前列腺癌诊断用基因组及其应用的制作方法
本发明涉及前列腺癌诊断用基因组及其应用,特别是涉及ar、hoxc6和nkx2-2基因及其在制备或筛选诊断的药物或试剂盒、或筛选治疗前列腺癌的药物中的应用。
背景技术:
恶性肿瘤是威胁人类健康的最危险因素之一,我国每年仅肿瘤死亡人数近200万人,恶性肿瘤造成的经济损失高达1400多亿元人民币。在我国,前列腺癌位于男性恶性肿瘤发病率第六位,在男性泌尿系肿瘤中排名第一位;而在美国前列腺癌发病率位居男性恶性肿瘤的第一位,死亡率仅次于肺癌位于第二位。虽然前列腺癌有比较好的预后,但对于男性来说仍然是造成死亡的重要癌症。随着男性防癌意识的增强和前列腺癌筛查方法的广泛应用,早期发现的前列腺癌中,只有约5%初步诊断为无法治愈。然而30%-40%的前列腺癌患者最终会死于前列腺癌复发。
预后是指发病后,疾病未来过程的一种预先估计。在医学上,“预后”是指根据经验预测的疾病发展情况。预后主要涉及到三个方面,将发生什么结果、发生不良结果的可能性有多大、什么时候发生。预后分析是对疾病发病后发展为各种不同结局的预测;是临床非常实用、对临床很有指导作用的临床研究。研究和评级预后的目的,为了便于了解各种疾病对人类危害性的大小、探索影响预后的因素、研究改善预后的具体措施。
肿瘤标识物是可以在血清、血浆、其他体液、组织提取物或石蜡固定的组织中检测到的自然生产的分子。肿瘤标识物可以为前列腺癌的诊断,选择治疗方案、预测疾病进程和复发以及治疗效果进行监测等提供有价值的信息。前列腺癌作为男性高发性恶性肿瘤,极为缺乏高效的肿瘤标记物。hpc1是最早被发现的高共显前列腺癌易感基因,但估计在全部前列腺癌中仅占不到5%,其他已知的前列腺癌易感基因如msr1、hpc2等也仅能说明约1%左右的前列腺癌。目前我国在临床上应用的前列腺癌标志物有psa、psca、psma等,在同一亚型中,不同病人的预后、存活时间、复发等都存在着显著差异。这些数据表明,如何对前列腺癌进行准确的分型、尽早判断肿瘤是否会复发、转移,进而选择合理的治疗手段,提高前列腺癌患者的生存率和生活质量是前列腺癌治疗的重要发展方向。
技术实现要素:
本发明的目的之一在于提供前列腺癌诊断用基因组ar、hoxc6和nkx2-2。
本发明的目的之二在于提供该基因组的筛选方法。
本发明的目的之三在于提供上述基因在制备筛选和诊断前列腺癌药物中的应用。
本发明的目的之四在于提供上述基因制备筛选和诊断前列腺癌试剂盒中的应用。
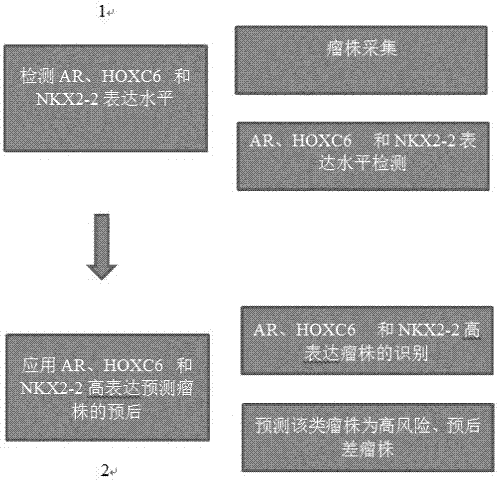
本发明发现ar、hoxc6和nkx2-2高表达与高风险前列腺癌的联系,通过检测前列腺癌样株中ar、hoxc6和nkx2-2的表达水平,识别出ar、hoxc6和nkx2-2高表达的前列腺癌的样株,并可进一步预测出该类前列腺癌可能具有较高的恶性程度,并表现出较差的预后。
为实现上述目的,本发明采用如下技术方案:
一种前列腺癌诊断用基因组,其特征在于该基因组为ar、hoxc6和nkx2-2基因。
一种前列腺癌的检测方法,采用上述的前列腺癌诊断用基因,其特征在于该方法的具体步骤为:
a.选取前列腺癌样本株组成3套数据;
b.采用simba算法寻找步骤a所得前列腺癌样本株中的互斥转录因子;
c.采用费舍尔精确检验推测转录因子和其配对基因的调节关系;
d.利用最小描述方法,即两个基因相关性最小情况,推测转录因子和相关的mirnas的调节网络;
e.通过qrt-pcr检测ar、hoxc6、nkx2-2的过表达。
上述的步骤b的具体步骤为:在r中调取simba程序包,对前列腺癌基因表达数据进行计算获得互斥转录因子。
上述的步骤c的具体步骤为:在r中调用simba程序包中的函数init_network对上述找到的互斥转录因子搜寻靶标并建立初步网络。
上述的步骤d的具体步骤为:在r中调用simba程序包中的函数filt_network对上一步构建的网络进行优化,获得最优化的mrna-mirna网络。
上述的步骤e的具体步骤为:提取前列腺癌细胞株totalrna,按照试剂盒方法反转录后做qrt-pcr实验检测ar、hoxc6、nkx2-2的表达。
一种上述的前列腺癌诊断用基因组在制备筛选和诊断前列腺癌药物中的应用。
一种上述的前列腺癌诊断用基因组在制备筛选和诊断前列腺癌试剂盒中的应用。
本发明的前列腺癌诊断用基因组通过下述方法来筛选:
a.选取前列腺癌样本株组成3套数据;
b.采用simba算法寻找步骤a所得前列腺癌样本株中的互斥转录因子;
c.采用费舍尔精确检验推测转录因子和其配对基因的调节关系;
d.利用最小描述方法,即两个基因相关性最小情况,推测转录因子和相关的mirnas的调节网络;
所述筛选诊断前列腺癌药物具体指使用ar、hoxc6和nkx2-2,通过监测ar、hoxc6和nkx2-2表达量对可能作为诊断前列腺癌药物使用的物质进行生物活性、药理作用或要用价值的评估。
所述筛选诊断前列腺癌的试剂盒具体指使用ar、hoxc6和nkx2-2,通过监测ar、hoxc6和nkx2-2表达量对可能作为诊断前列腺癌的试剂盒进行性能的评估,所述性能如准确性、特异性、稳定性等。
所述筛选治疗前列腺癌药物具体指使用具体指使用ar、hoxc6和nkx2-2,通过监测ar、hoxc6和nkx2-2表达量对可能作为治疗前列腺癌药物使用的物质进行生物活性、药理作用或药用价值的评估。
优选的,所述制备诊断前列腺癌的药物或试剂盒中,所述诊断前列腺癌具体指对前列腺癌恶性程度的预测和/或对前列腺癌预后的判断。
更优选的,所述前列腺癌恶性程度的预测具体指通过具体指通过ar、hoxc6和nkx2-2表达量的检测对前列腺癌样株的恶性程度进行预测。
更优选的,所述前列腺癌预后的判断具体指通过ar、hoxc6和nkx2-2表达量的检测对前列腺癌病人的病程和/或结局进行预后判断。
具体的,ar、hoxc6和nkx2-2高表达表示前列腺癌样株具有较高的恶性程度,并表现出较差的预后;ar、hoxc6和nkx2-2低表达表示前列腺癌样株具有较低的恶性程度,并表现出较好的预后。
本发明第二方面提供一种基于ar、hoxc6和nkx2-2高表达的高风险前列腺癌的新型诊断法,所述诊断方法具体为通过检测肿瘤样株中的ar、hoxc6和nkx2-2的表达水平对前列腺癌病人的预后进行判断。
具体的,所述检测肿瘤样株中的ar、hoxc6和nkx2-2的表达水平是指将现有的数据库中的肿瘤样株的ar、hoxc6和nkx2-2表达量信息进行数据分布分析,再检测患者前列腺癌样株中ar、hoxc6和nkx2-2的表达水平,根据数据分布识别出这三个基因互斥表达的前列腺癌样株。
在本发明实施例中,在统计学软件r工作平台中调用程序包rpackagelimma进行数据分布分析。
所述前列腺癌病人的预后进行判断具体指,ar、hoxc6和nkx2-2高表达的这类前列腺癌株可能具有较高的恶性程度,并表现出较差的预后。
本发明发明人发现通过检测瘤株中ar、hoxc6和nkx2-2的表达水平,能够识别出ar、hoxc6和nkx2-2高表达的瘤株,并可进一步预测该类瘤株为高风险、预后差的类型。此外,经公开可得知前列腺癌病人chip-seq、mrna和mirna-seq的基因表达数据和存活数据证实,通过ar、hoxc6和nkx2-2表达量的表征,能够有效预测出高风险、预后差的瘤株,可见ar、hoxc6和nkx2-2在制备或筛选诊断前列腺癌的药物或试剂盒、或筛选治疗前列腺癌的药物的用途中具有高度产业利用价值。
附图说明
图1显示为本发明的基于ar、hoxc6和nkx2-2高表达的高风险前列腺癌预后预测示意图;
图2显示为互斥转录因子的算法流程;
图3显示为通过算法所构建的转录因子与mirnas的调解网络图;
图4显示为公开可得的不同癌症或组织表达数据库中ar、hoxc6和nkx2-2互斥高表达分布图(a位tgca、b为robinson数据集,c为beltran数据集);
图5显示为ar、hoxc6和nkx2-2高表达的前列腺癌株的迁移与侵袭图;
图6显示为ar、hoxc6和nkx2-2高表达的四套前列腺癌株的存活分析曲线图(a为gse21032、b为glinskydataset、c为gse16560、d为tcgadatasets)。
具体实施方式
实施例一:
a.选取前列腺癌样本株组成3套数据;
b.采用simba算法寻找步骤a所得前列腺癌样本株中的互斥转录因子;
c.采用费舍尔精确检验推测转录因子和其配对基因的调节关系;
d.利用最小描述方法,即两个基因相关性最小情况,推测转录因子和相关的mirnas的调解网络;
e.从3套数据中都获得ar、hoxc6和nkx2-2过表达;
实施例二:
a.选取前列腺癌样本株组成3套数据;
b.采用simba算法寻找步骤a所得前列腺癌样本株中的互斥转录因子;具体步骤为:在r中调取simba程序包,对前列腺癌基因表达数据进行计算获得互斥转录因子。
c.采用费舍尔精确检验推测转录因子和其配对基因的调节关系;具体步骤为:在r中调用simba程序包中的函数init_network对上述找到的互斥转录因子搜寻靶标并建立初步网络。
d.利用最小描述方法,即两个基因相关性最小情况,推测转录因子和相关的mirnas的调解网络;具体步骤为:在r中调用simba程序包中的函数filt_network对上一步构建的网络进行优化,获得最优化的mrna-mirna网络。
实施例中所使用的前列腺癌样株信息取自ncbigeo,并采用两条标准对数据库进行数据筛选:1,瘤株样本数大于140例;2,具备临床信息特别是病人随访的存活时间数据,因此得到3套数据,具体如下:
1)seriesaccession:gse21034id:200021034samples(370)
2)seriesaccession:gse21035id:200021035samples(231)
3)seriesaccession:gse21036id:200021036samples(142)
数据处理:
我们首先运用simba算法,参见中国专利cn105512509a去寻找前列腺癌样株中的互斥转录因子,流程如图2示。首先参见a图,应用计算机二进制呈现出高斯混合分布离散连续的mrna和mirna表达谱成双峰分布,找出异常表达的基因;由于原发癌在转化成恶性癌症的过程中,其驱动的转录因子和靶mirnas的表达是异常的,为此用b图中费舍尔精确检验(fisher’sexacttests)推测转录因子和配对基因的调节关系;最后c图中利用最小描述方法(mdl)也就是两个基因相关性最小情况来推测转录因子和相关的mirnas的调解网络,驱动性转录因子比非驱动性转录因子可以调节更多的mirnas。
利用这个计算方法,我们重构了前面的三套前列腺癌基因表达数据库中转录因子的调节网络,如图3所示我们发现了三个互斥高表达的转录因子ar、hoxc6和nkx2-2及与之相关的mirnas,这些mirnas数量约占所有mirnas的数量的20%。随后我们通过qrt-pcr检测ar、hoxc6、nkx2-2的过表达,然后使用过表达的数据去检测癌细胞转移性的病人。
同时分析三套样本独立数据发现ar、hoxc6和nkx2-2互斥高表达在前列腺癌病人中是高度保守的,结果如图4所示,其中,a图数据库来源tgca;b图数据来源robinson数据集;c图数据来源beltran数据集。虚线是转录因子高表达的中断线。
同时如图5所示,高表达ar、hoxc6和nkx2-2后细胞的迁移与侵袭能力也增强了。相对于无ar、hoxc6和nkx2-21组而言,ar、hoxc6和nkx2-2高表达组前列腺癌样株均具有较高的迁移与侵袭能力。
依据瘤株样本的存活数据进行存活曲线分析,结果如图6所示。其中,横坐标表示的是瘤株样本来源的病人存活时间,纵坐标表示的是瘤株样本来源的病人前列腺癌复发几率(无转移生存率)。由图6中各图可见,相对于ar、hoxc6和nkx2-21低表达组而言(黑色曲线所示),ar、hoxc6和nkx2-2高表达表示前列腺癌样株均具有较高的恶性程度,并表现出较差的预后(浅蓝色、紫色、深蓝色和黄色曲线所示)。
综上所述,本发明发明人发现通过检测瘤株中ar、hoxc6和nkx2-2的表达水平,识别出ar、hoxc6和nkx2-2高表达的瘤株,并可进一步预测该类瘤株为高风险、预后差的类型。此外,经公开可得的前列腺癌病人chip-seq、mrna和mirna-seq的基因表达数据和存活数据证实,本发明能够有效预测出高风险、预后差的瘤株,具有高度产业利用价值。
上述实施例仅例示性说明本发明的原理及其功效,a.选取前列腺癌样本株组成3套数据;b.采用simba算法寻找步骤a所得前列腺癌样本株中的互斥转录因子;具体步骤为:在r中调取simba程序包,对前列腺癌基因表达数据进行计算获得互斥转录因子。c.采用费舍尔精确检验推测转录因子和其配对基因的调节关系;具体步骤为:在r中调用simba程序包中的函数init_network对上述找到的互斥转录因子搜寻靶标并建立初步网络。d.利用最小描述方法,即两个基因相关性最小情况,推测转录因子和相关的mirnas的调节网络;具体步骤为:在r中调用simba程序包中的函数filt_network对上一步构建的网络进行优化,获得最优化的mrna-mirna网络。而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
非另外说明,本发明中所公开的实验方法、检测方法、制备方法均采用本技术领域常规的分子生物学、生物化学、染色质结构和分析、分析化学、细胞培养、重组dna技术及相关领域的常规技术。这些技术在现有文献中已有完善说明,具体可参见sambrook等molecularcloning:alaboratorymanual,secondedition,coldspringharborlaboratorypress,1989andthirdedition,2001;ausubel等,currentprotocolsinmolecularbiology,johnwiley&sons,newyork,1987andperiodicupdates;theseriesmethodsinenzymology,academicpress,sandiego;wolffe,chromatinstructureandfunction,thirdedition,academicpress,sandiego,1998;methodsinenzymology,vol.304,chromatin(p.m.wassarmananda.p.wolffe,eds.),academicpress,sandiego,1999;和methodsinmolecularbiology,vol.119,chromatinprotocols(p.b.becker,ed.)humanapress,totowa,1999等。
- 还没有人留言评论。精彩留言会获得点赞!