奥贝胆酸的晶型及其制备方法与流程
本发明涉及奥贝胆酸的晶型及其制备方法,属于医药
技术领域:
。
背景技术:
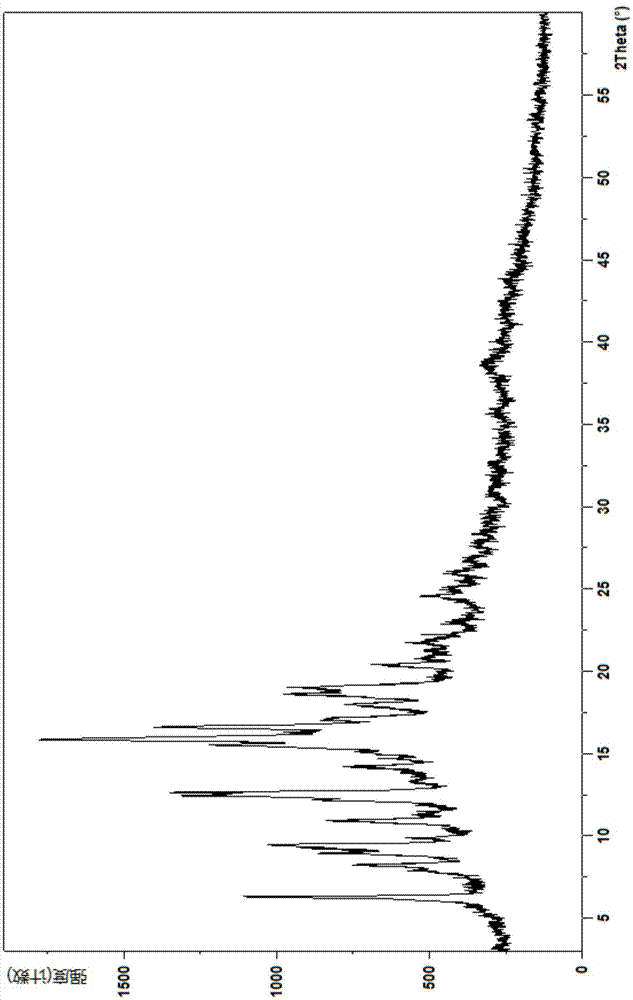
:奥贝胆酸是一种鹅脱氧胆酸衍生物,其化学名称为3α,7α-二羟基-6α-乙基-5β-胆烷-24酸,英文名称为obeticholicacid,其可用于治疗原发性胆汁性肝硬化和非酒精性脂肪性肝病,结构式如下:奥贝胆酸存在多种晶型,如cn104781272a中公开的晶型a、c、d、f和g及为非晶体形式的1型;其中,晶型a、c和d含有0.25mol当量水和不同量的各种有机溶剂的混合水合物/溶剂合物。加热时,这些晶型同时失去结晶水和溶剂,并且因其较低的熔解温度和较高的溶剂含量,不适合用于药物开发。另外,尽管晶型g、f具有较高的熔点,可能具有应用前景,但存在难以扩大化工业生产的问题,1型也存在含水量不易固定的问题,故这些晶型形式都存在难以用于制备药物过程中的问题。技术实现要素:术语定义术语“包含”或“包括”为开放式表达,即包括本发明所指明的内容,但并不排除其他方面的内容。术语“晶型”用来描述固体化合物的存在状态,描述晶体内部的离子、原子或分子组成、对称性质与周期排列规律的多种参量集合体。术语“相对结晶度”指样品中结晶部分在样品中所占的质量分数;术语“相对强度”是指将归属于某一晶型的一组衍射峰中的第一强峰的强度定义为100%时,其它峰的强度与第一强峰的强度的比值。在本发明的上下文中,x-射线粉末衍射图中的2θ(又称2theta或衍射峰)值均以度(°)为单位。当提及图谱和/或图中数据,术语“衍射峰”是指本领域的技术人员不会归属于背景噪音的一个特征。所述晶型的x-射线粉末衍射峰,其x-射线粉末衍射图谱的2θ或衍射峰的量度有实验误差,在一台机器和另一台机器之间以及一个样品和另一个样品之间,x-射线粉末衍射图谱的2θ或衍射峰的量度可能会略有差别,所述实验误差或差别的数值可能是+/-0.2个单位或+/-0.1个单位或+/-0.05个单位,因此所述2θ或衍射峰的数值不能视为绝对的。所述晶型的差示扫描量热曲线(dsc)有实验误差,在一台机器和另一台机器之间以及一个样品和另一个样品之间,吸热峰的位置和峰值可能会略有差别,实验误差或差别的数值可能小于等于5℃,或小于等于4℃,或小于等于3℃,或小于等于2℃,或小于等于1℃,因此所述dsc吸热峰的峰位置或峰值的数值不能视为绝对的。所述晶型的热重分析曲线(tga)有实验误差,在一台机器和另一台机器之间以及一个样品和另一个样品之间,失重温度和失重的量可能会略有差别,实验误差或差别的数值可能是大约+/-0.1个单位,大约+/-0.05个单位,或者大约+/-0.01个单位,因此所述失重温度和失重的量的数值不能视为绝对的。在本发明上下文中,无论是否使用“大约”或“约”等字眼,所有在此公开了的数字均为近似值。每一个数字的数值有可能会出现1%,2%,或5%等差异。“室温”是指温度在大约15℃-32℃或大约20℃-30℃或大约23℃-28℃或大约25℃。本发明中,晶型c指专利申请wo2013192097所述的c型,无定型指专利申请wo2013192097所述的1型。术语“良溶剂”可以是单一溶剂或混合溶剂,指样品在该单一溶剂或混合溶剂中的溶解度大于1g/l,或大于2g/l,或大于3g/l,或大于4g/l,或大于5g/l,或大于6g/l,或大于7g/l,或大于8g/l,或大于9g/l,或大于10g/l,或大于15g/l,或大于20g/l,或大于30g/l,或大于40g/l,或大于50g/l,或大于60g/l,或大于70g/l,或大于80g/l,或大于100g/l。在一些实施方案中,样品在良溶剂中的溶解度比不良溶剂大;在一些实施方案中,良溶剂和不良溶剂对样品的溶解度之差大约为10%,20%,30%,40%,50%,60%,70%,80%或90%;在一些实施方案中,良溶剂对样品的溶解度比不良溶剂大,大于10%,20%,30%,40%,50%,60%,70%,80%或90%。术语“不良溶剂”是指能促进溶液达到过度饱和状态或结晶的溶剂。在一些实施方案中,样品在不良溶剂中的溶解度小于0.001g/l,或小于0.01g/l,或小于0.1g/l,或小于0.2g/l,或小于0.3g/l,或小于0.4g/l,或小于0.5g/l,或小于0.6g/l,或小于0.8g/l,或小于1g/l,或小于2g/l,或小于3g/l,或小于4g/l,或小于5g/l,或小于6g/l,或小于7g/l,或小于8g/l,或小于9g/l,或小于10g/l。发明详述一方面,发明人通过研究开发了奥贝胆酸的晶型,称为晶型h。奥贝胆酸的晶型h,具有如下特性:其x-射线粉末衍射图中在2θ为6.28,8.26,9.46,10.97,15.50,15.88,16.59,17.91,18.56,和20.36度的位置有衍射峰,在2θ为2至6度间的位置无衍射峰。一些实施方案中,奥贝胆酸的晶型h的x-射线粉末衍射图中在2θ为6.28,8.26,8.91,9.46,9.83,10.97,12.32,12.48,12.65,14.19,15.50,15.88,16.59,17.91,18.56,19.01,20.36,和24.73度的位置有衍射峰,在2θ为2至6度间的位置无衍射峰。一些实施方案中,奥贝胆酸的晶型h的x-射线粉末衍射图如图1所示。奥贝胆酸的晶型h,还具有如下特性:其差示扫描量热曲线(dsc)在75℃-110℃处具有吸热峰。在一些实施方式中,奥贝胆酸的晶型h的差示扫描量热曲线(dsc)在87℃-105℃处具有吸热峰,峰顶值为99.1℃。奥贝胆酸的晶型h,还具有如下特性:其热重分析曲线(tga)显示在70℃-110℃间有失重,失重量约为4.46%。在一具体实施方案中,奥贝胆酸的晶型h的差示扫描量热曲线(dsc)和热重分析曲线(tga)如图2所示。另一方面,本发明还提供了奥贝胆酸晶型h的制备方法。一种制备奥贝胆酸的晶型h的方法,其包括:将奥贝胆酸与正庚烷和环己烷混合,室温搅拌12小时-36小时,收集固体,得到晶型h产物。根据晶型h的dsc和tga图谱,数据及结合其制备过程,认为晶型h中含有溶剂。发明人将所得晶型h延长时间真空干燥,检测xrpd,dsc和tga后发现,仍然含有溶剂,溶剂难以除去。发明人通过研究,还开发了奥贝胆酸的晶型,称为晶型i。奥贝胆酸的晶型i,具有如下特性:其x-射线粉末衍射图中在2θ为6.31,9.45,12.59,15.81,18.99,20.29度的位置有衍射峰,在2θ为2至6度间的位置无衍射峰。一些实施方案中,奥贝胆酸的晶型i的x-射线粉末衍射图中在2θ为6.31,8.19,9.45,12.59,15.81,16.61,18.61,18.99,20.29度的位置有衍射峰。一些实施方案中,奥贝胆酸的晶型i的x-射线粉末衍射图中在2θ为6.31,8.19,9.45,11.20,12.59,14.15,15.81,16.61,18.61,18.99,20.29,21.66,22.20度的位置有衍射峰。一些实施方案中,奥贝胆酸的晶型i的x-射线粉末衍射图如图3所示,其中,在2θ为12.59度的衍射峰的相对强度大于70%,或大于80%,或大于90%,或大于99%。一些实施方案中,奥贝胆酸的晶型i的x-射线粉末衍射图如图3所示。奥贝胆酸的晶型i,还具有如下特性:其差示扫描量热曲线(dsc)在95℃-110℃处具有吸热峰。在一些实施方式中,奥贝胆酸的晶型i的差示扫描量热曲线(dsc)在95℃-105℃处具有吸热峰,峰顶值为104.6℃。奥贝胆酸的晶型i,还具有如下特性:其热重分析曲线(tga)显示在70℃-110℃间有失重,失重量约为3.58%。在一具体实施方案中,奥贝胆酸的晶型i的差示扫描量热曲线(dsc)和热重分析曲线(tga)如图4所示。对本发明所述晶型i进行研究,发现晶型i在一定条件下向奥贝胆酸的晶型h转变。另一方面,本发明还提供了奥贝胆酸晶型i的制备方法。一种制备奥贝胆酸的晶型i的方法,其包括:将奥贝胆酸溶于良溶剂,然后加入不良溶剂,收集析出的固体,干燥,得到晶型i产物。在一些实施方式指,一种制备奥贝胆酸的晶型i的方法,其包括:将奥贝胆酸溶于良溶剂丙酮或乙酸乙酯,然后加入不良溶剂环己烷,收集析出的固体,在40℃-60℃真空干燥6小时-36小时,得到晶型i产物。发明人通过研究,开发了奥贝胆酸的晶型,称为晶型k。奥贝胆酸的晶型k,具有如下特性:其x-射线粉末衍射图中在2θ为7.2,10.5,12.8,13.9,14.6,15.7,16.0,16.6,17.5,18.0,18.6,18.9,19.3,19.5,21.2,22.9,24.4度的位置有衍射峰。一些实施方案中,奥贝胆酸的晶型k,具有如下特性:其x-射线粉末衍射图中在2θ为7.2,10.5,12.2,12.6,12.8,13.9,14.6,15.7,16.0,16.6,17.5,18.0,18.6,18.9,19.3,19.5,21.2,22.3,22.9,24.4,25.3,39.1度的位置有衍射峰。一些实施方案中,奥贝胆酸的晶型k,具有如下特性:其x-射线粉末衍射图中在2θ为7.2,10.5,12.2,12.6,12.8,13.9,14.6,15.7,16.0,16.6,17.5,18.0,18.6,18.9,19.3,19.5,21.2,21.7,21.9,22.3,22.9,23.4,23.9,24.4,25.3,27.0,29.4,30.1,31.2,32.0,35.1,36.9,39.1度的位置有衍射峰。一些实施方案中,奥贝胆酸的晶型k,具有如下特性:其x-射线粉末衍射图中在2θ为7.24,10.52,12.24,12.61,12.80,13.90,14.56,15.69,16.05,16.60,17.46,17.98,18.55,18.94,19.29,19.55,21.23,22.28,22.89,23.87,24.36,25.31,39.14度的位置有衍射峰。一些实施方案中,奥贝胆酸的晶型k,具有如下特性:其x-射线粉末衍射图中在2θ为7.24,10.52,12.24,12.61,12.80,13.90,14.56,15.69,16.05,16.60,17.46,17.98,18.55,18.94,19.29,19.55,21.23,21.66,21.91,22.28,22.89,23.45,23.87,24.36,25.31,39.14度的位置有衍射峰。一些实施方案中,奥贝胆酸的晶型k,具有如下特性:其x-射线粉末衍射图中在2θ为7.24,10.52,12.24,12.61,12.80,13.90,14.56,15.69,16.05,16.60,17.46,17.98,18.55,18.94,19.29,19.55,21.23,21.66,21.91,22.28,22.89,23.45,23.87,24.36,25.31,26.99,29.35,30.12,31.17,32.01,35.14,36.97,39.14度的位置有衍射峰。一些实施方案中,奥贝胆酸的晶型k,具有如下特性:其x-射线粉末衍射图中在2θ为7.24,12.69,13.93,14.56,17.49,17.97,22.82度的位置有衍射峰。一些实施方案中,奥贝胆酸的晶型k的x-射线粉末衍射图中在2θ为7.24,10.52,12.69,13.93,14.56,16.62,17.49,17.97,22.82,24.35度的位置有衍射峰。一些实施方案中,奥贝胆酸的晶型k的x-射线粉末衍射图中在2θ为7.24,10.52,12.69,13.93,14.56,15.86,16.62,17.49,17.97,18.65,19.34,22.82,24.35,39.16度的位置有衍射峰。一些实施方案中,奥贝胆酸的晶型k的x-射线粉末衍射图中在2θ为7.24,10.52,12.69,13.93,14.56,15.60,15.86,16.62,17.49,17.97,18.65,19.34,19.58,21.44,22.82,23.04,24.35,25.30,25.72,26.33,29.35,30.24,31.19,33.68,39.16度的位置有衍射峰。一些实施方案中,奥贝胆酸的晶型k的x-射线粉末衍射图中具有如下表1所示的以2θ值(度)表示的特征峰和对应的相对强度。表1:2θ值(度)相对强度%2θ值(度)相对强度%2θ值(度)相对强度%7.2450.619.2927.926.994.310.526.619.5523.427.703.312.245.621.237.228.143.912.6125.421.667.529.355.312.8031.721.915.630.124.713.9096.922.234.631.175.614.5610022.8936.532.014.515.6925.723.454.132.643.316.0513.823.874.733.703.316.6014.024.3610.835.143.617.4744.925.318.435.973.317.9813.425.755.736.884.918.5527.326.045.437.823.118.9418.026.373.639.148.0一些实施方案中,奥贝胆酸的晶型k的x-射线粉末衍射图如图5所示,其中,在2θ为13.93度的衍射峰的相对强度大于70%。一些实施方案中,奥贝胆酸的晶型k的x-射线粉末衍射图如图5所示。一些实施方案中,奥贝胆酸的晶型k的x-射线粉末衍射图如图6所示,其中,在2θ为13.93度的衍射峰的相对强度大于70%。一些实施方案中,奥贝胆酸的晶型k的x-射线粉末衍射图如图6所示。在一些实施方式中,所述晶型k的相对结晶度不低于95%。在一些实施方式中,所述晶型k的相对结晶度不低于97%。在一些实施方式中,所述晶型k的相对结晶度不低于99%。在一些实施方式中,所述晶型k基本不含无定型,或者无定型含量在5%以下,或者无定型含量在3%以下,或者无定型含量在1%以下。奥贝胆酸的晶型k,还具有如下特性:其差示扫描量热曲线(dsc)在85℃-110℃处具有吸热峰。在一些实施方式中,奥贝胆酸的晶型k,还具有如下特性:其差示扫描量热曲线(dsc)在90℃-105℃处具有吸热峰。在一些实施方式中,奥贝胆酸的晶型k,还具有如下特性:其差示扫描量热曲线(dsc)在95℃-105℃处具有吸热峰。在一些实施方式中,奥贝胆酸的晶型k的差示扫描量热曲线(dsc)在85℃-95℃处具有吸热峰,吸热峰峰顶值为93.8℃。在一些实施方式中,奥贝胆酸的晶型k的差示扫描量热曲线(dsc)在98℃-103℃处具有吸热峰,吸热峰峰顶值为101.2℃。在一些实施方式中,奥贝胆酸的晶型k的差示扫描量热曲线(dsc)在98℃-103℃处具有吸热峰,吸热峰峰顶值为101.6℃。奥贝胆酸的晶型k,还具有如下特性:其热重分析曲线(tga)显示在50℃-100℃间有失重,失重量约为3%-5%。在一些实施方式中,奥贝胆酸的晶型k,还具有如下特性:其热重分析曲线(tga)显示在80℃-100℃间有失重,失重量约为3%-5%。在一些实施方式中,奥贝胆酸的晶型k,具有如下特性:其热重分析曲线(tga)显示在50℃-100℃间有失重,失重量约为3.55%。在一些实施方式中,奥贝胆酸的晶型k,具有如下特性:其热重分析曲线(tga)显示在50℃-100℃间有失重,失重量约为4.5%。在一些实施方式中,奥贝胆酸的晶型k,具有如下特性:其热重分析曲线(tga)显示在50℃-100℃间有失重,失重量约为4%。在一具体实施方案中,奥贝胆酸的晶型k的差示扫描量热曲线(dsc)和热重分析曲线(tga)如图7所示。在一具体实施方案中,奥贝胆酸的晶型k的差示扫描量热曲线(dsc)如图8所示。根据奥贝胆酸的分子式,奥贝胆酸一水合物的水理论含量为4.10%。根据晶型k的差示扫描量热曲线(dsc)和热重分析曲线(tga)数据,认为所述晶型k为一水合物。发明人发现,当晶型k相对结晶度降低或晶型k中含有无定型时,其dsc和/或tga数据与相对结晶度高的晶型k的dsc和/或tga数据会略有差异,表现出存在不同的又相近的数据。基于晶型k样品中奥贝胆酸的总重量,在一些实施方式中,前述的晶型k含有至少95%的晶型k,可含有少量其它晶型或者无定型。基于晶型k样品中奥贝胆酸的总重量,在一些实施方式中,前述的晶型k含有至少97%的晶型k。基于晶型k样品中奥贝胆酸的总重量,在一些实施方式中,前述的晶型k含有至少99%的晶型k。在一些实施方式中,前述的晶型k的相对结晶度不低于95%。在一些实施方式中,前述的晶型k的相对结晶度不低于97%或者99%。对本发明所述晶型k进行研究,发现晶型k具有较好的稳定性,其虽然在60℃条件下长时间储存,有向无定型转化的趋势,但其在高湿度和光照下较为稳定,在40℃,相对湿度75%条件下也极为稳定,可以在常规储层条件下储层和用于制剂产品制备中。另外,发明人还发现,晶型k与专利申请wo2013192097中的晶型c及无定型相比,在一定条件下存在更好的稳定性和更强的竞争力。所述晶型k或制备成盐后可与药学上可接受的辅料,如填充剂,粘合剂,崩解剂,或润滑剂等混合,制备成各种剂型,如片剂,胶囊,颗粒剂等适合的剂型。一种药物组合物,包括前述的奥贝胆酸的晶型k和药学上可接受的辅料,其中,基于奥贝胆酸的总重量或按照奥贝胆酸的总重量计算,含有至少95%的晶型k,或至少97%的晶型k,或至少99%的晶型k。在一些实施方式中,一种药物组合物,包括前述的奥贝胆酸的晶型k和药学上可接受的辅料,其中,晶型k的相对结晶度不低于95%,或者相对结晶度不低于97%,或者相对结晶度不低于99%。另一方面,本发明还提供了奥贝胆酸晶型k的制备方法。一种制备奥贝胆酸的晶型k的方法,其包括:将奥贝胆酸粗品与甲苯混合,所得混悬液在室温搅拌一定时间,然后收集固体,干燥,得到晶型k产物。在一些实施方式中,一种制备奥贝胆酸的晶型k的方法,其包括:将奥贝胆酸粗品与甲苯混合,所得混悬液在室温搅拌12小时-48小时,然后收集固体,在25℃-40℃真空干燥8小时-20小时,然后再在40℃-60℃鼓风干燥6小时-24小时,得到晶型k产物。另一方面,一种制备晶型k的方法,包括:将奥贝胆酸粗品和晶型k晶种与水混合,在35℃-80℃搅拌10小时-30小时,然后过滤,得到晶型k。在一些实施方式中,一种制备晶型k的方法,包括:将奥贝胆酸粗品溶于乙腈得到澄清溶液,然后将所得溶液加入到水中,室温搅拌10小时-30小时,然后过滤,收集固体,真空干燥至恒重,得到晶型k。在一些实施方式中,一种制备晶型k的方法,包括:将奥贝胆酸粗品溶于乙腈得到澄清溶液,然后将所得溶液加入到水中,室温搅拌10小时-30小时,然后过滤,收集固体,在20℃-60℃真空干燥1小时-24小时,得到晶型k。在一些实施方式中,一种制备晶型k的方法,包括:将奥贝胆酸粗品溶于有机溶剂得到澄清溶液,然后将所得溶液加入到加有晶型k晶种的水中,室温搅拌10小时-30小时,然后过滤,收集固体,真空干燥至恒重,得到晶型k;其中,每一克粗品,有机溶剂的用量为3ml-20ml,水的用量为3ml-50ml,晶种用量为0.01g-0.5g,所述有机溶剂与水的体积比不高于1:2;所述有机溶剂选自甲醇,乙醇,丙酮,乙腈和n,n-二甲基甲酰胺中的至少一种。在一些实施方式中,一种制备晶型k的方法,包括:将奥贝胆酸粗品溶于有机溶剂得到澄清溶液,然后将所得溶液加入到加有晶型k晶种的水中,室温搅拌10小时-30小时,然后过滤,收集固体,在20℃-60℃真空干燥1小时-24小时,得到晶型k;其中,每一克粗品,有机溶剂的用量为3ml-20ml,水的用量为3ml-50ml,晶种用量为0.01g-0.5g,所述有机溶剂与水的体积比不高于1:2;所述有机溶剂选自甲醇,乙醇,丙酮,乙腈和n,n-二甲基甲酰胺中的至少一种。在一些实施方式中,所述有机溶剂与水的体积比不高于1:3。在一些实施方式中,所述有机溶剂选自甲醇,乙醇,丙酮和n,n-二甲基甲酰胺中的至少一种。在一些实施方式中,所述有机溶剂选自甲醇,乙醇和丙酮中的至少一种,有利于结晶时操作和成本控制。在一些实施方式中,所述有机溶剂选自乙醇和丙酮中的至少一种,有利于结晶时操作和成本控制。在一些实施方式中,一种制备晶型k的方法,包括:将奥贝胆酸粗品溶于有机溶剂得到澄清溶液,然后将所得溶液加入到加有晶型k晶种的水中,室温搅拌10小时-30小时,然后过滤,收集固体,在20℃-60℃真空干燥6小时-24小时,得到晶型k;其中,每一克粗品,有机溶剂的用量为5ml-15ml,水的用量为10ml-40ml,晶种用量为0.1g-0.5g,所述有机溶剂与水的体积比不高于1:2;所述有机溶剂选自甲醇,乙醇,丙酮,乙腈和n,n-二甲基甲酰胺中的至少一种。在一些实施方式中,一种制备晶型k的方法,包括:将奥贝胆酸粗品溶于有机溶剂得到澄清溶液,然后将所得溶液加入到加有晶型k晶种的水中,室温搅拌10小时-30小时,然后过滤,收集固体,在20℃-60℃真空干燥6小时-24小时,得到晶型k;其中,每一克粗品,有机溶剂的用量为8ml-12ml,水的用量为24ml-36ml,晶种用量为0.1g-0.5g,所述有机溶剂与水的体积比不高于1:3;所述有机溶剂选自甲醇,乙醇,丙酮,乙腈和n,n-二甲基甲酰胺中的至少一种。在一些实施方式中,一种制备晶型k的方法,包括:将奥贝胆酸粗品溶于有机溶剂得到澄清溶液,然后将所得溶液加入到加有晶型k晶种的水中,室温搅拌10小时-30小时,然后过滤,收集固体,在20℃-60℃真空干燥8小时-20小时,得到晶型k;其中,每一克粗品,有机溶剂的用量为10ml,水的用量为30ml,晶种用量为0.2g;所述有机溶剂选自甲醇,乙醇,丙酮,乙腈和n,n-二甲基甲酰胺中的至少一种。本发明提供的奥贝胆酸晶型k,其稳定,易于制备生产,且可以用于制备药物制剂的生产中。附图说明图1示奥贝胆酸的晶型h的x-射线粉末衍射图(xrpd)。图2示奥贝胆酸的晶型h的差示扫描量热曲线图(dsc)和热重分析曲线图(tga)。图3示奥贝胆酸的晶型i的x-射线粉末衍射图(xrpd)。图4示奥贝胆酸的晶型i的差示扫描量热曲线图(dsc)和热重分析曲线图(tga)。图5示实施例5所得奥贝胆酸的晶型k的x-射线粉末衍射图(xrpd)。图6示实施例8所得奥贝胆酸的晶型k的x-射线粉末衍射图(xrpd)。图7示实施例5所得奥贝胆酸的晶型k的差示扫描量热曲线图(dsc)和热重分析曲线图(tga)。图8示实施例8所得奥贝胆酸的晶型k的差示扫描量热曲线图(dsc)。图9示实施例8中所得奥贝胆酸的晶型k和按照体积比乙腈/水=1:1制备所得的晶型的x-射线粉末衍射图(xrpd);图中,按强度轴,从高至低,分别为晶型k和按照体积比乙腈/水=1:1制备所得的晶型。图10示实施例10晶型k样品在放置前和在40℃,相对湿度75%条件下放置,第0天,第30天,第60天和第90天检测的xrpd图;图中,按强度轴,从高至低,分别为晶型k样品在第90天,第60天,第30天,第0天的xrpd。图11示实施例10晶型k样品在放置前和在40℃,相对湿度75%条件下放置第0天,第30天,第60天和第90天检测的dsc图;图中,按照热流量轴,从高至低,分别为晶型k样品在第0天,第30天,第60天和第90天检测的dsc。图12示实施例10晶型k样品在光照、相对湿度90%、60℃条件下放置15天后的xrpd图谱;图中,按强度轴,从高至低,分别为晶型k样品在光照、相对湿度90%、60℃条件下放置15天后检测的xrpd。具体实施方式为了使本领域的技术人员更好地理解本发明的技术方案,下面进一步披露一些非限制实施例对本发明作进一步的详细说明。本发明所使用的试剂均可以从市场上购得或者可以通过现有技术的方法制备或者通过本发明所描述的方法制备而得。本发明中,℃表示摄氏度,g表示克,ml或ml表示毫升,h表示小时,dmf表示n,n-二甲基甲酰胺。以下实施例中,所述奥贝胆酸粗品为无定型,经tga检测含水量约2.5%。仪器参数除非参数中另行规定,以下所有分析都在室温下进行。x-射线粉末衍射(xrpd)在装配有自动化3*15零背景样品架的透射反射样品台的荷兰panalyticalempyreanx-射线衍射仪上收集x-射线粉末衍射(xrpd)图案。所用辐射源为(cu,kα,kα11.540598;kα21.544426;kα2/kα1强度比例:0.50),其中电压设定在45kv,电流设定在40max-射线的束发散度,即样品上x-射线约束的有效尺寸,为10mm。采用θ-θ连续扫描模式,得到3°~60°的有效2θ范围。取适量样品在环境条件(约18℃~32℃)下于零背景样品架圆形凹槽处,用洁净的载玻片轻压,得到一个平整的平面,并将零背景样品架固定。将样品以0.0167°的扫描步长在3°~60°2θ±0.2°范围内产生传统的xrpd图案,横坐标为2theta(°),纵坐标为强度计数(intensity(counts))。用于数据收集的软件为datacollector,数据用dataviewer和highscoreplus分析和展示。本发明中,相对结晶度是根据xrpd数据计算获得。差示扫描量热曲线(dsc)dsc测量在tainstrumentstm型号q2000中用密封盘装置进行。将样品(约1~3mg)在铝盘中称量,用tzero压盖,精密记录到百分之一毫克,并将样品转移至仪器中进行测量。仪器用氮气以50ml/min吹扫。在室温到300℃之间以10℃/min的加热速率收集数据。以吸热峰向下进行绘图,数据用tauniversalanalysis分析和展示。在dsc图中,横坐标表示温度(temperature,℃),纵坐标表示单位质量的物质放出的热流量(heatflow,w/g)。热重分析曲线(tga)tga测量在tainstrumentstm型号q500中进行。操作步骤为空坩埚去皮,取固体样品约10mg、于去皮空坩埚内,铺匀即可。待仪器运行稳定后,在氮气吹扫下,室温到300℃之间以10℃/min的加热速率收集数据,记录图谱。在tga图中,横坐标表示温度(temperature,℃),纵坐标表示质量百分数(weight,%)。以下实验中,晶型k样品纯度在99%以上,晶型k样品的相对结晶度不低于95%,含有至少97%的晶型k。实施例1将500mg的奥贝胆酸粗品加入3ml乙酸乙酯中,置于45℃油浴中搅拌得到澄清溶液后,再缓慢降温至室温(25℃),析出白色固体,抽滤并置于干燥箱内50℃真空干燥12小时,得到白色晶体约430mg。经测定,为晶型h,其x射线粉末衍射图谱见图1,其dsc和tga图谱见图2。将2g奥贝胆酸粗品按照上述相同比例、方法操作,发现降温至室温(25℃)难以析出固体,加入环己烷20ml后,可以析出固体,经测定,其x射线粉末衍射图谱与图1基本一致,其dsc和tga图谱与图2基本一致,为晶型h。实施例2将500mg的奥贝胆酸粗品加入5ml正庚烷和5ml环己烷的混合溶剂中,混悬搅拌24小时,抽滤并置于干燥箱内50℃真空干燥12小时,得到白色晶体约460mg。经测定,其x射线粉末衍射图谱与图1基本一致,其dsc和tga图谱与图2基本一致。实施例3将100mg的奥贝胆酸粗品加入1ml丙酮中得澄清溶液,随后缓慢滴加8ml环己烷,析出白色固体,抽滤并置于干燥箱内50℃真空干燥16小时,得到白色晶体约85mg。经测定,为晶型i,其x射线粉末衍射图谱见图3,其dsc和tga图谱见图4。实施例4将100mg的奥贝胆酸粗品加入1ml乙酸乙酯中得澄清溶液,随后缓慢滴加8ml环己烷,析出白色固体,抽滤并置于干燥箱内50℃真空干12小时燥,得到白色晶体约85mg。经测定,为晶型i,其x射线粉末衍射图谱与图3基本一致,其dsc和tga图谱与图4基本一致。实施例5将210mg的奥贝胆酸粗品加入2ml甲苯中得混悬搅拌24小时得到白色固体,抽滤并置于干燥箱内室温真空干燥12小时,得到白色晶体约180mg;然后将上述晶体于50℃真空干燥箱内干燥10小时,检测,发现为晶型k,其x射线粉末衍射图谱参见图5,其dsc和tga图谱参见图7。实施例6按照实施例5的方法,将2g奥贝胆酸粗品加入20ml甲苯中得混悬液,搅拌24小时,得到白色固体,抽滤,所得固体置于干燥箱内室温真空干燥12小时,得到白色晶体约1.83g;然后将上述晶体于50℃鼓风干燥箱内干燥20小时,检测,为晶型k。实施例7取无定型奥贝胆酸100mg置于一定量的水中打浆,分别加入晶型k为晶种或不加晶种,在一定温度混悬打浆搅拌24小时,然后过滤,所得固体于50℃真空干燥箱内干燥10小时,操作和结果如下表2所示。晶型k晶种可按照实施例6的方法获得。表2:根据结果可以看出,使用无定型原料在水中加晶型k晶种打浆可获得晶型k产物,优选在20℃~80℃区间混悬,更优选在35℃~65℃区间混悬,也可以得知,无定型在一定条件下可以转变为晶型k。另外,发明人根据上述方法,取无定型奥贝胆酸100mg置于1.0ml水中打浆,加入50mg晶型k为晶种,在50℃混悬打浆搅拌24小时,然后过滤,所得固体转移至xrpd测试仪直接检测xrpd,为晶型k;然后将所得固体于50℃真空干燥箱内干燥10小时,再检测xrpd,仍然为晶型k。根据实验结果也可以得出,晶型k在常规干燥处理前后无变化。实施例8取无定型奥贝胆酸50mg溶清于各溶剂(溶剂一)中,然后滴加至室温下的水(溶剂二)中,析出固体后搅拌24h,过滤,分离固液,固体于50℃真空干燥箱内干燥10小时,得到产物,各溶剂和所得固体产物的检测结果见下表3。表3:根据结果可知,在不含晶种的情况下,上述水/有机溶剂体系中,只有水/乙腈体系能获得晶型k,但乙腈毒性大,不适于工业生产中使用;获得的晶型k其xrpd图见图6,dsc图见图8。另外,将100mg无定型奥贝胆酸加入至1ml乙腈和1ml水的混合溶液中(即按照体积比乙腈/水=1:1制备),室温下未完全溶解,混悬搅拌24h,然后过滤,得到固体,所得固体直接检测xrpd,结果见附图9,图中,按强度轴,从高至低,分别为晶型k和按照体积比乙腈/水=1:1制备所得的晶型,根据xrpd图谱,可以看出两份样品的xrpd极为接近,但在2theta值为17.5-20度间和20-22.5度间,两种方法/条件获得的晶型有明显差别;而将此按照体积比乙腈/水=1:1制备所直接得到的固体于50℃真空干燥12h后再检测xrpd,发现其图谱已经变化,干燥后检测的xrpd与晶型k一致,即所得固体已经转变为晶型k。实施例9取无定型奥贝胆酸50mg溶清于各溶剂(溶剂三)中,然后滴加至室温下的预先加入了10mg晶型k晶种的水溶液(溶剂四)中,析出固体后搅拌24h,过滤,分离固液,所得固体于50℃真空干燥箱内干燥10小时,得到产物,各溶剂和所得固体产物的检测结果见下表4。表4:根据结果可以看出,在使用晶种的情况下,使用水/甲醇、水/乙醇、水/丙酮、水/乙腈、水/dmf等体系均可以制备出晶型k,工艺简单易操作,更优选在水/乙醇或水/丙酮中结晶制备晶型k,有利于操作控制和晶型k的获得。另外,也可以得出,在存在晶型k晶种的情况下,无定型在上述实施条件易转变为晶型k,一定条件下,无定型易转变为晶型k。实施例10:稳定性实验根据《中国药典》的相关规定,取晶型k样品置于pe袋中,扎口,然后铝箔袋封口包装,在40℃,相对湿度75%条件下放置3个月,分别在第0天,第30天,第60天,第90天取样检测xrpd,dsc和tga,结果如下表5,参照图9和图10。表5:根据结果可知,晶型k在40℃,相对湿度75%条件下,晶型、水分、纯度未发生明显变化,即晶型k在该条件下稳定。高温实验:取适量晶型k样品放入扁平称量瓶中,铺展成薄层(≤5mm),将扁平称量瓶置于60℃的培养箱中15天,然后在第16天检测样品;高湿度试验:将适量晶型k样品放入称量瓶中,铺展成薄层(≤5mm),将称量瓶置于湿度室中,25℃,相对湿度90%±5%放置15天;然后在第16天检测样品;光照实验:取适量晶型k样品放入称量瓶中,铺展成薄层(≤5mm),在开放式光照培养箱中使称量瓶在4500±500lx下光照,放置15天;然后在第16天检测样品;x-射线粉末衍射检测和检测纯度,结果如下表6,xrpd参见图11。表6:上述实验中,晶型k样品纯度在97%以上,晶型k样品的相对结晶度不低于95%,含有至少97%的晶型k;以上结果中,纯度基本不变是指纯度检测数据与放置实验前相差在0.5%以内。发明人发现,晶型k在60℃高温实验中,纯度降低是有杂质生成,暂未查找杂质结构和生成原因。根据上述实验结果可知,晶型k在高湿和光照条件下都较为稳定,晶型不变和纯度基本无改变,在高温下有一定变化,但主要变化不是在晶型方面,若控制包装和条件,仍然可以保持晶型k完全稳定而用于制剂中。实施例11:与晶型c和无定型的竞争实验分别取样品在50℃的水体系中混悬打浆24h,然后过滤,x-射线粉末衍射检测所得固体,结果如下表7。表7:根据结果可知,晶型k在与其它晶型或无定型混合打浆后,所得产物为晶型k,即其它晶型或无定型转变成晶型k,提示在该体系或条件下晶型k比晶型c或无定型更稳定。根据上述实验可知,晶型k是比较稳定的晶型,且在制备过程中,在某些条件更容易得到晶型k。本发明的方法已经通过较佳实施例进行了描述,相关人员明显能在本
发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1