化合物及其制备方法和应用与流程
本发明涉及多肽合成领域,特别涉及化合物及其制备方法和应用。
背景技术:
兰瑞肽(lanreotide)是一种生长抑素八肽类似物,其比天然生长抑素更具活性,与奥曲肽性质相似。长效制剂注射用于治疗肢端肥大症和促甲状腺激素腺瘤,也可用于控制神经内分泌肿瘤的症状,尤其是类癌综合征。最早由法国的益普生制药集团研制年,1994以注射用粉针剂首先在法国上市,2002年在中国批准上市,2007年经美国fda批准以长效缓释剂上市。
兰瑞肽的结构信息如下:
化学名:[3-(2-萘基)]-d-丙氨酰基-l半胱氨酰基-l-酪氨酰基-d-色氨酰基-l赖氨酰基-l-缬氨酰基-l半胱氨酰基-l-苏氨酰胺环状(3→7)-二硫化合物;
3-(2-naphthalenyl)-d-alanyl-l-cysteinyl-l-tyrosyl-d-tryptophyl-l-lysyl-l-valyl-l-cysteinyl-l-threoninamidecyclic(2-7)-disulfide;
结构如式ⅰ所示:
分子式:c54h69n11o10s2;分子量:1096.32;cas登记号:108736-35-2(兰瑞肽)。
目前多采用boc策略进行合成兰瑞肽,缺点在于:采用昂贵的二苯基甲胺-聚苯乙烯树脂作为载体,使用强腐蚀性的试剂(33%三氟乙酸)作为n端保护基的脱除试剂,对设备和操作具有较强的耐酸要求,同时废酸处理等不符合绿色化学的理念。
fmoc固相合成策略,固相合成在反应后处理上具有一定优势,但是有以下不足:a、需要3~5倍甚至更高倍数的fmoc-保护氨基酸投料。b、在偶联一个氨基酸后,需要极大量的dmf洗涤(约3~5次洗涤)。造成成本的增加和溶剂处理的困难。c、反应属于非均相反应。对于固相成环,偶联疏水性氨基酸等,由于位阻大,偶联效果欠佳,需要二次甚至三次投料。d、由于树脂体积较大,溶剂用量较大,导致反应釜效较低,一批次产量较低。
因此,提供一种操作简便、收率较高的兰瑞肽的合成方法具有重要的现实意义。
技术实现要素:
有鉴于此,本发明提供了化合物及其制备方法和应用。该化合物应用多肽合成,工艺操作简化,同时减少了粗肽的损失,提高了收率。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了化合物,结构如式ⅱ所示:
r选自c16~c25脂肪链。
在本发明的一些具体实施方案中,化合物结构如式ⅷ所示:
本发明还提供了所述的化合物的制备方法,以结构如式iv所示的化合物为原料,经取代反应、还原反应、氨解反应,再经水解反应,沉淀制得;
在本发明的一些具体实施方案中,所述取代反应采用的试剂为1-溴十六烷、1-溴十八烷、1-碘十八烷、1-溴二十五烷中的一种,其中优选1-溴十八烷;所述取代反应的缚酸剂为碳酸钾、碳酸钠、dipea,tea,优选碳酸钾;所述取代反应的温度为70~100℃,所述取代反应的时间为14~18小时。
在本发明的一些具体实施方案中,所述还原反应采用的催化剂为硼氢化钠、氰基硼氢化钠中的一种或两者的混合物,优选硼氢化钠;所述还原反应的溶剂为四氢呋喃、甲基四氢呋喃中的一种或两者的混合物,优选四氢呋喃;所述还原反应的试剂为甲醇、乙醇、异丙醇,优选甲醇,温度为50~65℃。
在本发明的一些具体实施方案中,所述氨解反应的试剂为甲烷磺酸和乌来糖;所述氨解反应的温度为100~110℃,所述氨解反应的时间为2~4min。
在本发明的一些具体实施方案中,所述水解反应的试剂为氢氧化钠、氢氧化钾中的一种或两者的混合物;所述水解反应的温度为90~110℃,所述水解反应的时间为14~17小时。
本发明还提供了所述的化合物或如所述的制备方法制得的化合物在多肽合成中的应用。
在本发明的一些具体实施方案中,所述多肽为兰瑞肽。
本发明还提供了兰瑞肽的合成方法,以式ⅱ所示化合物为原料,依次偶联氨基酸,脱保护,制得兰瑞肽粗肽,裂解。
在本发明的一些具体实施方案中,所述偶联和脱保护反应采用的反应溶剂为氯仿、二氯甲烷中的一种或两种的混合物,优选氯仿,以g/ml计,所述反应物与所述反应溶剂的质量体积比为8ml/g~15ml/g;所述偶联和脱保护后还包括浓缩反应液与溶剂混合打浆的步骤,所述溶剂为甲醇、乙醇、异丙醇、乙腈中的一种或两种以上的混合物,优选甲醇,所述溶剂的使用量为8ml/g-15ml/g。
在本发明的一些具体实施方案中,所述合成方法采用的溶剂为脂溶性溶剂;所述沉淀采用的溶剂为极性溶剂。作为优选,制备方法中的反应溶剂为氯仿,dcm等对该载体具有较好溶解能力的脂溶性溶剂;沉淀采用的溶剂(析出固体的溶剂):甲醇,乙腈等对保护氨基酸具有较强溶解能力的极性溶剂。
在本发明的一些具体实施方案中,制备方法中所用溶剂为三氯甲烷或二氯甲烷中的一种或两种,优选三氯甲烷。
在本发明的一些具体实施方案中,制备方法中偶联剂为hobt/dic、hobt/edc.hcl、edc.hcl,优选hobt/edc.hcl。
在本发明的一些具体实施方案中,制备方法中裂解液为tfa和水混合溶剂,混合溶剂的配比为tfa体积比为80~95%,水的体积比5-20%,其中优选的比例是tfa:h2o=95:5。
在本发明的一些具体实施方案中,兰瑞肽的合成工艺包括如下步骤:
1)偶联氨基酸:
将载体在氯仿溶剂体系中,溶解澄清,然后依次将fmoc保护氨基酸偶联至载体上,氨基酸投料当量为1.1~1.5eq,偶联试剂为edc.hcl(1.1~1.2eq),hobt(1.0~1.2eq),室温反应2~3小时,tlc监控反应。反应毕,浓缩氯仿,加入8~10eq甲醇,析出固体,抽滤,再用甲醇打浆3~5次洗涤过量的保护氨基酸和偶联试剂。烘干即可
2)脱fmoc:
上述fmoc-aa-nh-sieber用氯仿(10ml/g)加入dbu(1.0~1.1eq),将反应液冰浴冷却至5℃以下,缓慢滴加二乙胺(14~16eq),控温不超过5℃。滴加完后,将反应液升至室温继续搅拌2小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,将反应液在30℃条件下减压浓缩至成粘稠物,向粘稠物中加入乙腈(10ml/g),搅拌,过滤,滤饼用甲醇冲洗两次。将滤饼在40℃条件下真空干燥2小时,即可。
按照此偶联-脱fmoc循环偶联,最后一个氨基酸采用boc-nal-oh,不需n端-保护。
先成环,成环后再进行裂解脱除。
在两个cys中采用原料fmoc-cys(mmt)-oh,脱除方案为1%tfa~2%三氯甲烷溶液,脱除后,浓缩,至干,加入甲醇,洗涤2~3次(去除tfa),再采用0.3%的双氧水成环。浓缩。采用配方为tfa:h2o:tis:edt=91:3:3:3的裂解液裂解2小时,过滤,沉淀得粗肽。
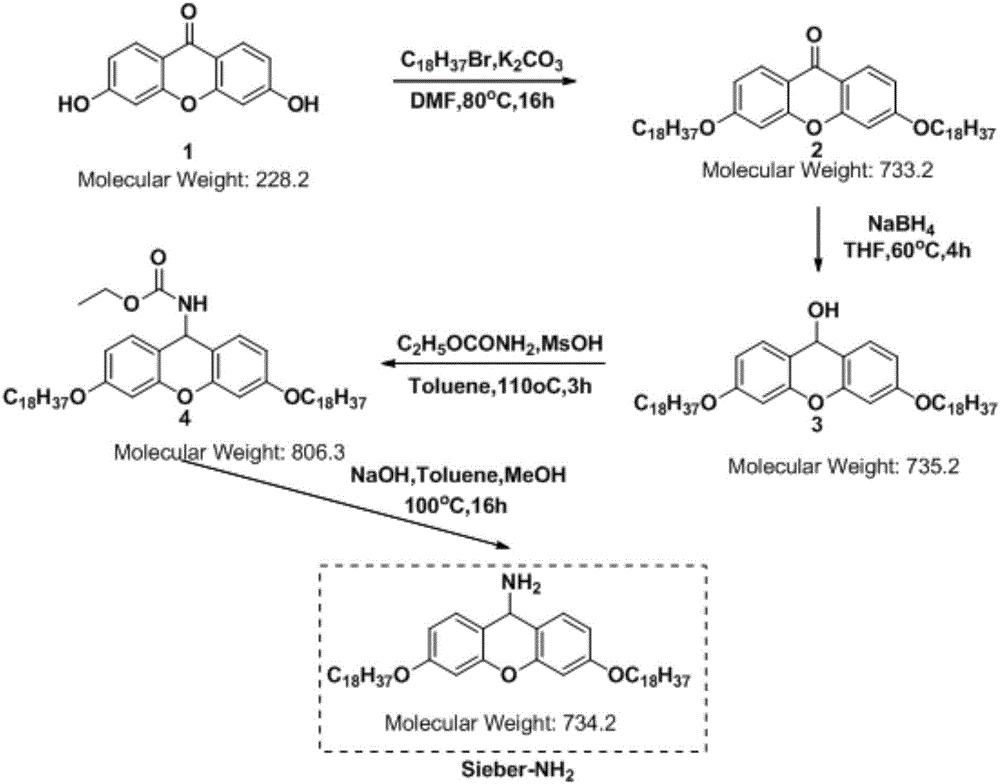
本发明巧妙的借助一个类似于树脂连接臂的载体(结构如式ⅱ所示,其合成工艺见图1)合成兰瑞肽,具体优势如下:
1、反应时,载体和肽溶解澄清,应属于均相反应,有利于困难氨基酸的偶联及成环等。反应完毕在甲醇中析出固体,而氨基酸原料和偶联试剂在甲醇中溶解。
2、具备固相合成树脂带来的优势,在后处理,通过过滤洗涤的方式分离原料和产品。
3、氨基酸原料投料比由5倍降低至1.2~1.3倍。不需要使用大量dmf,而使用少量的甲醇-三氯甲烷等。且三氯甲烷等可以回收。
4、因为载体在三氯甲烷体系是溶解的,需要溶剂量为正常液相反应的溶剂量(3ml/g~5ml/g),因为在相同的反应釜中,釜效较固相合成有较大提高,一批次产量提高。
5、利用cys(mmt)保护基,实现在定位脱除,在载体和保护基的环境下先成环,再进行裂解。相对于粗肽液相成环,操作简便且收率有较大提高。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
图1示式ⅷ所示化合物(反应载体)的合成工艺;
图2示实施例2制得的兰瑞肽粗肽的hplc谱图;
图3示实施例3制得的兰瑞肽粗肽的hplc谱图;
图4示实施例4制得的兰瑞肽粗肽的hplc谱图;
图5示对比例1制得的兰瑞肽粗肽的hplc谱图;
图6示对比例2制得的兰瑞肽粗肽的hplc谱图;
具体实施方式
本发明公开了化合物及其制备方法和应用,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
本发明提供了一种载体(sieber-nh2):
该载体(sieber-nh2)——式ⅷ所示化合物由式ⅲ所示化合物sieberamideresin结构修饰而来,暂时命名为sieber-nh2。
该载体(sieber-nh2)——式ⅷ所示化合物的合成路线如图1所示。
1)该载体合成难度及成本远低于常用多肽固相合成的树脂,如rinkresin等。
2)该载体因为侧链对称引入长链烷烃链,脂溶性增强,可以溶解于氯仿等脂溶性溶剂。使得反应由固相合成的非均相反应变成液相合成的均相反应。且该载体在甲醇等极性溶剂中可以析出,而保护氨基酸溶于甲醇等极性溶剂,利用这样的溶解度差异,通过过滤的方式除去过量的保护氨基酸和偶联试剂等。简而言之,该载体即借鉴了固相合成树脂在后处理方面的优势,由借助液相均相反应的优势。
3)由于该工艺在液相中进行,相比于固相合成常用的3~5倍投料,和大量的dmf等溶剂洗涤,该工艺采用1.1~1.5eq投料,甲醇洗涤。大幅度的节约了成本。
4)液相合成,批生产对设备要求较小,且相同反应釜,釜效能更大。
5)该工艺采用fmoc-cys(mmt)-oh为原料,代替fmoc-cys(trt)-oh,因为载体位阻较大,分子间成环概率较低。因此在偶联完成后,脱除mmt,可以在正常浓度的反应体系中进行氧化成环。而不需要像固相合成那样,先裂解,再制备,再配成极稀溶液(防止分子间成环)。工艺操作简化,同时减少了粗肽的损失,提高了收率。
本发明提供的化合物及其制备方法和应用中所用原料及试剂均可由市场购得。
缩写及英文含义:
缩写及英文含义
dcm二氯甲烷
tfa三氟乙酸
mmt4-甲氧基三苯甲基
edt乙二硫醇
tis三异丙基硅烷
nal3-(2-萘基)]-d-丙氨酸
edc·hcl1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐
dbu1,8-二氮杂二环十一碳-7-烯
fmoc-aa-ohfmoc保护氨基酸
下面结合实施例,进一步阐述本发明:
实施例1载体(sieber-nh2)合成
化合物2的制备例1:
称取22.8g(1mmol)化合物1与1-溴十八烷(2.1mmol)加入合适的三口烧瓶内,然后向反应瓶中加入dmf(适量),搅拌均匀,再加入碳酸钾(3.0mmol)。将反应液升温至80℃继续搅拌16小时。tlc监控反应。原料消耗完后,将反应液冰浴冷却至室温。在充分搅拌条件下,向反应液中缓慢滴加1m稀盐酸,和纯化水,滴加完后继续搅拌30分钟。过滤,滤饼依次用纯化水和甲醇冲洗。在40℃条件下真空干燥8小时,得到目标化合物2。
化合物2的制备例2:
称取22.8g(1mmol)化合物1与1-碘十八烷(2.1mmol)加入合适的三口烧瓶内,然后向反应瓶中加入dmf(适量),搅拌均匀,再加入dipea(3.0mmol)。将反应液升温至70℃继续搅拌18小时。tlc监控反应。原料消耗完后,将反应液冰浴冷却至室温。在充分搅拌条件下,向反应液中缓慢滴加1m稀盐酸,和纯化水,滴加完后继续搅拌30分钟。过滤,滤饼依次用纯化水和甲醇冲洗。在40℃条件下真空干燥8小时,得到目标化合物2
化合物2的制备例3:
称取22.8g(1mmol)化合物1与1-溴十六烷(2.1mmol)加入合适的三口烧瓶内,然后向反应瓶中加入dmf(适量),搅拌均匀,再加入碳酸钠(3.0mmol)。将反应液升温至100℃继续搅拌14小时。tlc监控反应。原料消耗完后,将反应液冰浴冷却至室温。在充分搅拌条件下,向反应液中缓慢滴加1m稀盐酸,和纯化水,滴加完后继续搅拌30分钟。过滤,滤饼依次用纯化水和甲醇冲洗。在40℃条件下真空干燥8小时,得到目标化合物2
化合物3的制备例1
称取化合物2加入三口烧瓶内,然后向反应瓶中加入thf和甲醇,搅拌均匀。将反应液升温至60℃后,缓慢加入硼氢化钠,加完后保持温度继续搅拌4小时。tlc(乙酸乙酯:正己烷=1:2,紫外灯显色)监控反应。原料消耗完后,将反应液冰浴冷却至10℃以下。在充分搅拌的条件下,缓慢滴加1mol/l稀盐酸,滴加完后,减压浓缩除去thf,向剩余反应液加入纯化水,并用1mol/l稀盐酸调ph值至5-7。过滤,滤饼滤饼依次用纯化水和甲醇冲洗。在60℃条件下真空干燥8小时,得到目标化合物3。
化合物3的制备例2
称取化合物2加入三口烧瓶内,然后向反应瓶中加入甲基四氢呋喃和乙醇,搅拌均匀。将反应液升温至65℃后,缓慢加入氰基硼氢化钠,加完后保持温度继续搅拌4小时。tlc(乙酸乙酯:正己烷=1:2,紫外灯显色)监控反应。原料消耗完后,将反应液冰浴冷却至10℃以下。在充分搅拌的条件下,缓慢滴加1mol/l稀盐酸,滴加完后,减压浓缩除去甲基四氢呋喃,向剩余反应液加入纯化水,并用1mol/l稀盐酸调ph值至5-7。过滤,滤饼滤饼依次用纯化水和乙醇冲洗。在60℃条件下真空干燥8小时,得到目标化合物3。
化合物3的制备例子3
称取化合物2加入三口烧瓶内,然后向反应瓶中加入thf和异丙醇,搅拌均匀。将反应液升温至50℃后,缓慢加入硼氢化钠,加完后保持温度继续搅拌4小时。tlc(乙酸乙酯:正己烷=1:2,紫外灯显色)监控反应。原料消耗完后,将反应液冰浴冷却至10℃以下。在充分搅拌的条件下,缓慢滴加1mol/l稀盐酸,滴加完后,减压浓缩除去thf,向剩余反应液加入纯化水,并用1mol/l稀盐酸调ph值至5-7。过滤,滤饼滤饼依次用纯化水和异丙醇冲洗。在60℃条件下真空干燥8小时,得到目标化合物3。
化合物4的制备:例1
称取化合物3加入三口烧瓶内,然后向反应瓶中加入甲苯,搅拌均匀,再依次加入甲烷磺酸、乌来糖。将反应液升温至100℃继续搅拌4小时。将反应液冷却至室温,加入碳酸钠,然后旋蒸至原体积的1/5除去溶剂。向残余物中加入甲醇,升温至70℃,回流反应30分钟,将反应液自然冷却至室温析晶。过滤。滤饼用甲醇和乙腈混合溶剂(1:1)冲洗,50℃真空干燥5小时,得到化合物4。
化合物4的制备:例2
称取化合物3加入三口烧瓶内,然后向反应瓶中加入甲苯,搅拌均匀,再依次加入甲烷磺酸、乌来糖。将反应液升温至110℃继续搅拌2小时。将反应液冷却至室温,加入碳酸钠,然后旋蒸至原体积的1/5除去溶剂。向残余物中加入甲醇,升温至70℃,回流反应30分钟,将反应液自然冷却至室温析晶。过滤。滤饼用甲醇和乙腈混合溶剂(1:1)冲洗,50℃真空干燥5小时,得到化合物4。
化合物4的制备:例3
称取化合物3加入三口烧瓶内,然后向反应瓶中加入甲苯,搅拌均匀,再依次加入甲烷磺酸、乌来糖。将反应液升温至105℃继续搅拌3小时。将反应液冷却至室温,加入碳酸钠,然后旋蒸至原体积的1/5除去溶剂。向残余物中加入甲醇,升温至70℃,回流反应30分钟,将反应液自然冷却至室温析晶。过滤。滤饼用甲醇和乙腈混合溶剂(1:1)冲洗,50℃真空干燥5小时,得到化合物4。
化合物5的制备(sieber-nh2的合成)例1:
将上述化合物4加入三口烧瓶内,向反应瓶中加入甲苯和乙醇,搅拌均匀,再加入氢氧化钠。将反应液升温至90℃继续搅拌17小时。向反应液中依次加水、正己烷、乙酸乙酯,搅拌30分钟后,过滤。滤饼用纯化水打浆两次,得到的滤液静置分层后收集有机相,将有机相在45℃条件下浓缩至干。将滤饼与浓缩后残余物合并,用甲醇与乙腈混合溶剂打浆。过滤,滤饼在45℃条件下真空干燥5小时,得到化合物5即目标载体化合物sieber-nh2。
化合物5的制备(sieber-nh2的合成)例2:
将上述化合物4加入三口烧瓶内,向反应瓶中加入甲苯和乙醇,搅拌均匀,再加入氢氧化钠。将反应液升温至100℃继续搅拌16小时。向反应液中依次加水、正己烷、乙酸乙酯,搅拌30分钟后,过滤。滤饼用纯化水打浆两次,得到的滤液静置分层后收集有机相,将有机相在45℃条件下浓缩至干。将滤饼与浓缩后残余物合并,用甲醇与乙腈混合溶剂打浆。过滤,滤饼在45℃条件下真空干燥5小时,得到化合物5即目标载体化合物sieber-nh2。
化合物5的制备(sieber-nh2的合成)例3
将上述化合物4加入三口烧瓶内,向反应瓶中加入甲苯和乙醇,搅拌均匀,再加入氢氧化钾。将反应液升温至110℃继续搅拌14小时。向反应液中依次加水、正己烷、乙酸乙酯,搅拌30分钟后,过滤。滤饼用纯化水打浆两次,得到的滤液静置分层后收集有机相,将有机相在45℃条件下浓缩至干。将滤饼与浓缩后残余物合并,用甲醇与乙腈混合溶剂打浆。过滤,滤饼在45℃条件下真空干燥5小时,得到化合物5即目标载体化合物sieber-nh2。
上述sieber-nh2的合成路线如图1所示。
其中,化合物1的结构如式iv所示:
化合物2的结构如式ⅴ所示:
化合物3的结构如式ⅵ所示:
化合物4的结构如式ⅶ所示:
化合物5:载体(sieber-nh2)的结构如式ⅷ所示:
实施例2
1、fmoc-thr(tbu)-nh-sieber(第一个氨基酸偶联):
称取化合物sieber-nh2(7.34g,10mmol)加入500ml三口烧瓶内,向反应瓶中加入氯仿(800ml约8ml/g相对于固体底物),再依次加入hobt(1.36g.10.05mmol)、fmoc-thr(tbu)-oh(3.99g,10.05mmol),搅拌,溶解,加入edc·hcl(1.93g,10.05mmol),在室温条件下继续搅拌3小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(40ml),搅拌2小时。过滤,滤饼用甲醇(15ml×3)冲洗三次。将滤饼在40℃条件下真空干燥5小时,得到fmoc-thr(tbu)-nh-sieber(10.71g,9.62mmol,收率96.16%)。
2、h-thr(tbu)-nh-sieber(脱fmoc):
称取化合物fmoc-thr(tbu)-nh-sieber(10.71g,9.62mmol)加入500ml三口烧瓶内,向反应瓶中加入适量氯仿(约100ml约10ml/g相对于化合物fmoc-thr(tbu)-nh-sieber)),搅拌溶清,再加入dbu(1.47g,9.62mmol)。将反应液冰浴冷却至5℃以下,缓慢滴加二乙胺(7.04g,96.2mmol),控温不超过5℃。滴加完后,将反应液升至室温继续搅拌2小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,将反应液在30℃条件下减压浓缩至成粘稠物,向粘稠物中加入乙腈(100ml),搅拌30分钟。过滤,滤饼用甲醇(30ml×2)冲洗两次。将滤饼在40℃条件下真空干燥2小时,得到类白色固体(8.59g,9.6mmol,100%)。
3、fmoc-cys(mmt)-thr(tbu)-nh-sieber(偶联第二个氨基酸):
将上述固体加入500ml三口烧瓶内,向反应瓶中加入适量氯仿(约100ml),再依次加入hobt(1.36g,10.1mmol)、fmoc-cys(mmt)-oh(6.22g,10.1mmol)。搅拌溶清。将反应液冷却至0℃。加入edc·hcl(1.93g,10.1mmol),在室温条件下继续搅拌3小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(50ml),搅拌2小时。过滤,滤饼用甲醇(20ml×3)冲洗三次。将滤饼在40℃条件下真空干燥3小时,得到(14.03g,收率98.01%)。
4、重复2~3进行第3、4、5、6、7个基酸的偶联和脱保护至最后一个boc-nal-oh的偶联:
将cys(mmt)-tyr(tbu)-d-trp(boc)-lys(boc)-val-cys(mmt)-thr(tbu)-nh-sieber(30.48g,8.50mmol)加入到500ml三口烧瓶,向反应瓶中加入适量氯仿(约200ml),再依次加入hobt(1.21g,8.92mmol)、boc-nal-oh(2.96g,32,8.92mmol)。搅拌溶解。将反应液冷却至0℃。加入edc·hcl(1.71g,8.92mmol),在室温条件下继续搅拌3小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(100ml),搅拌2小时。过滤,滤饼用甲醇(30ml×3)冲洗三次。将滤饼在40℃条件下真空干燥3小时,得到(31.35g,收率95.01%,总收率79~82%)。
5、兰瑞肽粗肽的合成:
boc-nal-cys(mmt)-tyr(tbu)-d-trp(boc)-lys(boc)-val-cys(mmt)-thr(tbu)-nh-sieber(31.0g8.15mmol)加入1000ml三口烧瓶内,向反应瓶中加入氯仿(200ml),搅拌溶清,再加入氯仿-三氟乙酸混合液(200ml-4ml)(三氟乙酸总浓度约1%),搅拌反应2小时。小样裂解,色谱监控mmt是否脱除完全监控反应。反应完全后,将反应液在30℃条件下减压浓缩至成粘稠物,向粘稠物中加入乙腈(110ml),搅拌30分钟。过滤,滤饼用甲醇(60ml×2)冲洗两次,再将滤饼溶解在氯仿300ml中,加入0.3%双氧水(约1ml)搅拌2小时,得到全保护的目标肽。
采用配方为tfa:phsme:phome:edt=90:5:3:2的裂解液裂解2小时,过滤,沉淀得粗肽7.26g,收率约82%,粗肽纯度92.07%(hplc谱图如图2所示)。总收率约:66.24%。
实施例3兰瑞肽线性肽树脂
(boc-nal-cys(mmt)-tyr(tbu)-d-trp(boc)-lys(boc)-val-cys(mmt)-thr(tbu)-nh-sieber)的合成
1、fmoc-thr(tbu)-nh-sieber(第一个氨基酸偶联):
称取化合物sieber-nh2(7.34g,10mmol)加入500ml三口烧瓶内,向反应瓶中加入氯仿(100ml),再依次加入hobt(1.49g.11mmol)、fmoc-thr(tbu)-oh(4.37g,11mmol),搅拌,溶解,加入edc·hcl(2.1g,11mmol),在室温条件下继续搅拌3小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(50ml),搅拌2小时。过滤,滤饼用甲醇(15ml×3)冲洗三次。将滤饼在40℃条件下真空干燥5小时,得到fmoc-thr(tbu)-nh-sieber(10.74g,9.64mmol,收率96.44%)。
2、h-thr(tbu)-nh-sieber(脱fmoc):
称取化合物fmoc-thr(tbu)-nh-sieber(10.74g,9.6mmol)加入500ml三口烧瓶内,向反应瓶中加入适量氯仿(约100ml),搅拌溶清,再加入dbu(1.47g,9.6mmol)。将反应液冰浴冷却至5℃以下,缓慢滴加二乙胺(7.05g,96.4mmol),控温不超过5℃。滴加完后,将反应液升至室温继续搅拌2小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,将反应液在30℃条件下减压浓缩至成粘稠物,向粘稠物中加入乙腈(100ml),搅拌30分钟。过滤,滤饼用甲醇(30ml×2)冲洗两次。将滤饼在40℃条件下真空干燥2小时,得到类白色固体(8.59g,9.6mmol,100%)。
3、fmoc-cys(mmt)-thr(tbu)-nh-sieber(偶联第二个氨基酸):
将上述固体加入500ml三口烧瓶内,向反应瓶中加入适量氯仿(约100ml),再依次加入hobt(1.44g,10.604mmol)、fmoc-cys(mmt)-oh(6.53g,10.604mmol)。搅拌溶清。将反应液冷却至0℃。加入edc·hcl(2.033g,10.604mmol),在室温条件下继续搅拌3小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(50ml),搅拌2小时。过滤,滤饼用甲醇(20ml×3)冲洗三次。将滤饼在40℃条件下真空干燥3小时,得到(14.063g,收率98.01%)。
4、重复2~3进行第3、4、5、6、7个基酸的偶联和脱保护至最后一个boc-nal-oh的偶联:
将cys(mmt)-tyr(tbu)-d-trp(boc)-lys(boc)-val-cys(mmt)-thr(tbu)-nh-sieber(30.56g,8.53mmol)加入到500ml三口烧瓶,向反应瓶中加入适量氯仿(约200ml),再依次加入hobt(1.26g,9.38mmol)、boc-nal-oh(2.96g,32,9.38mmol)。搅拌溶解。将反应液冷却至0℃。加入edc·hcl(1.80g,9.38mmol),在室温条件下继续搅拌3小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(100ml),搅拌2小时。过滤,滤饼用甲醇(30ml×3)冲洗三次。将滤饼在40℃条件下真空干燥3小时,得到(31.64g,收率95.62%,总收率70~81%)。
5、兰瑞肽粗肽的合成:
boc-nal-cys(mmt)-tyr(tbu)-d-trp(boc)-lys(boc)-val-cys(mmt)-thr(tbu)-nh-sieber(31.0g8.15mmol)加入1000ml三口烧瓶内,向反应瓶中加入氯仿(200ml),搅拌溶清,再加入氯仿-三氟乙酸混合液(200ml-4ml)(三氟乙酸总浓度约1%),搅拌反应2小时。小样裂解,色谱监控mmt是否脱除完全监控反应。反应完全后,将反应液在30℃条件下减压浓缩至成粘稠物,向粘稠物中加入乙腈(110ml),搅拌30分钟。过滤,滤饼用甲醇(60ml×2)冲洗两次,再将滤饼溶解在氯仿300ml中,加入0.3%双氧水(约1ml)搅拌2小时,得到全保护的目标肽。
采用配方为tfa:phsme:phome:edt=90:5:3:2的裂解液裂解2小时,过滤,沉淀得粗肽7.77g,收率约87%,粗肽纯度约93.71%(hplc谱图如图3所示)。总收率70.28%。
实施例4
1、fmoc-thr(tbu)-nh-sieber(第一个氨基酸偶联):
称取化合物sieber-nh2(7.34g,10mmol)加入500ml三口烧瓶内,向反应瓶中加入氯仿(150ml),再依次加入hobt(1.62g.12mmol)、fmoc-thr(tbu)-oh(4.77g,12mmol),搅拌,溶解,加入edc·hcl(2.3g,12mmol),在室温条件下继续搅拌3小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(100ml),搅拌3小时。过滤,滤饼用甲醇(30ml×3)冲洗三次。将滤饼在40℃条件下真空干燥5小时,得到fmoc-thr(tbu)-nh-sieber(10.74g,9.72mmol,收率97.16%)。
2、h-thr(tbu)-nh-sieber(脱fmoc):
称取化合物fmoc-thr(tbu)-nh-sieber(10.82g,9.72mmol)加入500ml三口烧瓶内,向反应瓶中加入适量氯仿(约150ml),搅拌溶清,再加入dbu(1.48g,9.72mmol)。将反应液冰浴冷却至5℃以下,缓慢滴加二乙胺(7.11g,97.2mmol),控温不超过5℃。滴加完后,将反应液升至室温继续搅拌2小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,将反应液在30℃条件下减压浓缩至成粘稠物,向粘稠物中加入乙腈(150ml),搅拌30分钟。过滤,滤饼用甲醇(60ml×2)冲洗两次。将滤饼在40℃条件下真空干燥2小时,得到类白色固体(8.65g,9.72mmol,100%)。
3、fmoc-cys(mmt)-thr(tbu)-nh-sieber(偶联第二个氨基酸):
将上述固体加入500ml三口烧瓶内,向反应瓶中加入适量氯仿(约150ml),再依次加入hobt(1.57,11.66mmol)、fmoc-cys(mmt)-oh(7.18g,11.66mmol)。搅拌溶清。将反应液冷却至0℃。加入edc·hcl(2.23g,11.66mmol),在室温条件下继续搅拌3小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(100ml),搅拌2小时。过滤,滤饼用甲醇(40ml×3)冲洗三次。将滤饼在40℃条件下真空干燥3小时,得到(14.063g,收率98.01%)。
4、重复2~3进行第3、4、5、6、7个基酸的偶联和脱保护至最后一个boc-nal-oh的偶联:
将cys(mmt)-tyr(tbu)-d-trp(boc)-lys(boc)-val-cys(mmt)-thr(tbu)-nh-sieber(32.21g,8.67mmol)加入到500ml三口烧瓶,向反应瓶中加入适量氯仿(约200ml),再依次加入hobt(1.41g,10.4mmol)、boc-nal-oh(3.28g,10.4mmol)。搅拌溶解。将反应液冷却至0℃。加入edc·hcl(2.0g,10.4mmol),在室温条件下继续搅拌3小时。tlc(dcm:meoh:hac=100:1:0.5)监控反应。反应完全后,反应液在30℃减条件下,减压浓缩至成粘稠物,向粘稠物中加入甲醇(200ml),搅拌2小时。过滤,滤饼用甲醇(60ml×3)冲洗三次。将滤饼在40℃条件下真空干燥3小时,得到(32.54g,收率96.61%,总收率70~81%)。
5、兰瑞肽粗肽的合成:
boc-nal-cys(mmt)-tyr(tbu)-d-trp(boc)-lys(boc)-val-cys(mmt)-thr(tbu)-nh-sieber(32.5g8.38mmol)加入1000ml三口烧瓶内,向反应瓶中加入氯仿(100ml),搅拌溶清,再加入氯仿-三氟乙酸混合液(300ml-4ml)(三氟乙酸总浓度约1%),搅拌反应2小时。小样裂解,色谱监控mmt是否脱除完全监控反应。反应完全后,将反应液在30℃条件下减压浓缩至成粘稠物,向粘稠物中加入乙腈(150ml),搅拌30分钟。过滤,滤饼用甲醇(80ml×2)冲洗两次,再将滤饼溶解在氯仿300ml中,加入0.3%双氧水(约1ml)搅拌2小时,得到全保护的目标肽。
采用配方为tfa:phsme:phome:edt=90:5:3:2的裂解液裂解2小时,过滤,沉淀得粗肽8.17g,收率约87%,粗肽纯度94.44%(hplc谱图如图4所示)。总收率71.89%。
对比例1
将二苯基甲胺-聚苯乙烯树脂置于肽合成器中,按下列顺序进行洗涤:1)二氯甲烷;2)33%三氟乙酸的二氯甲烷溶液;3)二氯甲烷4)乙醇5)二氯甲烷6)10%三乙胺的氯仿溶液。再该中性的树脂和叔丁氧羰基-o-苄基苏氨酸和二异丙基碳化二亚胺溶于二氯甲烷,搅拌反应,生成的氨基酸化的树脂按上面过程洗涤。依次接保护氨基酸:叔丁氧羰基-3-甲基苄基-cys、叔丁氧羰基-val、叔丁氧羰基-ne-苄氧羰基-lysine、叔丁氧羰基-d-trp、叔丁氧羰基-tyr、叔丁氧羰基-5-甲基苄基-cys和叔丁氧羰基-d-β-萘胺。该树脂水洗后干燥,和苯甲醚及无水氟化氢在0℃搅拌。通入干燥氮气吹走氟化氢,过滤收集沉淀的游离的肽,用乙醚洗,溶于90%乙酸,加入碘的甲醇溶液至呈棕色。搅拌片刻后,减压蒸出溶剂。油状物溶于最少体积的50%乙酸,进行柱层析分离,即得产物。收率约37%,粗肽纯度约59.7%,如图5所示。
对比例2
将rink酰胺树脂dcm和dmf洗涤溶胀后,脱除fmoc保护基,再依次偶联fmoc-thr(tbu)-oh、fmoc-cys(trt)-oh、fmoc-val-oh、fmoc-lys(boc)-oh、fmoc-d-trp(boc)-oh、fmoc-tyr(tbu)-oh、fmoc-cys(trt)-oh、fmoc-d-3-(2-萘基)-ala-oh,偶联体系均采用hobt/dic,dmf为溶剂,20%哌啶的dmf溶液脱除fmoc保护基。偶联毕,dmf洗涤3次,dcm洗涤3次,抽干,甲醇收缩,再采用tfa-水-捕获剂体系裂解,乙醚沉淀得到粗肽,粗肽再进行氧化成环,最后采用c18反相高效液相色谱柱纯化得到醋酸兰瑞肽。收率约62%,粗肽纯度约88.19%,如图6所示。
实施例5
实验组:实施例2~4制得的兰瑞肽粗肽;
对照组1:对比例1制得的兰瑞肽粗肽;
对照组2:对比例2制得的兰瑞肽粗肽;
比较结果见表1。
表1
注:*示与对照组相比,具有显著差异(p<0.05);#示与对照组相比,具有极显著差异(p<0.01)。
表1结果表明,实验组三组数据显示粗肽的平均收率约86%,其中实验4的收率最高,粗肽纯度也最高(即采用1.2倍氨基酸原料和1.2倍偶联试剂进行投料),但是三者无显著性差异。
而对照组,对照1即原研工艺boc策略,收率不足40%,且粗肽纯度不到60%;对照2即常用的fmoc策略,收率62%,纯度约88%,且需要3~5倍投料,并且合成规模较低。该两组数据与实验组相比,无论是收率还是粗肽纯度均存在显著性差异(p<0.05)。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!