一种用于检测染色体拷贝数变异的测序文库构建方法和试剂盒与流程
本发明涉及文库的构建方法和试剂盒,更具体地,本发明涉及用于检测染色体拷贝数变异的测序文库构建方法和试剂盒。
背景技术:
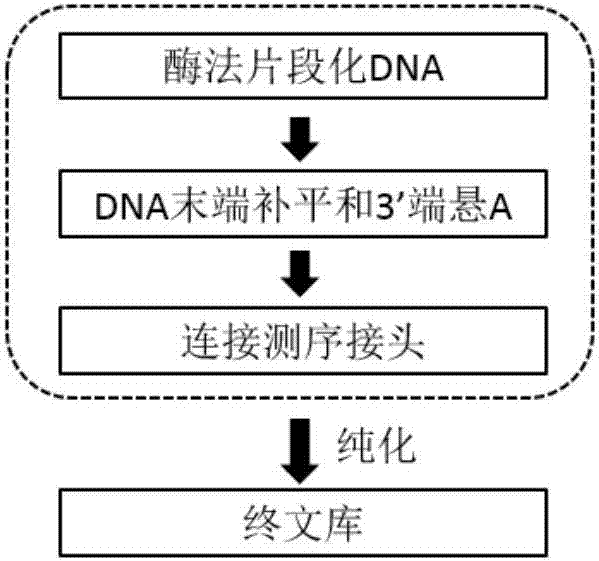
:研究表明,染色体数目的异常在已发现的染色体异常相关疾病中占有很大的比例(nagaoka,etal,2012)。染色体数目的异常除了可表现为整条染色体的数目改变(染色体非整倍体),也可表现为某染色体的某一片段的拷贝数变异(如染色体微缺失微重复综合征)。鉴于染色体异常疾病一般具有高发、危害性严重且缺乏有效的治疗手段等特点,所以利用技术手段准确检测染色体拷贝数是否存在异常具有重要的意义。采用传统核型分析技术(karyotyping)结合荧光原位杂交技术(fish)或染色体微列阵分析技术(chromosomemicroarrayanalysis,cma,如acgh和snp-array)是目前临床上用于检测染色体拷贝数变异的主要手段和标准(nord,etal,2015)。上世纪70年代建立起的高分辨率核型分析技术(yunis,1978;trask,2002)目前仍是检测染色体非整倍体、平衡或非平衡性结构重排、较大片段的缺失/重复以及嵌合体等染色体异常的金标准,但其分辨率较低(约为5mb),无法检出具有临床意义的染色体较小片段的拷贝数变异。而始于上世纪80年代后期的fish技术(trask,1991)将细胞遗传学、免疫学和分子生物学相结合,在临床研究上检测目标染色体非整倍体和结构异常等方面具有较好的应用,但是其特异性探针的设计受到对目标区域信息的限制,因而仅能够得到有限的染色体信息。近年来发展起来的染色体微阵列分析技术(cma)则是一种高通量检测基因组dna拷贝数变异的分子核型分析技术,其中相对成熟的比较基因组杂交(acgh)技术和单核苷酸多态性基因芯片(snparray)技术都属于cma技术(manningandhudgins,2010)。虽然cma技术相比于传统核型分析技术具有很高的分辨率(breman,etal,2012),但其在临床应用中需根据公共数据库中详尽的染色体异常及相关临床信息定制微阵列芯片,因此对某些罕见的或数据库中未涵盖的微缺失微重复(cnv)的检测存在不足。而且,目前cma技术的难度较高和成本相对较高等特点也限制了其在欠发达地区的广泛应用。因此,为了更广泛地排查和诊断染色体疾病,需要一种在保证检测通量和分辨率的前提下能够准确检测染色体拷贝数变异的经济、简单的方法。近些年日益成熟的高通量测序技术(nextgenerationsequencing,ngs)因其通量高、准确性高、灵敏性高、自动化程度高和运行成本低等突出的优势,使其在临床研究中得到广泛的应用(xuan,etal,2013)。目前通过ngs数据检测cnv最常用的原理是基于计算深度(read-depth)来实现的,即通过将某区段的深度与预先校正得到的理论值进行计算比较以判定该区段是否存在cnv,这种方法对于单端测序和双端测序都适用(yoon,etal,2009;mason-suares,etal,2016)。在ngs文库制备的过程中通常会采用标准的pcr来扩增随机片段,而pcr不可避免的扩增偏好则会对数据的准确性造成影响(vandijk,etal,2014;head,etal,2014;goodwin,etal,2016)。因此,建立一种简单的不依赖pcr的建库方法对于提高染色体非整倍性尤其是cnv的检测准确性至关重要。技术实现要素:本发明的目的在于克服传统检测染色体拷贝数变异的方法在分辨率、全基因组覆盖程度、通量和成本等方面的不足,同时克服ngs建库过程中pcr扩增偏好性带来的问题,提供了一种不依赖pcr的检测染色体拷贝数变异的测序文库构建方法和试剂盒。采用该方法和试剂盒对待测样本进行染色体拷贝数变异检测,可实现对染色体非整倍体和染色体微缺失微重复综合征的筛查和诊断。在第一个方面,本发明提供一种用于检测染色体拷贝数变异的测序文库构建方法,其主要包括以下步骤:(1)利用双链dna片段化酶将待测dna随机片段化;(2)对片段化后的dna进行末端补平和3’端悬a;(3)将末端补平并3’端悬a的dna与测序接头连接,获得连接产物;(4)纯化所述连接产物,获得测序文库,其中步骤(1)-(3)在单一反应管中完成。图1示出了根据本发明的方法构建测序文库的主要步骤。在一个实施方案中,待测dna的起始含量优选为10-260ng。本领域技术人员已知,为满足一定的上机测序要求,适合的文库总量应不低于2fmol。因此,为使所构建文库总量不低于2fmol,本发明人发现待测dna的起始含量最优为10-260ng。含量过低,将导致所得测序文库浓度不高,不能获得有效的测序数据;含量过高,则将导致待测dna不能被充分地随机片段化(即,将有部分待测dna未被片段化而仍然保持长片段的形式,这部分长片段dna不能被有效地测序),也会影响最终文库的浓度和测序的准确性(参见实施例2)。在一个实施方案中,双链dna片段化酶为非特异性切口核酸酶和t7内切酶突变体的混合酶。具体地,所述非特异性切口核酸酶是源自弧菌,例如嗜盐弧菌(vibriovulnificus)(vvn)的核酸酶,所述核酸酶可以是野生型或其突变体。所述t7内切酶突变体是指在美国公开号2007-0042379中描述的,例如在两个催化结构域之间的桥接区具有突变的t7内切酶。在一个实施方案中,包含非特异性切口核酸酶和t7内切酶突变体的双链dna片段化酶是本领域技术人员已知的,例如中国专利cn102301009a所述。在一个实施方案中,非特异性切口核酸酶和t7内切酶突变体的单位比例小于1:200,例如小于1:100或小于1:10,其范围可以为1:2-1:200。最优选地,非特异性切口核酸酶和t7内切酶突变体的单位比例为1:200。其中,1个单位t7内切酶突变体定义为在37℃下1个小时将90%的2μg线性切口的2.44kb的dsdna转化成2个片段(1.37kb和1.07kb)所需要的酶的量。1个单位vvn核酸酶和其突变体定义为在37℃下30min释放1a260单位的酸性可溶寡核苷酸所需要的酶的量。在一个实施方案中,根据dna是否落在大于60%gc(高gc含量)、40%-60%gc(标准gc含量)、或者小于40%gc(低gc含量)的范围内可确定dna片段化反应的最佳的温育时间。例如,温育时间范围可以典型地在5min至120min,例如,15min至60min范围内。在一个实施方案中,dna片段化反应的温度为35-40℃,最优选37℃。在一个实施方案中,待测dna的随机片段化是在缓冲液i存在下进行,其中缓冲液i主要包括20mm三羟甲基氨基甲烷-盐酸(tris-hcl)、15mm氯化镁(mgcl2)、50mm氯化钠(nacl)、0.1mg/ml牛血清白蛋白(bsa)、0.15%曲拉通(triton)。缓冲液i的存在能为随机片段化反应提供合适的反应环境,同时维持dna片段化酶的稳定性,并提高它的酶活力。在一个实施方案中,在步骤(1)和(2)之间还包括将所述双链dna片段化酶灭活的步骤。灭活所述双链dna片段化酶是指在60-70℃,例如65℃下温浴10-13min,优选在缓冲液ii存在下进行。所述缓冲液ii主要包括50mm氯化钠(nacl)、10mm三羟甲基氨基甲烷-盐酸(tris-hcl)、10mm氯化镁(mgcl2)、1mm二硫苏糖醇(dtt)。缓冲液ii的存在能为酶的灭活反应提供合适的离子浓度,同时也为接下来的末端补平和3’端悬a的反应提供合适的反应环境。在一个实施方案中,可以用本领域技术人员已知的任何适用于末端补平的酶对dna进行末端补平。这种酶的实例包括但不限于t4dna聚合酶、klenow酶等。在一个实施方案中,可以用本领域技术人员已知的任何适用于3’端悬a的酶对dna进行3’端悬a。这种酶的实例包括但不限于taq酶、klenowex-(newenglandbiolabs)(是一种改进的klenow酶,其3’-5’外切活性缺失)等。在本发明的文库构建方法中,末端补平和3’端悬a同时进行,这简化了操作步骤、节约成本,同时也降低了样本间的污染。在一个实施方案中,末端补平和3’端悬a所用的温育时间和温度可以根据具体需要由本领域技术人员根据常规技术确定。在一个实施方案中,可以用本领域技术人员已知的任何适用于连接测序接头的酶。这种酶的实例包括但不限于t4dna连接酶、t7dna连接酶或它们的混合物。本发明中的测序接头是与测序平台匹配的双链测序接头,是本领域技术人员根据常规技术可以选择的。优选地,该连接步骤是在缓冲液iii的存在下进行,所述缓冲液iii主要包括50mm氯化钠(nacl)、10mm三羟甲基氨基甲烷-盐酸(tris-hcl)、10mm氯化镁(mgcl2)、0.1%牛血清白蛋白(bsa)。缓冲液iii的存在能够为连接反应提供合适的反应环境,同时维持酶的稳定性。在一个实施方案中,在上机测序前可以用商购的试剂盒或本领域技术人员已知的任何试剂对根据本发明的文库构建方法所得的测序文库进行纯化。所述测序文库适用于第二代高通量测序平台,例如:illumina公司的hiseq/miseq/miniseq/myseq/novaseq测序平台、thermofisher公司的pgm/proton测序平台等。在第二个方面,本发明还提供一种用于构建检测染色体拷贝数变异的测序文库的试剂盒,其包括:双链dna片段化酶和缓冲液i、缓冲液ii、用于dna末端补平的酶和用于3’端悬a的酶、测序接头、连接酶和缓冲液iii,其中缓冲液i、ii和iii的成分如上所述。在一个实施方案中,本发明的试剂盒还包括用作阴性对照的dna样本。阴性对照是指不含染色体拷贝数变异的正常dna样本。本发明的文库构建方法基本上均通过酶反应完成,利用酶易失活的特点以及不同步骤中酶反应温度不同的特点使得可以在单一反应管中依次连续地完成dna随机片段化、末端补平和3’悬a、以及连接接头这三个步骤,步骤间无dna纯化过程。由于本发明的文库构建方法可以在单一反应管中完成,因此也避免了dna样本在转移之中造成的样本损失以及样本污染。此外,由于所涉及的试剂种类较少,本发明的文库构建方法和试剂盒操作流程较为简单,具有较好的易操作性。并且,本发明的文库构建方法不包含pcr步骤,从而避免了因pcr操作本身带来的扩增偏好性,最终提高检测的准确性。最后,本发明的文库构建方法和试剂盒可对低至10ng的样本dna实现准确的检测,即,dna样本的起始含量比较低。附图说明图1:本发明所述的文库构建方法的示意图。图2:采用本发明的方法构建文库后对待测dna样本(a)和对照正常样本(b)进行测序的结果。图3:采用本发明的方法构建文库后对第二个待测dna样本(a)和对照正常样本(b)进行测序的结果。具体实施方式实施例1:根据本发明的方法构建文库并用于测序dna样本根据本发明的用于检测染色体拷贝数变异的测序文库的构建方法和试剂盒,对以帕陶氏综合征(t13)为例的染色体非整倍体、以染色体22q11.2微缺失综合征为例的染色体微缺失进行检测。具体地,帕陶氏综合征是第13组染色体中出现第3个染色体引起的,是常见的三体综合征之一;染色体22q11.2微缺失综合征是指22号染色体22q11.21-q11.23微小缺失所造成的临床症候群,是人类最常见的微缺失综合征。根据以下方法检测样本dna:1.测定浓度:用qubit荧光计测定待测dna样本的浓度,使dna样本的起始含量优选为10-260ng。2.构建文库:按照本发明所述的用于构建检测染色体拷贝数变异的测序文库的方法进行文库构建,具体包括以下步骤:(1)取待测dna样本和阴性对照(即正常dna样本)在冰上按下表1分别配制反应混合物。表1:试剂加入量dna样本10ng双链dna片段化酶1μl缓冲液i1μleb缓冲液补至10μl混合均匀后,将反应体系置于37℃温育10min。(2)将反应混合物转至冰上,加入7μl的缓冲液ii,充分混匀后置于65℃下温育10min,以灭活双链dna片段化酶。(3)将反应混合物转至冰上,加入1.5μlt4dna聚合酶和1.5μltaq酶,用eb缓冲液将体积补至50μl。充分混匀后,在室温下瞬间离心5s,轻弹去除气泡。然后将上述反应混合物放入普通pcr仪(或恒温金属浴)中开始下列温育步骤:首先37℃,20min,然后72℃,20min,最后4℃,5min。(4)在pcr仪(或恒温金属浴)中完成温育后,将反应混合物取出并置于冰上。同时,根据下表2配制预混液:表2:试剂加入量缓冲液iii25μlt4dna连接酶1μl将预混液进行充分混匀后,在室温下瞬间离心5s。然后将该预混液加入已经完成温育的反应混合物,并加入2μl的测序接头在反应管壁上,然后立即在室温下瞬间离心5s,快速混匀后,在室温下瞬间离心5s,并轻弹去除气泡,再次在室温下瞬间离心5s。然后将上述反应混合物放入pcr仪(或恒温金属浴)中开始如下温育步骤:首先20℃,15min,然后65℃,10min,最后4℃,5min。温育完成后,即可获得测序文库。3.文库纯化和上机测序:使用高通量测序文库构建dna纯化试剂盒(磁珠法)(杭州贝瑞和康基因诊断技术有限公司生产,货号r0022)纯化所得测序文库,纯化所得测序文库,使其总量应不低于2fmol。使用nextseqcn500(国械注准20153400460)测序仪,根据制造商的说明进行上机测序。4.分析测序结果:将测序结果与人类基因组参考序列进行比对,待测样本和正常样本的染色体拷贝数检测结果分别如图2和图3所示。在图2和图3中,纵坐标log2(ratio)中ratio是指检测区域的拷贝数与2(二倍体)的比值,横坐标代表整条染色体区域。其中整条染色体被均匀地分成若干个区域(bin),每个点代表一个bin的log2(ratio)值,然后根据所有点的分布得到趋势线(灰色实线)。图中的黑色实线表示染色体的着丝点位置,将染色体分为短臂和长臂。由于目前人类基因组参考序列中尚无可供数据分析用的13号染色体短臂和22号染色体短臂的参考序列,因此无法分析13号染色体短臂和22号染色体短臂的相关测序数据,导致在这些区域没有相应的log2(ratio)值。具体地,图2示出了待测dna样本(a)和正常样本(b)的13号染色体区域的拷贝数检测结果。从结果可以看出,待测dna样本的13号染色体区域长臂的log2(ratio)值为0.585(=log21.5)左右,说明至少13号染色体的长臂部分有三个拷贝,即待测dna样本为帕陶氏综合征(t13);而正常样本的13号染色体区域的log2(ratio)值均为0(=log21),说明13号染色体为二倍体。用snp-array芯片检测该待测dna样本,证实确实存在三个拷贝的13号染色体长臂。换言之,本发明的方法和试剂盒建库测序的检测结果与临床检测结果一致。此外,还使用本发明的方法和试剂盒对第二个待测dna样本和正常样本进行建库并测序,结果如图3所示。从检测结果可以看出,待测dna样本的22号染色体在第20m左右的区域其log2(ratio)值约为-1(=log20.5),说明该区域为单倍体。将染色体的横坐标与人类基因组参考序列进行比对,发现上述单倍体区域为22q11.21-q11.23区域,即待测dna样本为染色体22q11.2微缺失综合征;而正常样本的22号整条染色体区域的log2(ratio)值均为0(=log21),说明不存在22号染色体22q11.2区域的缺失。用snp-array芯片检测该待测dna样本,证实确实在22号染色体22q11.2区域具有微小缺失。换言之,本发明的方法和试剂盒建库测序的检测结果与临床检测结果一致。以上实施例说明,本发明的文库构建方法以及相应的试剂盒可以用于通过高通量测序来准确地检测染色体拷贝数的变异。实施例2:dna起始含量对测序文库浓度的影响根据实施例1所述的构建文库和纯化文库的方法,用起始含量不同的dna样本制备测序文库,并测定所得文库的总量,结果如下表3所示。表3.dna的起始含量与最终所得文库的总量。起始dna量(ng)文库总量(fmol)<50.142<50.2<50.21<50.108<50.129<50.16951.05351.592102.776102.401502.97502.9881002.3181003.111502.4771502.7522002.3482001.7822202.4622201.7582402.0782401.9352602.1492602.2282801.4052801.5852901.9822901.8123001.043000.568从上表可以看出,当dna起始含量为10-260ng时,所得文库的最终总量基本上大于2fmol,满足测序的需要。当浓度过低或过高,所得文库的浓度均低于2fmol,不是最优适合用于测序。因此,在本发明中,用于制备文库的起始dna含量优选为10-260ng。以上所述仅为本发明的实施例,并不用于限制本发明,对于本领域的技术人员来讲,本发明可以有更改和变化。凡在本发明的精神和原则之内,所作的任何修改、同等替换、改进等,均应包含在本发明的保护范围之内。参考文献nagaokasi,hassoldtj,andhuntpa.humananeuploidy:mechanismsandnewinsightsintoanage-oldproblem.nat.rev.genet.2012,13:493e504norda,salipantesj,pritchardc.chapter11–copynumbervariantdetectionusingnext-generationsequencing.clinicalgenomics,2015,8:165-187yunisj.high-resolutionchromosomeanalysisinclinicalmedicine.prog.clin.pathol.,1978,7:267-288traskbj.humancytogenetics:46chromosomes,46yearsandcounting.nat.rev.genet.2002,3:769e778traskbj.fluoresenceinsituhybridization:applicationincytogeneticsandgenemapping.trendsgnent.1991,7:149-154.manningm,andhudginsl.array-basedtechnologyandrecommendationsforutilizationinmedicalgeneticspracticefordetectionofchromosomalabnormalities.genet.med.2010,12(11):742-745.bremana,pursleyan,hixsonp,biw,wardp,bacinoca,shawc,lupskijr,beaudeta,patela,cheungsw,vandenveyveri:prenatalchromosomalmicroarrayanalysisinadiagnosticlaboratory:experiencewith>1000casesandreviewoftheliterature.prenat.diagn.2012,32:351e361.xuanj,yuy,qingt,guol,shil.next-generationsequencingintheclinic:promisesandchallenges.cancerlett.2013,340(2):284-295.yoons,xuanz,makarovv,yek,sebatj.sensitiveandaccuratedetectionofcopynumbervariantsusingreaddepthofcoverage.genomeres.2009,19(9):1586-1592.mason-suaresh,landryl,leboms.detectingcopynumbervariationvianextgenerationtechnology.curr.genet.med.report.2016,4(3):1-12.vandijkel,jaszczyszyny,thermesc.librarypreparationmethodsfornext-generationsequencing:tonedownthebias.exp.cellres.2014,322(1):12-20.headsr,komorihk,lameresa,whisenantt,vannieuwerburghf,salomondr,ordoukhanianp1.libraryconstructionfornext-generationsequencing:overviewsandchallenges.biotechniques.2014,56(2):61-64.goodwins,mcphersonjd,mccombiewr.comingofage:tenyearsofnext-generationsequencingtechnologies.nat.rev.genet.2016,17(6):333-351.当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1