一种靶向猪TTN基因的打靶载体及其应用的制作方法
本发明属于生物技术领域,具体地说,涉及一种用于敲除猪ttn基因的打靶载体及其应用。
背景技术:
扩张型心肌病(dcm)是全世界最常见的心肌病,患病率为1:250,约占所有心衰患者的30%~40%,且dcm是一类可遗传的心肌病。扩张型心肌病(dcm)主要病症为左心室或双室扩张和收缩功能障碍,能够引起进行性心力衰竭,心律失常,猝死,是心脏移植最常见的指征。
引起dcm最重要的致病基因为ttn,ttn基因编码人体最大的蛋白质,丰度第三的横纹肌蛋白质。是肌节组装所必需的,并且在横纹肌中提供大部分被动力并调节主动的收缩力。目前研究表明杂合ttn截短突变(ttntv)是dcm最常见的原因,ttntv占家族性dcm病例的25%。在细胞或个体水平上对猪ttn基因进行敲除或编辑,可为进一步解析dcm分子机理提供基础。
因此,急需提供一种可高效敲除ttn基因的方法,构建ttn基因敲除的猪疾病模型,为dcm分子机理的研究提供良好的材料和基础。
技术实现要素:
为了解决现有技术中存在的问题,本发明的目的是提供一种靶向猪ttn基因的打靶载体及其应用,所述打靶载体可实现对猪ttn基因的高效敲除。
为了实现本发明目的,本发明的技术方案如下:
第一方面,本发明提供一种靶向猪ttn基因的打靶载体,所述打靶载体是基于crispr/cas9系统的sgrna表达载体,其中,sgrna的靶标序列位于猪ttn基因的第278个外显子上。
所述靶标序列符合5′-n(20)ngg-3′的排列规则,其中n(20)表示20个连续的碱基,每个n表示a或t或c或g中的任意一个碱基。
所述sgrna表达载体是将sgrna的靶标序列的5′端加上bbsi酶切位点序列,并人工合成其互补序列,将两条互补序列形成的双链dna,与经过bbsi酶切的初始载体相连接。
作为优选,所述初始载体为px330。
更具体地,sgrna的序列如下:
sgrna1-f:5′-caccggtcattgcaaagaatgcggc-3′;
sgrna1-r:5′-aaacgccgcattctttgcaatgacc-3′。
第二方面,本发明提供了前述打靶载体在制备猪ttn基因敲除细胞或ttn基因敲除猪方面的应用。
具体表现为:利用所述打靶载体转染猪体细胞系,筛选获得ttn基因敲除的细胞,或进一步将获得的ttn基因敲除的细胞,通过体细胞核移植技术制备ttn基因敲除猪。
在本发明的具体实施方式中,采用成纤维细胞进行示例性说明,但本发明所述方案在实际应用中并不局限于此。
第三方面,本发明提供一种制备猪ttn基因敲除细胞的方法,将前述打靶载体转染猪体细胞系,筛选获得ttn基因敲除的细胞。
本发明进一步提供一种制备ttn基因敲除猪的方法,将前述打靶载体转染猪成纤维细胞系,筛选获得ttn基因敲除的成纤维细胞,并将所获得的ttn基因敲除的成纤维细胞,通过体细胞核移植技术制备ttn基因敲除猪。
上述方法和应用中,可采用本领域常规方法筛选获得ttn基因敲除的细胞。本发明仅提供一种可选方法如下,并不对筛选方法构成限定:
在所述打靶载体转染猪成纤维细胞48h后,提取细胞基因组dna,用上游引物ttn1-f:tgtgggtgtaggcaaagc;下游引物ttn1-r:taaagccttgaaatccgtgt对提取的基因组dna进行pcr扩增,并纯化产物;纯化的ttn1片段与pmdtm19-tvectorcloningkit载体进行连接,取5μl连接产物转化到感受态细胞中,克隆测序检测是否打靶成功。
本发明涉及到的原料或试剂均为普通市售产品,涉及到的操作如无特殊说明均为本领域常规操作。
在符合本领域常识的基础上,上述各优选条件,可以相互组合,得到具体实施方式。
本发明的有益效果至少在于:
首先,本发明通过大量客观实验研究发现了具有高打靶敲除效率的靶标序列位置,发现了将靶标序列定位于ttn基因的第278个外显子上,相比定位于其他外显子上,可更为有效地模拟dcm疾病,并实现ttn基因的敲除。
第二,本发明通过针对ttn基因的第278个外显子,设计了不同的sgrna,筛选出了打靶效率更高的靶标序列。
本发明采用新型基因组编辑技术crispr/cas9系统,利用构建好的ttn基因敲除打靶载体,通过电击法转染猪体细胞系,筛选获得ttn基因敲除的猪体细胞,敲除效率达到11.1%。在此基础上,还可进一步筛选ttn基因敲除的成纤维细胞系,通过体细胞核移植技术获得ttn基因敲除猪,为制备ttn基因突变导致的猪dcm疾病模型奠定基础。
附图说明
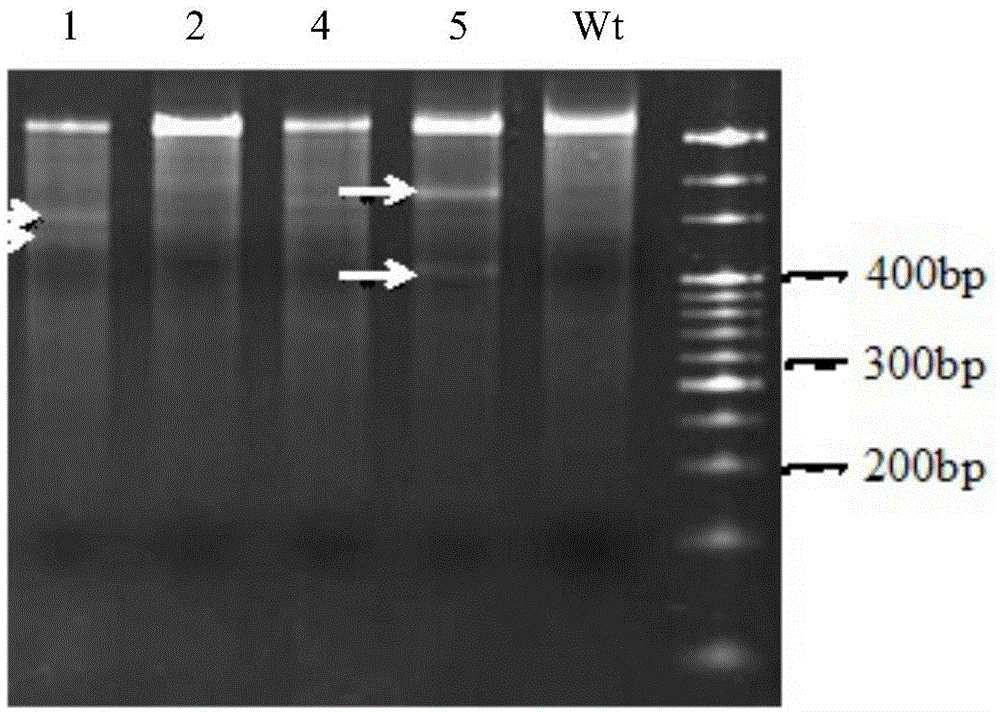
图1为打靶载体px330-sgrna1转染猪纤维细胞系48h,提取细胞dna并扩增ttn基因靶标序列的pcr产物经过t7e1酶切割的电泳图。
图2为打靶载体px330-sgrna1转染细胞后的ttn1克隆测序结果。
具体实施方式
下面结合实施例对本发明做进一步的解释说明。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
下述实施例中的px330载体为addgene公司产品,序列通过北京六合华大基因科技股份有限公司合成。
实施例1
1、sgrna的设计
根据猪ttn基因的mrna序列(ncbixm_021075901.1),在ttn基因第278外显子上设计sgrna,在sgrna序列f链的5′端加上接头序列cacc,其反向互补序列r链的5′端添加接头序列aaac,如果sgrna序列的5′端第一个碱基不是g,那么应先在sgrna序列f链的5′端添加一个g,再加上接头序列cacc,相应地,其反向互补序列r链的3′端再增加一个c,以便能够与经bbsⅰ酶切的px330载体的粘性末端互补。四对sgrna的正义链序列如下:
sgrna1:gtcattgcaaagaatgcggc;
sgrna2:gtgatttgcctgagggtcgc;
sgrna4:agtaaaccttctgacagtac;
sgrna5:tactggaccaattactgcca。
根据sgrna序列通过北京六合华大基因科技股份有限公司合成相应的单链寡核苷酸,具体序列如下:
针对sgrna-1合成的单链寡核苷酸:
f:5′-caccggtcattgcaaagaatgcggc-3′;
r:5′-aaacgccgcattctttgcaatgacc-3′;
针对sgrna-2合成的单链寡核苷酸:
f:5′-caccggtgatttgcctgagggtcgc-3′;
r:5′-aaacgcgaccctcaggcaaatcacc-3′;
对sgrna-4合成的单链寡核苷酸:
f:5′-caccgagtaaaccttctgacagtac-3′;
r:5′-aaacgtactgtcagaaggtttactc-3′;
针对sgrna-5合成的单链寡核苷酸:
f:5′-caccgtactggaccaattactgcca-3′;
r:5′-aaactggcagtaattggtccagtac-3′。
分别合成上述正向和反向sgrna寡聚核苷酸,经94℃5min,37℃10min,冰点5min,退火之后形成具有粘性末端的双链dna。
2、px330-sgrna载体构建
用bbsⅰ酶切px330质粒,50μl酶切体系包括质粒5μg,bbsⅰ4.5μl,buffer5μl,添加ddh2o至50μl体系,37℃水浴至少5h。
将酶切后的px330质粒分别与上述退火的双链dna进行连接,连接体系为10μl:px330酶切片段50ng,t4连接酶0.5μl,t4连接buffer1μl,退火产物补足10μl,在16℃连接3h。
分别将上述连接产物转化至trans1-t1phageresistant感受态细胞中,具体操作如下:感受态细胞在冰上融化,向其中加入连接产物px330-sgrna,弹匀后冰浴30min;42℃水浴热击90s之后再冰浴2-3min;向感受态中加入1ml的无抗性的lb液体培养基,37℃摇床1h使菌体复苏;4000rpm离线2min,弃去800μl上清,然后吹打混匀剩余菌液;将剩余菌液涂布在含有氨苄青霉素抗性的lb固体培养基中,将菌板倒置于37℃培养箱中过夜培养。挑选3个菌落摇菌测序(北京六合华大基因科技股份有限公司)。
3、px330-sgrna打靶效率的验证
将连接成功的px330-sgrna转染猪成纤维细胞。具体操作如下:提前将4μg的质粒与100μl电击液制成混液;将6孔板中的10%dmem吸走,用pbs冲洗一遍;加入500μl的胰酶消化细胞,待多数细胞变圆后,加入1ml10%dmem终止消化,吹打细胞并将细胞转移至15ml离心管中,1000g离心5min;尽可能将上清吸干净,用电击液和质粒的混液重悬细胞,转移至电击杯中;而后将电击杯放在核电转仪中电击,程序结束后立即加入1000ml10%dmem,吹打细胞并转移至6孔板中,添加10%dmem至2ml,37℃培养。
4、转染细胞的dna提取
转染24h后,换新的10%dmem培养基,继续培养,在48h后收集细胞提取细胞基因组。用qiagen基因组提取试剂盒,具体操作如下:在细胞沉淀中加入200μlpbs,20μl蛋白酶k,200μlalbuffer,涡旋混匀,56℃水浴10min;向上述裂解液中加入200μl无水乙醇,涡旋混匀,并将混液转移至spincolumn中,10000rpm离心1min,弃滤液;在spincolumn中加500μlaw1,10000rpm离心1min,弃滤液;继续加500μlaw2,10000rpm离心1min,弃滤液;之后在离心机中空离,14000rpm离心2min;打开spincolumn盖,使酒精挥发干,加入50μlddh2o至膜上室温静置1min,10000rpm离心1min;最后重复洗脱一次以提高基因组的浓度。
5、ttn基因片段的pcr扩增
以上述提取的细胞基因组为模板,ttn1为引物进行pcr扩增(ttn1-f:tgtgggtgtaggcaaagc;下游引物ttn1-r:taaagccttgaaatccgtgt),扩增产物的片段长度为541bp。然后对ttn1片段进行胶回收。
6、ttn基因片段的t7e1酶切
胶回收的ttn1片段需要先进行变性退火。pcr梯度变性体系:400ng胶回收产物,1μl10×taqbuffer,ddh2o补足10μl。pcr梯度变性程序为95℃10min,每秒降低2℃;95℃1min,每秒降低0.3℃;85℃1min,每秒降低0.3℃;75℃1min,每秒降低0.3℃;65℃1min,每秒降低0.3℃;55℃1min,每秒降低0.3℃;45℃1min,每秒降低0.3℃;35℃1min,每秒降低0.3℃;25℃1min,每秒降低0.3℃;4℃-/-。退火产物进行t7e1酶切,酶切体系为:退火产物10μl,10×buffer2μl,t7e10.5μl,ddh2o补至20μl。然后进行8%的suryeror丙烯酰胺凝胶电泳。由图1可以看出转染px330-sgrna2和px330-sgrna4的ttn1片段经过t7e1酶切后未出现预期大小的切割条带,表明sgrna2和sgrna4未成功打靶。而px330-sgrna1和px330-sgrna5出现预期的切割条带,表明sgrna1和sgrna5打靶成功。
7、ttn1片段的克隆测序
将转染px330-sgrna1的纯化ttn1片段与pmdtm19-tvectorcloningkit载体(北京全式金,货号:6013)进行连接。连接体系为10μl,包括solutioni5μl,pmd19-t载体1μl,dna0.1pmol-0.3pmol,将ddh2o补至10μl。连接条件16℃2h。取5μl连接产物转化到感受态细胞中,步骤与px330-sgrna的转化步骤一致。通过蓝白斑筛选方法挑选18个白斑进行测序(北京六合华大基因科技股份有限公司),计算px330-sgrna1载体对ttn基因的切割效率。结果显示2个克隆在预期切割位点附近出现突变(图2),px330-sgrna1切割效率为11.1%。
实施例2
将转染px330-sgrna5的纯化ttn1片段与pmdtm19-tvectorcloningkit载体(北京全式金,货号:6013)进行连接。连接体系为10μl,包括solutioni5μl,pmd19-t载体1μl,dna0.1pmol-0.3pmol,将ddh2o补至10μl。连接条件16℃2h。取5μl连接产物转化到感受态细胞中,步骤与px330-sgrna的转化步骤一致。通过蓝白斑筛选方法挑选白斑进行测序(北京六合华大基因科技股份有限公司),计算px330-sgrna5载体对ttn基因的切割效率。结果显示2个克隆在预期切割位点附近出现突变,px330-sgrna5切割效率为8.7%。
综上,本发明所述px330-sgrna1打靶载体对猪ttn基因进行靶向敲除,敲除效率最高,约为11.1%。
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
序列表
<110>中国农业大学
<120>一种靶向猪ttn基因的打靶载体及其应用
<141>2018-07-11
<160>14
<170>siposequencelisting1.0
<210>1
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>1
caccggtcattgcaaagaatgcggc25
<210>2
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>2
aaacgccgcattctttgcaatgacc25
<210>3
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>3
tgtgggtgtaggcaaagc18
<210>4
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>4
taaagccttgaaatccgtgt20
<210>5
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>5
gtcattgcaaagaatgcggc20
<210>6
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>6
gtgatttgcctgagggtcgc20
<210>7
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>7
agtaaaccttctgacagtac20
<210>8
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>8
tactggaccaattactgcca20
<210>9
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>9
caccggtgatttgcctgagggtcgc25
<210>10
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>10
aaacgcgaccctcaggcaaatcacc25
<210>11
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>11
caccgagtaaaccttctgacagtac25
<210>12
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>12
aaacgtactgtcagaaggtttactc25
<210>13
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>13
caccgtactggaccaattactgcca25
<210>14
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>14
aaactggcagtaattggtccagtac25
- 还没有人留言评论。精彩留言会获得点赞!