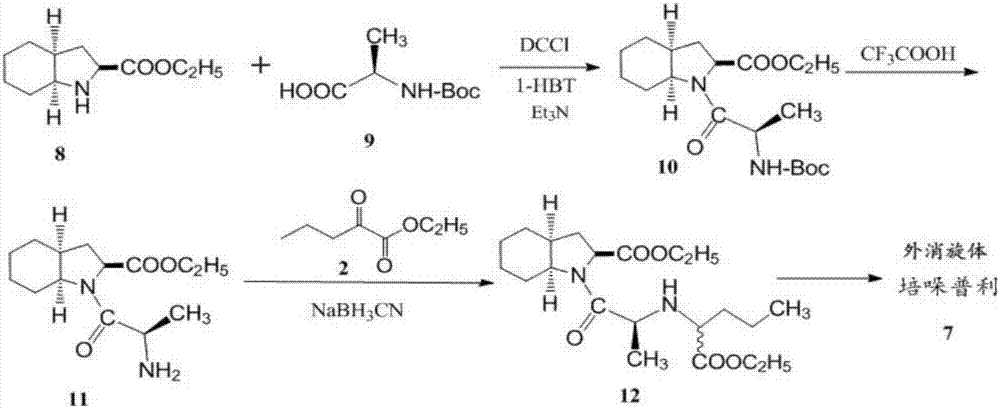
目前制备培哚普利主要合成工艺主要有两类:第一类合成工艺(如图2所示):以(2s,3αs,7αs)-八氢化吲哚-2-羧酸乙酯(8)为原料与n-boc-丙氨酸(9)为原料,在dcci与1-hbt(1-羟基苯并三唑)作用下缩合产生八氢化吲哚-2-羧酸-n-boc-丙氨酸酯(10),然后在三氟乙酸催化下脱除保护基boc获得八氢化吲哚-2-羧酸-丙氨酸酯(11),该化合物再与2-酮基戊酸乙酯(2)进行缩氨化反应并在氰基硼氢化钠还原下,获得消旋体化合物(12),然后,生成外消旋体的培哚普利。该路线使用三氟乙酸作为脱除保护基的试剂,酸性太强,工业上使用不便;其二,最终产物是外消旋体的培哚普利,未解决外消旋中间体(12)的分离问题;其三,合成关键的外消旋中间体(12)原料2-酮基戊酸乙酯(2)合成也比较复杂,所以该种合成方法并未在工业生产实际中应用。
目前工业上常使用的是第二类合成工艺路线(如图3所示),该方法主要通过丙酮酸(13)与l-正缬氨酸乙酸盐酸盐(14)原料进行缩胺化反应后,继而进行不对称氢化反应,获得关键性中间体光活性异构体n-[(s)-乙氧羰基丁基]-(s)-丙氨酸(15),然后再与不对称光学异构体(2s,3αs,7αs)-八氢化吲哚-2-羧酸乙酯(8)进行各种酰胺化反应获得最终产物。在该类合成工艺中,各种工艺的不同点主要在于如何将关键中间体光活性异构体丙氨酸衍生物(15)的羧基转换成各类活性基团,最后与光学原料(8)进行温和的酰胺化反应,最终获得光活性培哚普利产物。但这类工艺也有很大弱点,首先是使用的关键光学活性中间体(15)的合成原料为具有光学活性的l-正缬氨酸乙酯,是非天然氨基酸,需要专门的合成,合成路线复杂;其次,在与(2s,3αs,7αs)-八氢化吲哚-2-羧酸乙酯(8)缩合成酰胺过程中,为合成活性酯或内酯或酰氯,使用了大量高活性的酰氯或有毒试剂如光气、亚硫酰氯或其类似物,这不仅造成环境空气污染,而且也比较危险,对生产设备腐蚀也比较强,加上该类方法是先生成活性中间体再进行酰胺化反应,人为也增加了反应步骤,反应路线长,生产成本也高。为此有必要进行新的绿色合成工艺研究。
技术实现要素:
本发明在总结前人发明制备工艺的基础上,通过使用3-酮酸已酸乙酯(1)为原料氧化脱亚甲基方法,合成2-酮戊酸乙酯(2),继而利用光活性的l-丙氨酸甲酯盐酸盐(3)与其进行缩合氨化反应与不对称氢化催化反应,获得制备培哚普利的重要关键中间体不对称光学活性化合物n-[(s)-乙氧羰基丁基]-(s)-丙氨酸甲酯(4),后者与制备培哚普利的另一种重要原料(2s,3αs,7αs)-八氢化吲哚-2-羧酸(5),在路易斯酸催化下,直接进行酰胺化反应获得产物培哚普利(6),然后与叔丁胺成盐,获得医用培哚普利叔丁胺盐(7)无定形晶体。该合成工艺路程短(只有四步),所应用方法与原料均无毒,无强腐蚀性,对环境绿色友好。新工艺方法制备简单,条件温和,步骤少,产率高,费用低,是一种制备培哚普利叔丁胺盐的绿色合成新工艺。
本发明涉及到的绿色合成培哚普利的工艺路线主要特征为如图1制备培哚普利叔丁胺盐的绿色合成新工艺路线图所示:
1.本发明新制备工艺特征之一在于,所采取反应路线为,首先以易得的3-酮基己酸乙酯(1)为反应原料,在lewis酸存在下通过无机氧化剂在水溶液里氧化生成2-酮基-戊酸乙酯(2),后者与l-丙氨酸甲酯盐酸盐(3)混合,在弱碱性条件下,进行相转移催化缩合氨化反应,并在pt-c催化剂下进行催化氢化还原,获得不对称光活性异构体l-丙氨酸甲酯衍生物(4),继而在lewis酸作用及相转移催化剂催化下,与(2s,3αs,7αs)-八氢化吲哚-2-羧酸(5)直接进行酰胺化反应,将l-丙氨酸甲酯衍生物(4)转换成酰胺产物具有光学活性的培哚普利粗品(6),后者与叔丁胺在一定温度下在有机溶剂中成盐,析出产物(7)无定形晶体粉末。
2.本发明新制备工艺特征之二在于,权利要求第1项所涉及反应中,使用了路易斯酸催化方法,将n-[(s)-乙氧羰基丁基]-(s)-丙氨酸甲酯(4)与(2s,3αs,7αs)--氢化吲哚-2-羧酸(5)直接反应转化成(2s,3αs,7αs)--氢化吲哚-2-羧酸(5)的酰胺衍生物,获得不对称光学产物培哚普利粗品(6)。而不是采取先将n-[(s)-乙氧羰基丁基]-(s)-丙氨酸(15)转化为活性酯或酰氯等活性物后再与(2s,3αs,7αs)--氢化吲哚-2-羧酸酯(8)反应的方式进行酰胺化反应。
3.本发明新制备工艺特征之三在于,权利要求书第1项中所涉及反应中,在直接酰胺化反应里,使用的(2s,3αs,7αs)--氢化吲哚-2-羧酸(5)作为直接酰胺化反应的重要反应原料,而不是用(2s,3αs,7αs)--氢化吲哚-2-羧酸酯(8)作为重要的直接酰胺化反应的反应原料。
4.本发明新制备工艺特征之四在于,权利要求书第1项中所涉及反应中,使用了2-酮基-戊酸乙酯(2)为原料与l-丙氨酸甲酯盐酸盐(3)在弱碱性条件下,进行相转移催化缩合氨化反应,并在pt-c催化剂下进行催化氢化还原,获得不对称光活性异构体l-丙氨酸甲酯衍生物(4),而不是使用丙酮酸与非天然l-正缬氨酸甲酯盐酸盐反应合成不对称光活性异构体l-丙氨酸甲酯衍生物(4)。
5.本发明新制备工艺特征之五在于,权利要求书第1项中所涉及反应中,不但使用了路易斯酸为催化剂,通过氧化反应合成化合物(2),而且还使用了路易斯酸为催化剂,通过直接酰胺化反应合成化合物(6),这是本发明工艺路线的重要特征之一。
6.本发明新制备工艺特征之六在于,权利要求第1项所涉及使用的路易斯酸主要包含如下类型:ticl4,alcl3,bf3,sncl2,zncl2,fecl3,sbcl5,sbf5,aucl3,三氟甲磺酸盐类,如三氟甲磺酸钾,三氟甲磺酸钠,三氟甲磺酸铁,三氟甲磺酸铝。
7.本发明新制备工艺特征之七在于,由化合物(4)在路易斯酸催化下进行的直接酰胺化反应生成化合物(6)时,所用路易斯酸催化剂与所用有机碱性调节剂如三乙胺的摩尔比的范围在1-10倍之间,所用的有机碱包括三乙胺,三丙胺,二异丙胺,六氢哌啶,哌嗪,1-甲基哌嗪,吗啉,甲基吗啉,三丁基胺,叔丁基胺。
8.本发明新制备工艺特征之八在于,所有制备工艺路线中各步反应温度均在0-50℃,反应条件温和,可操作性强。
9.本发明新制备工艺特征之九在于,所有制备工艺路线中各步反应时间均在10-1200分钟内完成。
10.本发明新制备工艺特征之十在于,权利要求第1项所涉及的制备化合物(4)时所涉及的不对称氢化催化试剂为1-10%pt-c,催化时反应釜内氢气压力为0.1-10mpa。
11.本发明新制备工艺特征之十一在于,权利要求第1项所涉及的制备不对称光学活性化合物(4)时所涉及的缩合氨化与不对称氢化催化反应中以及在制备不对称光学活性化合物(6)的直接酰胺化反应中,均使用了相转移催化剂,其用量范围为l-丙氨酸甲酯盐酸盐(3)的摩尔数的1-10%,相转移催化剂类型为四丁基氯化铵、苄基三乙基氯化铵(teba)、四丁基溴化铵(tbab)、四丁基硫酸氢铵、三辛基甲基氯化铵、十二烷基三甲基氯化铵、十四烷基三甲基氯化铵、环糊精及聚乙二醇(分子量400-1200)。
12.本发明新制备工艺特征之十二在于,权利要求第1项所涉及合成化合物(2)的氧化反应中,所用的无机氧化剂包括高锰酸钾,重铬酸钾,高碘酸钠,过硫酸钠,双氧水,过硫酸氢钾,氯酸钾,次氯酸钠,过碳酸钠、过硼酸钠、过硼酸钾,过硫酸铵,过氧乙酸。
附图说明
图1制备培哚普利叔丁胺盐的绿色合成新工艺路线图;
图2第一类培哚普利合成工艺路线图;
图3第二类培哚普利合成工艺路线图。
实施例
1.2-酮戊酸乙酯制备(2):
3-酮己酸乙酯(1)(1.58g,10mmol)与50ml蒸馏水搅拌混合,,然后分别加入过硫酸氢钾复合盐(12.91g,21mmol)和无水zncl2(3.14g,23mmol),在室温下搅拌25分钟,反应完毕后,加入饱和酒石酸钾钠溶液50ml,然后用叔丁基甲醚萃取3次(100mlx3),合并萃取液然后用无水硫酸镁干燥,蒸去溶液,蒸馏,得到油状物2-酮戊酸乙酯(2)1.37g(96%)。
1h-nmr(300mhz;cdcl3)δ0.94(t,j=7.4hz,3h),1.34(t,j=7.1hz,3h),1.66-1.78(m,2h),2.78(t,j=6.9hz,2h),4.34(q,j=7.1hz,2h);
2.n-[(s)-乙氧羰基丁基]-(s)-丙氨酸甲酯合成(4)
向高压反应釜加入l-丙氨酸甲酯盐酸盐(3)(30g,0.165mol),2-酮戊酸乙酯(2)(26.2g,0.182mol),5%pd-c催化剂3g,以20%nah2po4水溶液调节ph至弱碱性(ph=7.5-8),加入1%mol的四丁基氯化铵为相转移催化剂,合上釜盖,以氮气置换3次后充氢至0.5mpa,在20℃下进行催化加氢,直到吸氢至理论量,停止反应过滤得到固体,以浓盐酸调节至ph=3.1,冷却至0-5℃,过滤得到固体,以冰乙睛淋洗后,0℃真空干燥,得油状物产品l-丙氨酸甲酯衍生物(4)28.6g,收率79%。[α]20d=+4.6(c=1,etoh)。1h-nmr(300mhz;cdcl3)δ0.86(t,j=7.4hz,3h),1.14-1.20(m,3h),1.24-1.29(m,2h),1.34(t,j=7.1hz,3h),1.36-1.53(m,2h),3.16(m,1h),3.21(m,1h),3.42(s,3h),4.06-4.13(m,2h)。
3.以lewis酸为催化剂合成酰胺化产物培哚普利(6)
将n-[(s)-乙氧羰基丁基]-(s)-丙氨酸甲酯(4)(4.95,21.4mmol)、无水zncl2(2.63g,19.3mmol)和无水二氯甲烷(100ml)加至反应瓶中,搅拌至zncl2溶解,加入1%mol的四丁基氯化铵为相转移催化剂,向反应瓶中滴加(2s,3αs,7αs)-八氢化吲哚-2-羧酸(5)(4.35,25.7mmol)、三乙胺(5.9g,57.9mmol)和无水二氯甲烷(100ml)的混合溶液。约5min滴加完毕,室温下搅拌0.5小时。反应毕,搅拌下加入0.5m盐酸,调节ph=3.8-4.0左右,静置分液。水相用二氯甲烷(40ml×3)萃取,合并有机相,经无水硫酸钠干燥后过滤,滤液经减压浓缩后得油状物培哚普利(6)7.02g(89.02%)。
4.制备培哚普利叔丁胺盐结晶物(7)
在24℃温度并搅拌下,向含有培哚普利(6)(4g,10.8mmol)的24ml乙酸乙酯溶液中滴加特丁基胺(0.88g,12mmol),经15分钟加完后,培哚普利叔丁胺盐结晶体开始从溶液里析出,该反应混合物加热到回流状态,直到获得澄清液体,然后趁热过滤,然后冷却滤液到20-25℃,等产物析出后,抽滤获得产物在40℃下减压干燥6小时,获得4.1g(85.5%)培哚普利叔丁胺盐(7)。
m.p.=152-154℃,[α]20d=-64.5,(c=0.5%etoh)。文献为-64.8。1h-nmr(300mhz;cdcl3),δppm:0.89(t,3h),1.13-1.73(m,2h),2.03(m,1h),2.10(m,1h),2.15(m,1h),3.10(m,1h),3.35(m,0.6h),3.53(m,1h),3.79(m,1h),4.12-4.23(m,2h),4.29(t,1h,j=18.2hz)。(与对照品1hnmr谱相一致)。13c-nmr(300mhz;cdcl3),δppm:13.8,13.9,14.3,19.0,20.0,24.0,25.9,28.3,28.6(3xch3),30.6,35.6,37.5,50.3,52.6,58.4,59.6,60.6,61.1,173.3,174.8,177.0(3xc=o)。msm/z:(m+h)+369(100%),(m-h)-367(100%)。
- 还没有人留言评论。精彩留言会获得点赞!