一种吲哚啉类化合物及其用途的制作方法
本发明涉及一种染料化合物,具体是一种吲哚啉类化合物及其用途。
背景技术:
伴随着人类工业文明的发展,煤、石油和天然气等矿物资源日益枯竭,由此引发的能源危机和环境污染已成为亟待解决的严重问题。因此人们迫切需要寻找其他新的可替代能源。有机薄膜太阳能电池,诸如染料敏化太阳能电池、钙钛矿太阳能电池以及有机聚合物电池等,因其低成本,具有潜在的实用价值。在上述光伏器件中,有机分子作为敏化染料、空穴传输材料以及电子传输材料等极大影响着器件的性能。吲哚啉类化合物由于具有较好的荧光性能以及合适的能级结构,在上述材料中得到广泛应用。但是现有吲哚啉类化合物仍然存在两个重要缺陷:1)化合物的光捕获范围仍然较窄;2)化合物平面性较差,导致其自组装能力较弱,使其作为空穴传输材料或电子传输材料时迁移率/电导率较低。这两个问题直接影响了基于吲哚啉类化合物的有机分子在上述器件中的性能,限制了其在上述器件中的应用。因此,通过分子工程,调控吲哚啉类染料分子结构,拓展其光捕获范围,增强其分子平面性,是当今吲哚啉类染料研究的热点。
技术实现要素:
本发明目的之一在于提供一种吲哚啉类化合物,其具有较强的光捕获能力并且具有较好平面性。
本发明的目的还在于提供一种所述吲哚啉类化合物的用途。
本发明所说的吲哚啉类化合物,其具有如下式(i)所示结构:
式(i)中:r1为c1~c12烃基或c1~c12烷氧基或c4~c20杂环基,其中所说杂环基的杂原子为n或s或o;r2为c1~c12烃基。
本发明的一个优选方案,r1为c1~c6烷基或c1~c6烷氧基或c4~c10杂环基,其中所说杂环基的杂原子为n或s或o;r2为c1~c8烃基。
更优选的方案,r1为c1~c3烷基或c1~c3烷氧基或c5~c6杂环基,其中所说杂环基的杂原子为n;r2为c4~c8烃基。
最佳的方案,r1为甲基;r2为c8烃基。
制备本发明所说的吲哚啉类化合物,其合成路线反应式如下:
上述合成路线r1和r2的含义与前文所述相同。
具体的,制备本发明所说的吲哚啉类化合物的方法,包括如下步骤:
(1)2,3-环戊基吲哚类化合物[式(a)所示化合物]和邻溴苯甲酸甲酯[式(b)所示化合物]在碘化亚铜催化下,在邻二氯苯中回流反应12小时得到式(c)所示化合物;
(2)将式(c)所示化合物与r2基溴化镁通过格氏反应得到式(d)所示化合物;
(3)将式(d)所示化合物和三氟化硼的乙醚溶液在有惰性存在条件下反应5小时得粗产物,所得产物用乙酸乙酯/石油醚重结晶可得目标产物,即式(i)所示化合物。
本发明所说的吲哚啉类化合物,即式(i)所示化合物可作为合成敏化染料、空穴传输材料以及电子传输材料的重要中间体。
附图说明
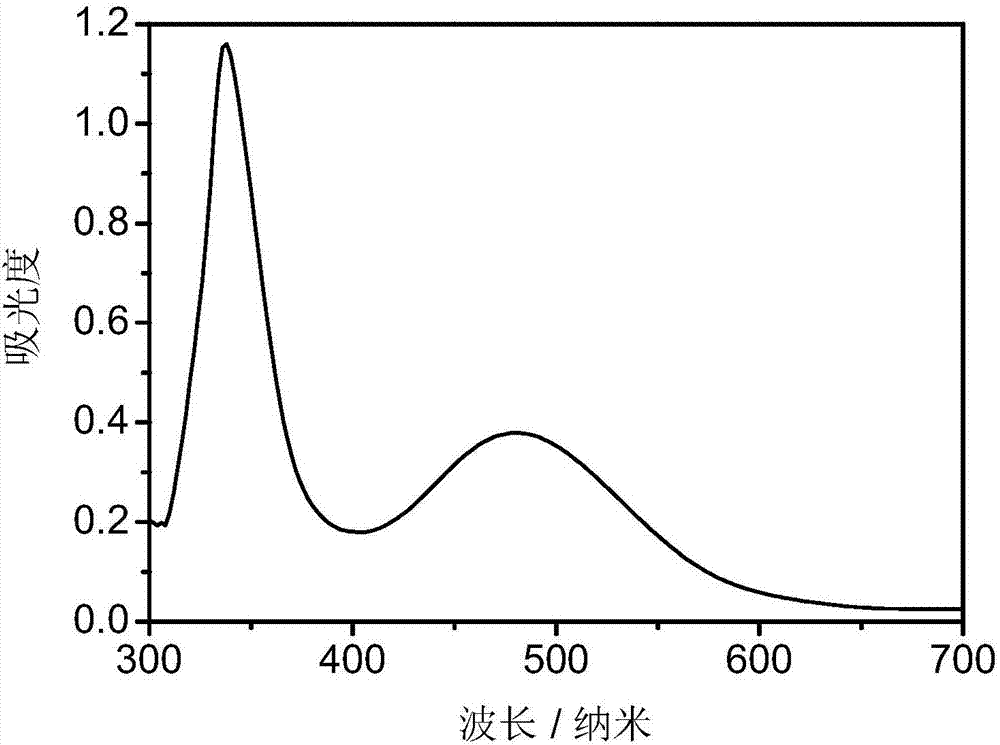
图1为实施例3所制备的吲哚啉类化合物14在甲醇溶液中的紫外吸收谱图(3×10-4mol/l)。
图2为实施例3所制备的吲哚啉类化合物14吸附在二氧化钛电极之后的紫外吸收谱图。
图3为实施例3所制备的吲哚啉类化合物14作为敏化剂应用于染料敏化太阳电池的电流-电压曲线图。
图4为实施例4所制备的吲哚啉类化合物19作为空穴传输材料应用于钙钛矿太阳电池的电流-电压曲线图。
具体实施方式
下面通过实例对本发明作进一步的说明,目的在于更好理解本发明的内容。因此,所举之例并不限制本发明的保护范围。
实施例1化合物7的合成(其中,r1=r2=ch3)
在250ml单口烧瓶中加入5.00g(23.26mmol)化合物1与6.64g(28.99mmol)化合物2,加入150ml甲醇,安装分水器在70°下回流搅拌24小时,停止反应。旋干溶剂后,用水分多次洗反应物,乙酸乙酯萃取,乙酸乙酯层旋干柱层析方法提纯得到5.5g油状化合物2。1hnmr(500hz,cdcl3):7.61(s,1h),7.54(d,j=8.0hz,1h),7.15(d,j=8.0hz,1h),3.94(s,3h),2.35(s,3h)。
在100ml单口圆底烧瓶中加入2.29g(1.00mmol)化合物2和1.57g(1.00mmol)化合物3,
cu粉0.64g(1.00mmol)、1.9gcui(1.00mmol)、k2co3固体2.07g(1.5mmol)、50ml邻二氯苯,氮气保护下升温至180°回流搅拌24-48小时。反应完毕减压蒸馏掉溶剂。用ch2cl2300ml萃洗固体3次,抽滤得到滤液旋干,用石油醚柱层析得到1.88g产物,产率61.56%。1hnmr(500hz,cdcl3):7.83(s,1h),7.46(t,j=7.5hz,2h),7.32(d,j=8.0hz,1h),7.01-7.11(m,3h),3.95(s,3h),2.94(t,j=7.5hz,2h),2.66-2.80(m,2h),2.53-2.58(m,2h),2.50(s,3h)。
在250ml单口烧瓶内加入4.2g(13.75mmol)化合物4和冰乙酸150ml,搅拌下分批少量加入硼氢化钠固体共20.8g(550.12mmol),保持温度在40°,加完后70°反应7小时,降温至50°反应过夜,冷却至室温。反应液水洗多次,乙酸乙酯萃取(直至冰乙酸水洗干净),萃取液旋干石油醚过柱提纯得到3.89g化合物5,产率91.90%。1hnmr(500hz,cdcl3):7.61(s,1h),7.30(d,j=7.5hz,1h),7.23(d,j=7.5hz,1h),7.06(d,j=7.5hz,1h),6.89(t,j=7.5hz,1h),6.61(t,j=7.5hz,1h),6.22(d,j=7.5hz,1h),4.80(t,j=7.5hz,1h),3.81(t,j=7.5hz,1h),3.95(s,3h),2.94(t,j=7.5hz,2h),2.66-2.80(m,2h),2.53-2.58(m,2h),2.37(s,3h)。
在单口圆底烧瓶内加入0.96g(3.12mmol)化合物5,取8ml甲基溴化镁,除氧并氮气保护下搅拌回流12h,反应液用饱和氯化铵溶液洗乙酸乙酯萃取,旋干过柱提纯得到0.52g淡黄色固体化合物6。1hnmr(500hz,cdcl3):7.89(s,1h),7.18(d,j=7.5hz,1h),7.12(d,j=8.5hz,1h),6.98-7.04(m,2h),6.88(t,j=7.5hz,1h),6.24(d,j=8.5hz,1h),4.53(t,j=7.5hz,1h),3.98(t,j=7.5hz,1h),2.40(s,3h),1.99-2.16(m,2h),1.86-1.90(m,1h),1.81-1.84(m,1h),1.74-1.79(m,2h),1.71(s,3h),1.66(s,3h)。
在25ml单口瓶中加入0.11g(0.36mmol)化合物6,加入0.4g阳离子交换树脂amberlyst15,加入15ml甲苯并安装分水器装置,搅拌回流12h,反应物抽滤,滤液旋干,石油醚柱层析得到0.06g化合物7,产率61.93%。1hnmr(500hz,acetone-d6):7.23(s,1h),7.12(d,j=7.5hz,1h),6.92-6.99(m,2h),6.75(t,j=7.5hz,1h),6.65(d,j=7.5hz,1h),4.62(t,j=8.5hz,1h),3.92(t,j=8.5hz,1h),2.27(s,3h),1.89-2.02(m,2h),1.79-1.86(m,2h),1.72(s,3h),1.63-1.69(m,2h),1.42(s,3h)。
实施例2化合物9的合成(其中,r1=ch3,r2=c8h17)
在单口圆底烧瓶内加入1.00g化合物5,取10ml正辛基基溴化镁,除氧并氮气保护下搅拌回流12h,反应液用饱和氯化铵溶液洗乙酸乙酯萃取,旋干过柱提纯得到0.60g化合物8。1hnmr(500hz,cdcl3):7.89(s,1h),7.18(d,j=7.5hz,1h),7.12(d,j=8.5hz,1h),6.99-7.02(m,2h),6.88(t,j=7.5hz,1h),6.24(d,j=8.5hz,1h),4.53(t,j=8.5hz,1h),3.98(t,j=8.5hz,1h),2.40(s,3h),2.06-2.08(m,1h),1.80-1.85(m,5h),1.46-1.52(m,10h),1.31-1.38(m,18h),0.90(t,j=8.5hz,6h)。
在25ml单口瓶中加入0.11g化合物8,加入0.4g阳离子交换树脂amberlyst15,加入15ml甲苯并安装分水器装置,搅拌回流12h,反应物抽滤,滤液旋干,石油醚柱层析得到0.06g化合物9,产率61.93%。1hnmr(500hz,acetone-d6):7.23(s,1h),7.12(d,j=7.5hz,1h),6.94-6.99(m,2h),6.75(t,j=7.5hz,1h),6.65(d,j=8.0hz,1h),4.62(t,j=8.5hz,1h),3.92(t,j=8.5hz,1h),2.27(s,3h),2.03-2.05(m,1h),1.76-1.80(m,5h),1.42-1.46(m,10h),1.31-1.35(m,18h),0.90(t,j=8.5hz,6h)。
实施例3将实施例1中化合物7应用于合成敏化染料化合物14
在100ml单口瓶中加入0.51g化合物7,并用ch2cl2溶解,在冰浴下恒压滴液漏斗加入用乙腈溶解的0.41gn-溴代琥珀酰亚胺溶液,避光反应3h,水洗反应液,乙酸乙酯萃取旋干过柱提纯得到0.43g化合物10,产率66.48%。1hnmr(500hz,acetone-d6):7.25-7.28(m,2h),7.12(s,1h),6.99(d,j=8.0hz,1h),6.70(d,j=8.0hz,1h),4.70(t,j=8.5hz,1h),3.98(t,j=8.5hz,1h),2.29(s,3h),2.05-2.14(m,2h),1.98-2.01(m,2h),1.82-1.87(m,2h),1.70(s,3h),1.43(s,3h)。
将0.2g(0.54mmol)化合物10用干燥的thf溶解加入到50ml三口瓶中,氮气保护下搅拌反应底物至温度降到-78°,缓慢滴加2ml的n-buli后搅拌1-2h,缓慢滴加0.51g(2.71mmol)硼酸三异丙酯,搅拌30min后取出,室温反应12h。水洗乙酸乙酯萃取后旋出溶剂,得到化合物11的粗产物,不经提纯直接进行下一步反应。
在100ml单口烧瓶中加入50ml甲苯,加入2.5mol/l的碳酸钾溶液4ml,除去溶剂中的氧气,加入上一步反应产物11,加入0.21g(0.54mmol)的化合物12,无氧条件下反应12h,水洗后蒸除溶剂,硅胶柱层析提纯得到红色粘稠状化合物13,两步总产率49%。1hnmr(400hz,cdcl3):8.21(d,j=8.5hz,2h),8.10(d,j=8.5hz,2h),7.85-7.87(m,2h),7.80(d,j=8.0hz,1h),7.72(s,1h),7.29(s,1h),7.06(d,j=8.0hz,1h),6.74(d,j=8.0hz,1h),4.78(t,j=8.5hz,1h),4.13(t,j=8.5hz,1h),2.39(s,3h),2.12-2.22(m,2h),1.96-2.08(m,2h),1.83-1.87(m,4h),1.69-1.72(m,10h),1.59(s,3h)。
取化合物13于25ml圆底烧瓶中,加入1mlcf3cooh,搅拌1h后水洗反应液,二氯甲烷萃取得到的有机层旋干,硅胶柱层析提纯得到红色固体化合物14,产率91%。1hnmr(400hz,dmso):13.0(s,1h),8.13(d,j=8.0hz,2h),8.09(d,j=8.0hz,2h),7.98(d,j=7.5hz,1h),7.90(d,j=7.5hz,1h),7.86(s1h),7.70(s,1h),7.25(s,1h),6.97(d,j=8.0hz,1h),6.69(d,j=8.0hz,1h),4.73(t,j=8.5hz,1h),4.02(t,j=8.5hz,1h),2.25(s,3h),2.03-2.11(m,1h),1.82-1.96(m,3h),1.68-1.72(m,4h),1.43-1.18(m,4h).
实施例4将实施例1化合物7应用于合成空穴传输材料化合物19
在100ml单口瓶中,加入3.84g(30mmol)化合物15,3.14g(10mmol)化合物16,加入2mol/l的碳酸钾4ml,加入60ml的四氢呋喃,然后鼓泡30min除去氧气,然后加入0.1g(0.09mmol)四三苯基膦钯,在氮气保护下80℃回流搅拌12h,冷却后加入乙酸乙酯20ml,有机相用冰水洗涤3次,用无水硫酸钠干燥,然后过滤蒸发滤液得到粗产物,硅胶柱层析提纯得到2.36g化合物17,产率73%。1hnmr(500hz,cdcl3)δ8.18(s,3h),7.78(d,j=2.5hz,3h),7.51(d,j=2.5hz,3h),7.23(t,j=2.5hz,3h)。
在100ml单口瓶中,将2.36g上一步产物化合物17溶于50ml二氯甲烷,并将单口瓶置于冰浴中且避光,然后将4.5gnbs用乙腈溶解倒入恒压滴液漏斗中缓慢滴入单口瓶中,氮气保护下冰浴搅拌4h。反应物倒入100ml水中,用二氯甲烷萃取三次,得到的液体旋干,硅胶柱层析提纯得到3.38g化合物18,产率83%。1hnmr(500hz,cdcl3)δ8.14(s,3h),7.13(d,j=2.5hz,3h),7.08(d,j=2.5hz,3h)。
将前述化合物11和0.06g化合物18,加入2mol/l的碳酸钾4ml,加入50ml的四氢呋喃,然后鼓泡30min除去氧气,然后加入0.1g(0.09mmol)四三苯基膦钯,在氮气保护下80℃回流搅拌12h,冷却后加入乙酸乙酯20ml,有机相用冰水洗涤3次,用无水硫酸钠干燥,然后过滤蒸发滤液得到粗产物,硅胶柱层析得到133mg化合物19,产率56%。1hnmr(500hz,acetone-d6):8.33(s,3h),7.22-7.31(m,12h),7.16(s,3h),6.96(d,j=8.0hz,3h),6.77(d,j=8.0hz,3h),4.65(t,j=8.5hz,3h),3.96(t,j=8.5hz,3h),2.29(s,9h),2.05-2.14(m,6h),1.98-2.01(m,6h),1.82-1.87(m,6h),1.70(s,9h),1.43(s,9h).
实施例4将实施例3所制备的吲哚啉化合物14应用于染料敏化太阳电池并对其进行光伏性能测试
以化合物14为光敏化剂制备染料敏化太阳电池器件:将烧制好的纳米二氧化钛电极(二氧化钛厚度:6微米)加热到80℃后,在化合物14配制成的3×10-4mol/l甲醇溶液中浸泡敏化6小时。敏化完成后取出,用无水甲醇冲洗掉未吸附的染料,吹干得到染料敏化的二氧化钛电极。将此敏化电极和镀铂的玻璃电极面对面组合得到染料敏化太阳电池器件。向其注入事先配好的电解质溶液并对其进行测量。电解质溶液的组成:0.6mol/l的3-甲基-1-丁基咪唑碘、0.05mol/l的i2、0.1mol/l的lii、0.5mol/l的叔丁基吡啶和用乙腈作为溶剂。在太阳模拟器100mw/cm-2光源照射下测定其开路电压为0.694v,短路电流为18.02macm-2,填充因子为0.707,光电转化效率为8.84%。其光电流和光电压曲线见图3。图3表明:相比传统结构的吲哚啉类敏化剂,本发明所说的吲哚啉类化合物14应用于染料敏化太阳电池能够在较薄的二氧化钛电极条件下即可产生更高的短路电流(chem.soc.rev.,2013,42,2039-2058;chem.sci.,2017,8,2115-2124)和较高的开路电压,从而得到较高的光电转换效率,证明其相比传统结构的吲哚啉敏化剂更适合作为染料敏化太阳电池中的敏化剂使用。
实施例5将实施例3所制备的吲哚啉类化合物19作为空穴传输材料应用于钙钛矿太阳电池并进行光伏性能测试
以化合物19作为空穴传输材料制备钙钛矿太阳电池器件:将烧制好的纳米二氧化钛电极在手套箱中以3000-4500rpm转速旋涂钙钛矿层,110℃下加热30分钟,放置于手套箱自然冷却。将化合物19配置为0.3mol/l的氯仿溶液,以4000-6500rpm转速旋涂于上述旋涂完钙钛矿层的电极上,然后在手套箱中放置8小时,使之固化,再放入低湿度干燥塔中氧化12小时。氧化完成的电极放入真空蒸镀机,依次蒸镀8纳米的moo3和100纳米银,得到制备完成的钙钛矿太阳电池器件。在太阳模拟器100mw/cm-2光源照射下测定其开路电压为1007mv,短路电流为19.37macm-2,填充因子为0.792,光电转化效率为15.44%。其光电流和光电压曲线见图4。图4表明:由于具有较好的平面性,证明吲哚啉化合物19具有较好的成膜性,能够成功地作为空穴传输材料应用于钙钛矿太阳电池中,并可产生较高的短路电流和开路电压,从而得到较高的光电转换效率,证明其适合作为钙钛矿太阳电池中的空穴传输材料使用。
- 还没有人留言评论。精彩留言会获得点赞!