VDR基因多态性检测试剂盒的制作方法
本发明涉及生物技术领域,具体为一种vdr基因多态性检测试剂盒。
背景技术
np,单核苷酸多态性(全称singlenucleotidepolymorphisms),是指在基因组上单个核苷酸的变异(包括置换、颠换、缺失和插入)形成的遗传标记,其数量很多,多态性丰富。从理论上来看,每一个snp位点都可以有4种不同的变异形式,但实际上发生的只有两种,即转换和颠换,二者之比为1:2。snp在cg序列上出现最为频繁,而且多是c转换为t,原因是cg中的c常为甲基化的,自发地脱氨后即成为胸腺嘧啶。一般而言,snp是指变异频率大于1%的单核苷酸变异。在人类基因组中大概每1000个碱基就有一个snp,人类基因组上的snp总量大概是3×106个。因此,snp成为第三代遗传标志,人体许多表型差异、对药物或疾病的易感性等等都可能与snp有关。
维生素d受体(vdr)为亲核蛋白,是介导1,25(oh),d发挥生物效应的核内生物大分子,属于超家族成员。维生素d的许多生物学功能都是通过vdr介导调节靶基因转录来实现的1,25(oh),d激素信号分子在靶细胞与vdr结合形成激素一受体复合物,该复合物作用于靶基因上的特定dna序列,对结构基因的表达产生调节作用。vdr在本质上是一种配体依赖的核转录因子,它在维持机体钙一磷代谢,调节细胞增殖、分化等方面起重要作用。
vdr基因序列上存在多个内切酶酶切位点,证实了vdr基因具有明显的多态性,到目前为止,至少有25个vdr多态性位点被发现,其中研究较多的为bsmi、apai、foki这三个位点,是参与骨代谢的主要位点。不同区域的多态性位点对vdr基因表达产生的影响不同,bsmi和apal酶切位点位于第viii内含子上,其多态性不影响vdr的氨基酸序列;foki酶切位点位转录起始部位,其多态性可导致氨基酸序列长度发生改变。
技术实现要素:
本发明所解决的技术问题在于提供一种vdr基因多态性检测试剂盒,以解决上述背景技术中的问题。
本发明所解决的技术问题采用以下技术方案来实现:vdr基因多态性检测试剂盒,包括:mgcl2:2.0~5.0mmol,dntps:0.2~0.8mmol,各条引物:0.1~1.0μmol,各条探针:0.1~1.0μmol,abifastmasterpremix:6~10μl,模板:10μl,总体积:40μl。
vdr基因多态性检测试剂盒使用方法:包括以下步骤:
步骤(1),提取的dna样品;
步骤(2),dna样品扩增:2份基因组dna,用te稀释液稀释到浓度1ng/μl,在abi7300荧光定量pcr仪上进行扩增;荧光定量pcr反应体系为:
pcr反应液:mgcl2:2.0~5.0mmol,dntps:0.2~0.8mmol,各条引物:0.1~1.0μmol,各条探针:0.1~1.0μmol,abifastmasterpremix:6~10μl,模板:10μl,总体积:40μl。
pcr反应条件为:第一阶段:95℃5min;第二阶段:95℃5s,58℃30s,10个循环;第三阶段:95℃5s,58℃30s,72℃30s,35个循环;第三阶段:35个循环时,收集fam和joe信号;
步骤(3),结果检测,检测方法如下:
(1)每次pcr反应中,同时进行非模板对照(notemplatecontrol;ntc)、阳性对照和待测样本的检测;
(2)将阳性对照取出解冻后震荡混匀,并离心30s;
(3)将6联pcr反应管取出解冻后离心30s,防止反应液在开盖时溅出;
(4)将ddh2o(ntc)、待测样本、阳性对照分别加入6联pcr反应管,每孔10μl;
(5)将以上6联pcr反应管盖好管盖于离心机上离心30s,确保管壁上不沾有液滴;
(6)将6联pcr反应管放于实时pcr仪器中;
pcr反应条件为:第一阶段:95℃4min;第二阶段:95℃5s,58℃30s,10个循环;第三阶段:95℃30s,58℃30s,72℃30s,35个循环;信号收集:第三阶段72℃时收集fam和joe信号。
所述提取的dna样品为临床样本dna的提取,包括以下步骤:
1.处理血液材料:
a.当血液样品体积小于200μl时,可加缓冲液gs补足体积至200μl,再进行下一步实验(如血液样品体积为200μl,可直接进行下一步实验,不需加入gs);b.当血液样品体积超过200μl时,需用细胞裂解液cl处理,具体步骤如下:在样品中加入1~2.5倍体积的细胞裂解液cl,颠倒混匀,10,000rpm(~11,500×g)离心1min,吸去上清,留下细胞核沉淀(如果裂解不彻底,可重复以上步骤一次),向离心收集到的细胞核沉淀中加200μl缓冲液gs,振荡至彻底混匀;
2.加入20μlproteinasek溶液,混匀;
3.加200μl缓冲液gb,充分颠倒混匀,56℃放置10min,其间颠倒混匀数次,溶液应变清亮;
4.加200μl无水乙醇,充分颠倒混匀,此时可能会出现絮状沉淀;
5.将上一步所得溶液和絮状沉淀都加入一个吸附柱cb3中(吸附柱cb3放入收集管中),12,000rpm(~13,400×g)离心30sec,倒掉收集管中的废液,将吸附柱cb3放入收集管中;
6.向吸附柱cb3中加入500μl缓冲液gd(使用前请先检查是否已加入无水乙醇),12,000rpm(~13,400×g)离心30sec,倒掉收集管中的废液,将吸附柱cb3放入收集管中;
7.向吸附柱cb3中加入600μl漂洗液pw(使用前请先检查是否已加入无水乙醇),12,000rpm(~13,400×g)离心30sec,倒掉收集管中的废液,将吸附柱cb3放入收集管中;
8.重复操作步骤7;
9.12,000rpm(~13,400×g)离心2min,倒掉废液。将吸附柱cb3置于室温放置数分钟,以彻底晾干吸附材料中残余的漂洗液;
10.将吸附柱cb3转入1.5ml离心管中,向吸附膜中间位置悬空滴加50-200μl洗脱缓冲液tb,室温放置2~5min,12,000rpm(~13,400×g)离心2min,将溶液收集到离心管中;定量:将提取的dna用紫外分光光度计进行定量,稀释dna浓度到1ng/μl。
所述提取的dna样品为口腔拭子样本,包括以下步骤:
1.将口腔拭子的棉签用剪刀从杆上剪下,置于2ml的离心管(自备)中,加入400μlbuffergr;如需无rna污染的基因组dna,可加入4μl浓度为100mg/ml的rnasea溶液(货号:cw0601),震荡混匀;
2.加入20μlproteinasek和400μlbuffergl,立即涡旋震荡15秒,充分混匀;加入buffergl后立即充分混匀;不可将proteinasek直接加入buffergl中使用;
3.56℃放置10分钟,短暂离心,使管壁上的溶液收集到管底;
4.加入400μl无水乙醇,涡旋震荡充分混匀,短暂离心,使管壁上的溶液收集到管底;
5.将上步所得溶液和沉淀分两次加入到已装入收集管(collectiontube2ml)的吸附柱(spincolumnds)中,一次最多不超过700μl。12,000rpm(~13,400×g)离心1分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中;
6.向吸附柱中加入500μlbuffergw1(使用前检查是否已加入无水乙醇),12,000rpm离心1分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中;
7.向吸附柱中加入500μlbuffergw2(使用前检查是否已加入无水乙醇),12,000rpm离心3分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中;如需进一步提高dna纯度,可重复此步骤;
8.12,000rpm离心1分钟,倒掉收集管中的废液。将吸附柱置于室温数分钟,以彻底晾干;
9.将吸附柱置于一个新的1.5ml离心管中,向吸附柱的中间部位悬空加入50μlbufferge或灭菌水,室温放置2~5分钟,12,000rpm离心1分钟,收集dna溶液,-20℃保存;
10、定量,将提取的dna用紫外分光光度计进行定量,稀释dna浓度到1ng/μl。
与已公开技术相比,本发明存在以下优点:
(1)灵敏度高,可以准确检出低至1ng的基因组dna。对于保存时间过长以及口腔拭子这些提取浓度很低的样本能准确检出。
(2)特异性强,对于高至500ng及以上的基因组dna不会出现非特异性的结果。可以减少稀释样本的过程,提取出的样本直接上母液都能准确检测而不会出现非特异性。
(3)适用于多种样本类型:本发明不仅能检测常规的edta抗凝的全血细胞dna,还能检测提取质量差的口腔拭子,检测结果准确度高。
(4)成本低,arms分型法相对于taqman探针分型法,不仅能保留taqman的高灵敏度和高特异性,而且还能节约成本,arms引物合成快速简单,合成成本低,扩增效果更佳。
(5)检测速度快,整个检测过程只需要90分钟。
(6)整个试剂盒不包含有毒有害物质,对操作人员和环境无危害。
附图说明
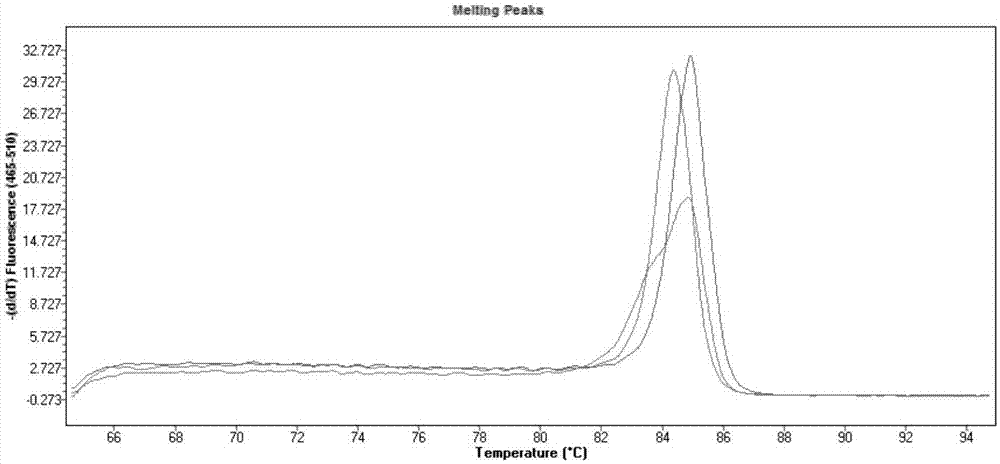
图1为本发明的bsmi位点对照基因型的高分辨熔解曲线图;
图2为本发明的apai位点对照基因型的高分辨熔解曲线图;
图3为本发明的foki位点对照基因型的高分辨熔解曲线图。
图4为本发明的bsmi位点对照基因型以及样本①②的高分辨熔解曲线图
图5为本发明的apai位点对照基因型以及样本①②的高分辨熔解曲线图
图6为本发明的foki位点对照基因型以及样本①②的高分辨熔解曲线图
图7为本发明的样本①②对应三个位点bsmi、apai、foki的基因型。
具体实施方式
为了使本发明的技术手段、创作特征、工作流程、使用方法达成目的与功效易于明白了解,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,在本发明的描述中,需要说明的是,除非另有明确的规定和限定,术语“安装”、“连”、“连接”应做广义理解,例如,可以是固定连接,也可以是可拆卸连接,或一体地连接以是机械连接,也可以是电连接;可以是直接相连,也可以通过中间媒介间接相连,可以两个元件内部的连通,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
临床样本dna的提取
一、edta抗凝血
本实施例是从edta抗凝血中提取基因组dna,并对其进行定量,作为pcr检测的模板。采用天根(tiangen)公司的血液基因组dna提取试剂盒(tianampdnabloodminikit),按照试剂盒说明进行操作,具体详述如下。
1.处理血液材料:
a.当血液样品体积小于200μl时,可加缓冲液gs补足体积至200μl,再进行下一步实验(如血液样品体积为200μl,可直接进行下一步实验,不需加入gs)。
b.当血液样品体积超过200μl时,需用细胞裂解液cl处理,具体步骤如下:在样品中加入1~2.5倍体积的细胞裂解液cl,颠倒混匀,10,000rpm(~11,500×g)离心1min,吸去上清,留下细胞核沉淀(如果裂解不彻底,可重复以上步骤一次),向离心收集到的细胞核沉淀中加200μl缓冲液gs,振荡至彻底混匀。
2.加入20μlproteinasek溶液,混匀。
3.加200μl缓冲液gb,充分颠倒混匀,56℃放置10min,其间颠倒混匀数次,溶液应变清亮,
4.加200μl无水乙醇,充分颠倒混匀,此时可能会出现絮状沉淀。
5.将上一步所得溶液和絮状沉淀都加入一个吸附柱cb3中(吸附柱cb3放入收集管中),12,000rpm(~13,400×g)离心30sec,倒掉收集管中的废液,将吸附柱cb3放入收集管中。
6.向吸附柱cb3中加入500μl缓冲液gd(使用前请先检查是否已加入无水乙醇),12,000rpm(~13,400×g)离心30sec,倒掉收集管中的废液,将吸附柱cb3放入收集管中。
7.向吸附柱cb3中加入600μl漂洗液pw(使用前请先检查是否已加入无水乙醇),12,000rpm(~13,400×g)离心30sec,倒掉收集管中的废液,将吸附柱cb3放入收集管中。
8.重复操作步骤7。
9.12,000rpm(~13,400×g)离心2min,倒掉废液。将吸附柱cb3置于室温放置数分钟,以彻底晾干吸附材料中残余的漂洗液。
10.将吸附柱cb3转入1.5ml离心管中,向吸附膜中间位置悬空滴加50-200μl洗脱缓冲液tb,室温放置2~5min,12,000rpm(~13,400×g)离心2min,将溶液收集到离心管中。
定量:将提取的dna用紫外分光光度计进行定量,稀释dna浓度到1ng/μl。
实时荧光pcr法扩增临床样本dna
本实施例为采用本发明所提供的引物、探针扩增实施例1所提取的dna样品。2份基因组dna,用te稀释液稀释到浓度1ng/μl,用本发明的试剂盒在abi7300荧光定量pcr仪上进行扩增。
荧光定量pcr反应体系为:
pcr反应液:
mgcl2:2.0~5.0mmol
dntps:0.2~0.8mmol
各条引物:0.1~1.0μmol
各条探针:0.1~1.0μmol
abifastmasterpremix:6~10μl
模板:10μl
总体积:40μl。
优选pcr反应管件为:
第一阶段:95℃5min;
第二阶段:95℃5s,58℃30s,10个循环;
第三阶段:95℃5s,58℃30s,72℃30s,35个循环;
第三阶段:35个循环时,收集fam和joe信号。
检测方法如下:
(1)每次pcr反应中,同时进行非模板对照(notemplatecontrol;ntc)、阳性对照和待测样本的检测。
(2)将阳性对照取出解冻后震荡混匀,并离心30s。
(3)将6联pcr反应管取出解冻后离心30s,防止反应液在开盖时溅出。
(4)将ddh2o(ntc)、待测样本、阳性对照分别加入6联pcr反应管,每孔10μl。
(5)将以上6联pcr反应管盖好管盖于离心机上离心30s,确保管壁上不沾有液滴。
(6)将6联pcr反应管放于实时pcr仪器中。
反应条件的设置
第一阶段:95℃4min;
第二阶段:95℃5s,58℃30s,10个循环;
第二阶段:95℃30s,58℃30s,72℃30s,35个循环;
信号收集:第三阶段72℃时收集fam和joe信号。
实施例2
口腔拭子样本
从口腔拭子中提取基因组dna,并对其进行定量,作为pcr检测的模板。采用康为世纪的口腔拭子基因组dna提取试剂盒,具体详述如下。
1.将口腔拭子的棉签用剪刀从杆上剪下,置于2ml的离心管(自备)中,加入400μlbuffergr。
注意:如需无rna污染的基因组dna,可加入4μl浓度为100mg/ml的rnasea溶液(货号:cw0601),震荡混匀。
2.加入20μlproteinasek和400μlbuffergl,立即涡旋震荡15秒,充分混匀。
注意:加入buffergl后立即充分混匀;不可将proteinasek直接加入buffergl中使用。
3.56℃放置10分钟,短暂离心,使管壁上的溶液收集到管底。
4.加入400μl无水乙醇,涡旋震荡充分混匀,短暂离心,使管壁上的溶液收集到管底。
5.将上步所得溶液和沉淀分两次加入到已装入收集管(collectiontube2ml)的吸附柱(spincolumnds)中,一次最多不超过700μl。12,000rpm(~13,400×g)离心1分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中。
6.向吸附柱中加入500μlbuffergw1(使用前检查是否已加入无水乙醇),12,000rpm离心1分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中。
7.向吸附柱中加入500μlbuffergw2(使用前检查是否已加入无水乙醇),12,000rpm离心3分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中。
注意:如需进一步提高dna纯度,可重复步骤7。
8.12,000rpm离心1分钟,倒掉收集管中的废液。将吸附柱置于室温数分钟,以彻底晾干。
9.将吸附柱置于一个新的1.5ml离心管中,向吸附柱的中间部位悬空加入50μlbufferge或灭菌水,室温放置2~5分钟,12,000rpm离心1分钟,收集dna溶液,-20℃保存。
10、定量
将提取的dna用紫外分光光度计进行定量,稀释dna浓度到1ng/μl。
本发明针对不同基因位点分别设计野生型和突变型arms引物和taqman-mgb探针,结合荧光定量pcr反应,对从人外周血细胞或口腔拭子中提取的基因组dna进行检测,可以一次性在一条6联pcr反应条上实现对基因多态性进行检测,通过实时荧光pcr仪上收集信号,计算野生型和突变型的△ct值来确定样本dna的基因型。
以上显示和描述了本发明的基本原理、主要特征及本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明的要求保护范围由所附的权利要求书及其等效物界定。
- 还没有人留言评论。精彩留言会获得点赞!
- ERCC1基因多态性检测体系及其试剂盒的制造方法与工艺
- 用于检测CYP2C19基因多态性的引物、探针及检测试剂盒的制造方法与工艺
- 高脂血症风险关联基因多态性检测方法与制造工艺
- 用于检测AGTR1基因的A1166C多态性位点的核酸、试剂盒及方法与制造工艺
- 一种检测与结直肠癌靶向用药西妥昔单抗治疗敏感性相关基因的多态性的试剂盒及其应用的制造方法与工艺
- 线粒体基因组9bp序列基因多态性在评价苯中毒风险中的应用的制造方法与工艺
- 一种rs12979860基因分型双色荧光PCR快速检测试剂盒的制造方法与工艺
- CYP2C19基因多态性检测引物、探针及试剂盒的制造方法与工艺
- 检测CYP2C9和VKORC1基因多态性的引物、探针和试剂盒的制造方法与工艺
- 一种检测CYP2C9基因多态性的ARMS引物的制造方法与工艺