一锅法制备厄贝沙坦的方法与流程
本发明属于医药技术领域,具体涉及一锅法制备厄贝沙坦的方法。
背景技术
厄贝沙坦(化学名称:2-丁基-3-{[2'-(1h-四唑-5-基)-[1,1'-联苯]-4-基]甲基}-1,3-二氮杂螺[4,4]壬-1-烯-4-酮)为血管紧张素ⅱ(angiotensinvⅱ,angⅱ)受体拮抗剂,能特异性地拮抗血管紧张素ⅱ受体(at1)。对at1的拮抗作用大于血管紧张素ⅱ2型受体(at2)8500倍,通过选择性地阻断angⅱ与at1受体的结合,抑制血管收缩和醛固酮的释放,产生降压作用。厄贝沙坦具有高效、长效、安全、可口服以及耐受性好等特点,并且有心、脑、肾保护作用,是心血管药物中的一个里程碑。
美国专利us5270317首次公开了厄贝沙坦及其相关化合物的制备方法。us5270317专利的制备方法中,咪唑盐酸盐与溴联苯在强碱性试剂氢化钠、溶剂n,n-二甲基甲酰胺(dmf)中发生烃化反应,联苯咪唑啉,再经烷基氯化锡和叠氮钠或者烷基叠氮化锡进行环合反应,形成四氮唑,制得厄贝沙坦。(参考专利文献:ep0454511、fr2659967、fr2665702、jp1992506222、jp1998279566、us5270317、wo9114679、cn1415614a。)此方法有如下缺点:
(1)氢化钠为危险试剂,遇水、湿空气放出氢气可爆炸,使用和存储危险;
(2)反应溶剂为dmf,dmf与水互溶,且沸点高,反应结束后,因dmf与水互溶,难以处理,容易污染环境,而且不能回收使用,导致成本较高;
(3)环合反应采用烷基氯化锡和叠氮钠或者烷基叠氮化锡,锡容易残留在产品中,造成重金属超标,而且价格贵,增加了产品的成本;
(4)需要色谱分离,产率低。
在us5629331记载的专利公开的合成方法中,其中使用三乙胺盐酸盐与叠氮化钠代替三丁基叠氮化锡进行四唑反应,所使用溶剂为1-甲基吡咯烷-2-酮,其价格昂贵且不易回收,因此使得该方法不适于工业生产。从反应混合物中分离厄贝沙坦的过程冗长,需要一些关键的层分离和层过滤,不能得到令人满意的收率和纯度。
专利cn101648945、w02004065383中,通过suzuki偶联的方法制备厄贝沙坦,反应过程中使用了价格昂贵的金属钯催化,因此该方法在工业生产上成本过高。相同地,专利us5412102、cn1070193专利中,使用了昂贵的催化剂氢化铝锂、氯化镍,应用于生产中成本过高。
特瓦公司于cn1668612专利中,公开了第一和第二相的反应体系中在相转移催化剂条件下2-丁基-1,3-二氮杂螺环[4.4]壬烷-1-烯-4酮与5-(4’-溴甲基联苯-2-基)-1-三苯甲基-1h四唑反应合成厄贝沙坦的方法。该方法克服了以往生产工艺上叠氮化合物带来的安全风险,但产物的纯度也不高。在该合成工艺中,由于解保护后得到三苯基甲醇是固体,需要和厄贝沙坦产物固体完全分离开还需要经过酸,碱,酸的处理步骤,为了使厄贝沙坦中三苯基甲醇降至足够低,还需要足够的重结晶次数,用以提纯产品,除去残留三苯基甲醇。同时三苯基甲醇转化为三苯基氯甲烷的难度大,不易回收。
目前,使用一锅法制备厄贝沙坦的方法主要有wo2010116380及其同族专利。
专利wo2010116380公开了使用一锅法制备厄贝沙坦的方法,在该方法中,以咪唑盐酸盐与溴联苯为原料制备联苯咪唑啉,该步骤是在水中进行,反应时间长达33-36小时,在反应完成后,分离的水层加入有机溶剂和亚硝酸钠溶液,析晶离心之后,滤饼还使用极性溶剂乙酸乙酯回流过滤,才得到厄贝沙坦粗品,厄贝沙坦粗品精制后得到厄贝沙坦精品,相当于厄贝沙坦经2步精制,总收率为69-71%。使用该方法制备厄贝沙坦,反应时间长,收率不高。
技术实现要素:
本发明公布了一种一锅法制备厄贝沙坦的方法,该方法以咪唑盐酸盐与溴联苯为原料,在碱性的极性有机溶剂中反应,在相转移催化剂的作用下,进行烃化反应,制得联苯咪唑啉;联苯咪唑啉与叠氮化钠在环合催化剂作用下,在极性有机溶剂中发生环合反应,制得厄贝沙坦。
本发明所述的厄贝沙坦的制备方法有如下有益效果:烃化反应和环合反应的反应时间短,操作简单,粗品收率高,经一步精制,成品纯度高的优点,适合工业化生产。
本发明是通过以下技术方案实现目的。
一锅法制备厄贝沙坦的方法,包括以下步骤:
(1)以2-丁基-1,3-二氮杂螺环[4,4]壬-1-烯-4-酮盐酸盐(以下简称咪唑盐酸盐)与2-氰基-4'-溴甲基联苯(以下简称溴联苯)为原料,在含有碱性溶液的非质子性溶剂中反应,在相转移催化剂的作用下,进行烃化反应,制得2-丁基-3-[(2'-氰基-1,1'-联苯-4-基)甲基]-1,3-二氮杂螺[4,4]壬-1-烯-4-酮(以下简称联苯咪唑啉);
(2)联苯咪唑啉与叠氮化钠在环合催化剂作用下,在非质子性溶剂中发生环合反应,制得厄贝沙坦,其反应式如下:
(3)反应完成后,加入碱性溶液,加水分层,分离水层和非质子性溶剂层;
(4)水层加入有机溶剂,亚硝酸钠溶液,用盐酸调节ph值为4-4.5,搅拌结晶,离心,得到厄贝沙坦粗品;
优选的,所述咪唑盐酸盐与溴联苯的摩尔比为0.95-1.05:1。
优选的,所述相转移催化剂选自季铵盐。
优选的,所述季铵盐为四丁基溴化铵,四丁基氢氧化铵,四丁基碘化铵或三丁基苄基氯化铵。
优选的,步骤(1)的反应温度为20-45℃,反应时间为3-4小时。
优选的,步骤(1)所述的非质子性溶剂选自甲苯、二甲苯、二氯甲烷、三氯甲烷中的一种,更优选的,所述非质子性溶剂为甲苯。所述碱性溶液选自氢氧化钠、氢氧化钾、碳酸钠、碳酸钾中的一种或多种混合物。
优选的,所述非质子性溶剂与溴联苯的重量比为2.5-3.5:1。
优选的,步骤(2)所述的环合催化剂为三乙胺盐酸盐。
优选的,步骤(2)的反应温度为105±5℃,反应时间为24-28小时。
优选的,步骤(3)的反应温度为10-40℃。
优选的,步骤(4)的有机溶剂选自甲醇、乙醇、异丙醇、乙酸乙酯中的任一种,所述有机溶剂的与溴联苯的重量比为0.6-1.2:1。
采用上述方案,本发明所述的厄贝沙坦的制备方法具有烃化反应和环合反应的反应时间短,操作简单,粗品收率高,经一步精制,成品纯度高的优点,适合工业化生产。
附图说明
为了更清楚的说明实施例或现有技术中的技术方案,下面对实施例或现有技术描述中所需使用的附图作简单介绍。
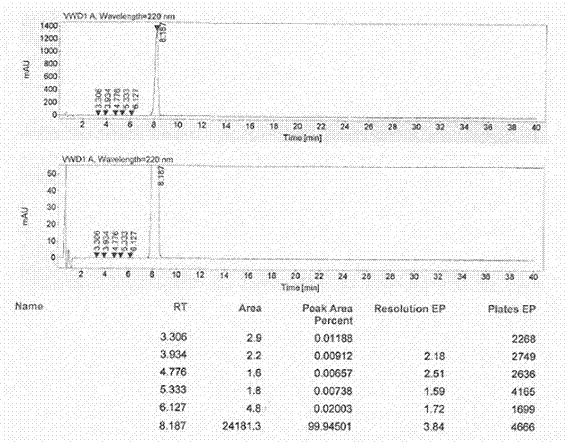
图1为本发明厄贝沙坦成品的液相色谱图。
具体实施方式
下面对本发明实施例中的技术方案进行清楚、完整的描述,显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1:
(1)联苯咪唑啉的制备:(烃化反应)
向圆底烧瓶中加入640ml甲苯,加入咪唑盐酸盐175g,控温15℃,缓慢加入10%氢氧化钠溶液125ml,加毕搅拌0.5h。加入四丁基溴化铵2.5g,溴联苯200g,控温15℃,缓慢加入10%氢氧化钠溶液125ml。控温15℃,保温反应4.5h。加入饮用水250ml,提取分层,甲苯层加入饮用水350ml,提取甲苯层,继续加入饮用水350ml,提取分层,甲苯层转入圆底烧瓶,加热回流,停止加热。降温至35℃,得联苯咪唑啉甲苯液。
(2)厄贝沙坦粗品制备:(环合反应)
联苯咪唑啉甲苯液在搅拌下,加入200g三乙胺盐酸盐,95g叠氮化钠,加热升温,控温在100℃反应,反应24h,降温至30℃,缓慢加入配制的10%氢氧化钠溶液,提取分层,甲苯层加入饮用水500ml,提取分层,下层水层转入分液漏斗中,加入150ml甲苯,提取分层,下层水层转入烧杯,烧杯中加入200ml乙醇、98g亚硝酸钠溶液,控温15℃,缓慢加盐酸调节溶液ph=4~4.5,加入饮用水250ml,继续搅拌结晶,将物料过滤,得厄贝沙坦粗品(湿)。将厄贝沙坦粗品(湿)干燥26h之后停止加热,降温至室温,收料得厄贝沙坦粗品295.6g,取样送检,纯度≥99%,摩尔收率93.9%(以溴联苯计算)。
(3)厄贝沙坦精制
烧杯中加入750ml乙醇,搅拌,加入厄贝沙坦粗品100g,10g活性炭,控温80℃,保温30min。溶液过滤,滤饼用100ml乙醇洗涤,洗液和滤液降温至10±5℃,保温析晶2h,过滤,得厄贝沙坦精品湿品,干燥18h,收料得厄贝沙坦干品95.9g,总收率90.0%,纯度99.94%。
厄贝沙坦成品核磁分析:
hnmr(400mhz,d-dmso)δ:0.82(t,j=7.2hz,3h,ch3),1.26-1.31(m,2h,ch2),1.46-1.52(m,2h,ch2),1.69-1.71(m,2h,ch2),1.85-1.87(m,6h,3ch2),2.32(t,j=7.2hz,2h,ch2),4.71(s,2h,nch2),7.12(s,4h,arh),7..54-7.61(m,2h,arh),7.68-7.70(m,2h,arh);
厄贝沙坦成品质谱结果:
ms(+c,esi):m=428,found(429,m+1),(451,m+na)
实施例2:
(1)联苯咪唑啉的制备:(烃化反应)
向圆底烧瓶中加入640ml二氯甲烷,加入咪唑盐酸盐175g,控温15℃,缓慢加入10%氢氧化钠溶液125ml,加毕搅拌0.5h。加入四丁基溴化铵3.0g,溴联苯200g,控温20℃,缓慢加入10%氢氧化钠溶液130ml。控温20℃,保温反应5h。加入饮用水250ml,提取分层,甲苯层加入饮用水350ml,提取甲苯层,继续加入饮用水350ml,提取分层,甲苯层转入圆底烧瓶,加热回流,停止加热。降温至40℃,得联苯咪唑啉二氯甲烷液。
(2)厄贝沙坦粗品制备:(环合反应)
联苯咪唑啉甲苯液在搅拌下,加入200g三乙胺盐酸盐,95g叠氮化钠,加热升温,控温在105℃反应,反应24h,降温至30℃,缓慢加入配制的10%氢氧化钠溶液,提取分层,甲苯层加入饮用水500ml,提取分层,下层水层转入分液漏斗中,加入150ml甲苯,提取分层,下层水层转入烧杯,烧杯中加入250ml乙醇、98g亚硝酸钠溶液,控温15℃,缓慢加盐酸调节溶液ph=4~4.5,加入饮用水250ml,继续搅拌结晶,将物料过滤,得厄贝沙坦粗品(湿)。将厄贝沙坦粗品(湿)干燥26h之后停止加热,降温至室温,收料得厄贝沙坦粗品274.5g,取样送检,纯度≥99%,摩尔收率87.2%(以溴联苯计算)。
(3)厄贝沙坦精制
烧杯中加入750ml乙醇,搅拌,加入厄贝沙坦粗品100g,10g活性炭,控温80℃,保温30min。溶液过滤,滤饼用100ml乙醇洗涤,洗液和滤液降温至10±5℃,保温析晶2h,过滤,得厄贝沙坦精品湿品,干燥18h,收料得厄贝沙坦干品95.5g,总收率83.3%,纯度99.94%。
厄贝沙坦成品核磁分析:
hnmr(400mhz,d-dmso)δ:0.82(t,j=7.2hz,3h,ch3),1.26-1.31(m,2h,ch2),1.46-1.52(m,2h,ch2),1.69-1.71(m,2h,ch2),1.85-1.87(m,6h,3ch2),2.32(t,j=7.2hz,2h,ch2),4.71(s,2h,nch2),7.12(s,4h,arh),7..54-7.61(m,2h,arh),7.68-7.70(m,2h,arh);
厄贝沙坦成品质谱结果:
ms(+c,esi):m=428,found(429,m+1),(451,m+na)
实施例3:
(1)联苯咪唑啉的制备:(烃化反应)
向反应罐加入1280l甲苯,加入咪唑盐酸盐350kg,控温15-20℃,缓慢加入10%氢氧化钠溶液250l,加毕搅拌0.5h。加入四丁基溴化铵5kg,溴联苯400kg,控温15-20℃,缓慢加入剩余的10%氢氧化钠溶液250l。控温15-20℃,保温反应4.5h,直至反应终点(溴联苯≤0.5%)。反应毕,加入饮用水500l,提取分层,下层碱水层入废液管。甲苯层加入饮用水750l,提取分层,下层水层入废液管。继续加入饮用水750l,提取分层,水层入废液管。甲苯层转入环合罐,加热至回流,共沸带水至分水器中未见水分出为止,停止加热。降温至40±5℃,得联苯咪唑啉甲苯液,纯度≥97%。
(2)厄贝沙坦粗品制备:(环合反应)
环合反应罐开启搅拌,加入400kg三乙胺盐酸盐,190kg叠氮化钠,加热升温,控温在100±5℃反应,反应24h后,每2h取样检测直至反应终点。反应毕,稍降温,将物料转至提取罐。开启搅拌,降温至30℃,控温30±5℃缓慢加入配制的10%氢氧化钠溶液,提取分层,下层碱水层排入废水池。甲苯层加入饮用水1000l,提取分层,下层水层转入洗涤罐,上层甲苯液转入储罐回收处理。洗涤罐中加入300l甲苯,提取分层,下层水层转入析晶罐,上层甲苯液转入储罐回收处理。析晶罐中加入400l乙醇、195kg亚硝酸钠溶液,控温10-20℃,缓慢加盐酸调节溶液ph=4~4.5,加入饮用水500l,继续搅拌结晶2h将结晶物料分次放入离心机,离心、洗涤,得厄贝沙坦粗品(湿)。将厄贝沙坦粗品(湿)转入双锥真空干燥机,控制真空度≥0.08mpa,控温热水温度90±5℃,干燥26h之后每4h取样检测,至干燥失重≤2%。停止加热,降温至室温,收料得厄贝沙坦粗品,取样送检,纯度≥99%,摩尔收率93%(以溴联苯计算)。
(3)厄贝沙坦精制
向精制溶解罐中加入计算量的乙醇,开启搅拌,加入厄贝沙坦粗品和回收品,10kg活性炭,控温80±5℃,保温30min。溶解液经粗过滤器、精过滤器过滤,滤饼用100l乙醇洗涤,洗液和滤液至洁净区10000l结晶罐。降温至10±5℃,保温析晶2h。将物料分次放至离心机中,离心,每次离心所得滤饼用乙醇淋洗一次,离心,收料,得厄贝沙坦湿品。将厄贝沙坦湿品投入双锥干燥机中,控制真空度≥0.08mpa,热水控温70±5℃,干燥18h。每2h取样检测,直至干燥失重≤0.5%。降温,收料得厄贝沙坦干品,总收率90%。
厄贝沙坦成品核磁分析:
hnmr(400mhz,d-dmso)δ:0.82(t,j=7.2hz,3h,ch3),1.26-1.31(m,2h,ch2),1.46-1.52(m,2h,ch2),1.69-1.71(m,2h,ch2),1.85-1.87(m,6h,3ch2),2.32(t,j=7.2hz,2h,ch2),4.71(s,2h,nch2),7.12(s,4h,arh),7..54-7.61(m,2h,arh),7.68-7.70(m,2h,arh);
厄贝沙坦成品质谱结果:
ms(+c,esi):m=428,found(429,m+1),(451,m+na)
综上所述,本发明烃化反应和环合反应的反应时间短,操作简单,粗品收率高,成品纯度高的优点,适合工业化生产。
需要说明的是,以上所述,仅是本发明的较佳实施例而已,对于本领域技术人员而言,显然本发明不限于上述示范性实施例的细节,在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。
- 还没有人留言评论。精彩留言会获得点赞!