一种基于Sec63的不同重组蛋白表达水平的毕赤酵母菌株的分选方法与流程
本发明涉及基因工程技术领域,特别涉及一种基于sec63的不同重组蛋白表达水平的毕赤酵母菌株的分选方法。
背景技术:
巴斯德毕赤酵母(pichiapastoris)是一种甲醇营养型酵母,其既具有原核生物易培养、快繁殖、好操作、高密度发酵的优点,又具有真核生物对产物正确折叠的加工系统,且能够将产物分泌到发酵液中,易于后续分离纯化。基于上述优点,毕赤酵母已成为目前应用最为广泛的重组蛋白表达系统之一,已有超过5000种重组蛋白在毕赤酵母中成功表达。
在工业生产中,通常希望可以尽可能地提高重组蛋白的表达水平以获得最大的经济效益。为实现这一目的,需要对重组菌株进行改造以提高重组蛋白的表达水平,其中最常用的方法是增加重组蛋白基因的基因剂量即增加重组蛋白基因表达盒的拷贝数。但经过改造后的菌株仍需要检测重组蛋白的表达水平,分选出不同重组蛋白表达水平的菌株以进行下一步的改造。对于不同重组蛋白,检测其表达量的方法有所不同,如对于重组酶一般是检测其酶活,这些过程一般需要一定的反应时间且比较繁琐。另外,当利用体内构建法构建多拷贝菌株时,由于抗性与基因拷贝数的对应并不严格,很多高抗性菌株鉴定后发现并不是高拷贝菌株,这无疑更加大了分选出高重组蛋白表达水平菌株的工作量与难度。综上,目前没有较好的快速分选出不同重组蛋白表达水平的菌株的方法。所以,寻找快速、简单分选出不同重组蛋白表达水平菌株的方法显得十分重要。
技术实现要素:
为了克服现有技术的缺点与不足,本发明的目的在于提供一种基于sec63的不同重组蛋白表达水平的毕赤酵母菌株的分选方法。该方法具有简便快速的特点,适用于菌株育种过程中不同蛋白表达水平的菌株的分选。转膜蛋白sec63是内质网的标记蛋白,经常被用于观察内质网的形态。
本发明的目的通过下述技术方案实现:
一种基于sec63的不同重组蛋白表达水平的毕赤酵母菌株的分选方法,包括如下步骤:
将毕赤酵母内质网膜蛋白融合表达荧光蛋白,通过分选出荧光值高低不同的菌株而分选出重组蛋白表达水平不同的菌株;
具体包括如下步骤:
(1)构建内质网膜蛋白-荧光蛋白的基因片段,转化进入毕赤酵母的感受态细胞中;
(2)制备内质网膜蛋白融合表达荧光蛋白的毕赤酵母感受态细胞;
(3)将表达重组蛋白的载体转化进入步骤(2)中制备的感受态细胞中,涂布于相对应的筛选平板上,获得阳性转化子;
(4)将阳性转化子接种于发酵培养基中进行发酵培养,利用流式细胞仪或酶标仪检测细胞的荧光值高低并分选出荧光值高低不同的细胞群落或单克隆;
(5)高荧光值的细胞群落或单克隆即为高表达重组蛋白菌株,低荧光值细胞群落或单克隆即为低表达重组蛋白菌株。
所述的内质网膜蛋白为sec63;
所述的荧光蛋白为增强型绿色荧光蛋白egfp;
所述的毕赤酵母为毕赤酵母(pichiapastoris)gs115。
优选的,本发明的实施例以内质网膜蛋白sec63、增强型绿色荧光蛋白egfp、毕赤酵母gs115为例:
采用在毕赤酵母内源sec63蛋白融合表达增强型绿色荧光蛋白egfp,通过分选egfp荧光值高低不同的细胞群落或单克隆,从而分选出不同重组蛋白表达水平的菌株。具体包括如下步骤:
1)使用重叠延伸pcr制备sec63-egfp片段,并将该片段转化毕赤酵母gs115得到sec63融合表达egfp的重组菌株gs115-e;
2)制备gs115-e的感受态细胞,分别将携带一、六拷贝来源于柠檬酸杆菌(citrobacteramalonaticus)cgmcc1696的植酸酶基因的载体线性化后转化进入其中,分别获得表达一拷贝与六拷贝植酸酶基因的重组菌株。
3)将步骤2)中得到的两种表达植酸酶的重组菌株混合培养,在诱导发酵96h之后,利用分选型流式细胞仪分选出egfp荧光值高低不同的细胞群落。
4)将步骤3)中分选得到的细胞群落分别涂布于ypd平板,然后分别挑选生长出的20个单菌落进行诱导发酵并检测植酸酶酶活与egfp表达情况,并在其中各随机挑选2株检测植酸酶基因拷贝数。
5)将携带来源于耐盐芽孢杆菌(bacillushalodurans)c125的木聚糖酶基因的载体线性化后转化进入gs115-e的感受态细胞中,检测诱导期间菌体egfp荧光值与木聚糖酶酶活,根据菌体egfp荧光值分选出木聚糖酶表达情况不同的菌株。
本发明相对于现有技术具有如下的优点及效果:
本发明使用egfp荧光值可以快速地分选出不同重组蛋白表达水平的毕赤酵母菌株,无需繁琐的操作,有利于不同重组蛋白表达水平的毕赤酵母重组菌株的快速分选。
附图说明
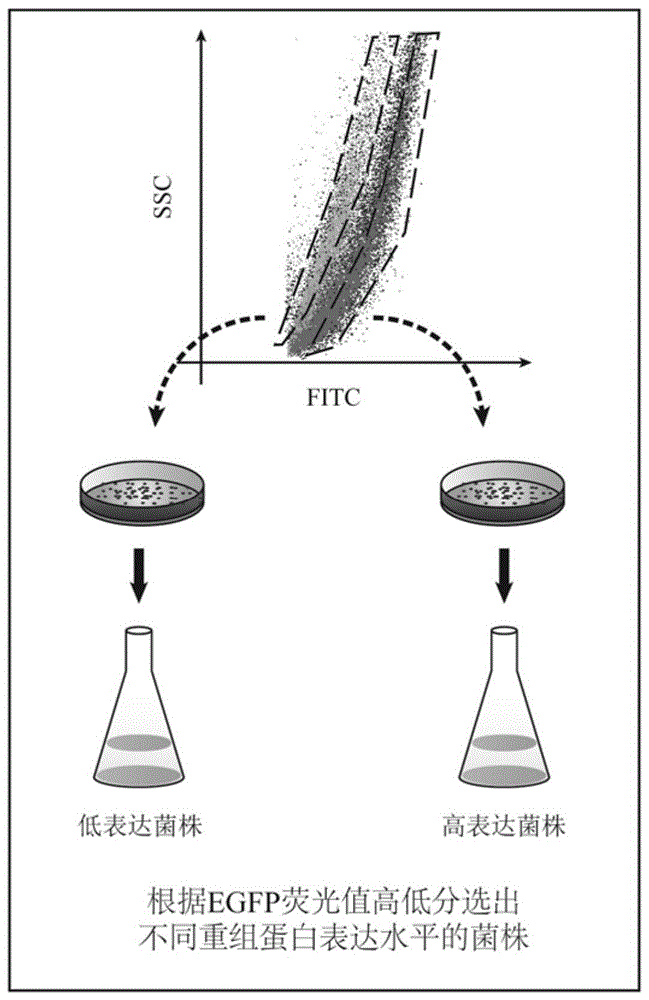
图1是本发明根据egfp荧光值高低分选出不同重组蛋白表达水平的菌株的示意图。
图2是sec63融合表达egfp菌株gs115-e的细胞生长检测;其中,(a)菌株gs115-e诱导96h时的荧光显微镜观察结果,其中实线箭头代表内质网的核膜层,虚线箭头代表内质网的皮质层;(b)菌株gs115与gs115-e的生长曲线。
图3是不同菌株的流式细胞仪分析图。(a)菌株gs115-e/6c-phy和gs115-e/1c-phy的混合发酵菌株的流式细胞仪分析图;(b)菌株gs115-e/6c-phy和gs115-e/1c-phy的混合发酵菌株与菌株gs115-e/1c-phy相叠加的流式细胞仪分析图;(c)菌株gs115-e/6c-phy和gs115-e/1c-phy的混合发酵菌株与菌株gs115-e/1c-phy、gs115-e/6c-phy相叠加的流式细胞仪分析图。
图4是20株高efgp荧光值菌株与20株低efgp荧光值菌株的植酸酶酶活均值曲线。
图5是甲醇诱导120h时20株高efgp荧光值菌株与20株低efgp荧光值菌株的植酸酶酶活
图6是各重组菌株phy拷贝数鉴定。(a)从20株低efgp荧光值的菌株随机挑选的第一个菌株的phy基因拷贝数;(b)从20株高efgp荧光值的菌株随机挑选的第一个菌株的phy基因拷贝数;(c)从20株低efgp荧光值的菌株随机挑选的第二个菌株的phy基因拷贝数;(d)从20株高efgp荧光值的菌株随机挑选的第二个菌株的phy基因拷贝数。
图7是表达木聚糖酶的单克隆的木聚糖酶酶活与菌体荧光值曲线。(a)各菌株的菌体egfp荧光值曲线;(b)各菌株的木聚糖酶酶活曲线。
具体实施方式
下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
下列实施例中未注明具体实验条件的试验方法,通常按照常规实验条件或按照制造厂所建议的实验条件。所使用的材料、试剂等,如无特殊说明,为从商业途径得到的试剂和材料。
本发明根据egfp荧光值高低分选出不同重组蛋白表达水平的菌株的示意图,如图1所示。
实施例中涉及如下生物材料:
质粒ppicza-egfp在文献doi:10.1007/s10529-012-1055-8”中公开。
质粒ppicza-creg357c在文献“doi:10.1038/s41598-017-11494-5”中公开。
质粒ppiczαa-phy、ppichka、ppiczαa-phy-g均在文献“doi:10.1186/s12896-015-0204-2”中公开。
质粒phka-xyn10在文献“林小琼.基于itraq技术的高效表达木聚糖酶重组毕赤酵母细胞的蛋白组学研究[d].华南理工大学,2013”中公开。
实施例1sec63融合表达增强型绿色荧光蛋白宿主菌株的构建
(1)以毕赤酵母pichiapastorisgs115基因组作为模板,使用引物p1和p2与p3和p4扩增sec63基因(genbankaccessionxp_002493824.1)的上下游部分。以质粒ppicza-egfp(在参考文献“doi:10.1007/s10529-012-1055-8”中公开)作为模板,使用引物p5与p6扩增增强型绿色荧光蛋白egfp的基因片段。以质粒ppicza-creg357c(在参考文献“doi:10.1038/s41598-017-11494-5”中公开)作为模板,使用引物p7与p8扩增cre表达盒片段。以上述胶回收纯化后的四种pcr产物为模板,使用引物p1与p4扩增片段sec63-egfp。
p1:5′-gctaataaatgggctggtcgat-3′;
p2:5′-tttatcttcaccctcatcttcc-3′;
p3:5′-tacattatacgaacggtagtgatagtgacagatcaaa-3′;
p4:5′-ggagggaagattcactccaaaa-3′;
p5:5′-gaagatgagggtgaagataaagtgagcaagggcgaggagct-3′
p6:5′-agatctataacttcgtataatgtatgctatacgaacggtattacttgtacagctcgtcca-3′;
p7:5′-taccgttcgtatagcatacattatacgaagttatagatctaacatccaaagacg-3′;
p8:5′-taccgttcgtataatgtatgctatacgaagttatgtctccagcttgcaaattaa-3′;
(2)将上述sec63-egfp片段胶回收纯化后,电转化pichiapastorisgs115(购自美国invitrogen英杰生命技术有限公司)感受态细胞,并在ypdz博来霉素抗性平板上进行阳性转化子筛选。
(3)挑选鉴定正确的转化子接种到ypm培养基,30℃,250rpm培养24h。将10μl培养基划线于ypd平板上,30℃,250rpm培养36h。将获得的转化子命名为gs115-e。
(4)将重组菌株gs115-e与野生型毕赤酵母gs115分别同时接种到bmgy培养基,30℃,250rpm培养20h。6000rpm,4℃离心5min收集细胞,然后将收集的细胞重悬到bmmy培养基至起始od600接近0.5,30℃,250rpm震荡培养,每天补加终浓度1%甲醇并取样。测定菌体od600并使用激光共聚焦显微镜观察细胞。
使用激光共聚焦显微镜观察甲醇培养96h时菌株gs115-e的细胞,发现这些细胞的内质网核膜层具有连续的荧光外观及皮质层具有断续的荧光外观(图2-a),说明sec63已成功融合表达egfp。甲醇培养期间,菌株gs115-e与gs115的生长曲线类似(图2-b),说明sec63融合表达egfp后并不影响细胞生长。
实施例2表达一拷贝植酸酶基因的内质网内源sec63蛋白融合表达增强型绿色荧光蛋白egfp的重组菌株gs115-e/1c-phy的构建
一.制备sec63融合表达增强型绿色荧光蛋白宿主菌株的gs115-e感受态细胞。
a.接种毕赤酵母菌株于100mlypd中,30℃,200rpm培养至od600=1.5±0.3;
b.分装到2个50ml离心管,室温、1500g离心5min;
c.弃上清,加入40ml新配的ldst溶液【(100mmliac,10mmdithiothreitol,0.6msorbitoland10mmtris-hcl,ph7.5)】(25℃),然后30℃孵育30min;
d.室温、1500g离心5min。弃上清,用1ml冰浴1m山梨醇重悬菌体,转至1.5mlep管中;
e.用1ml冰浴1m山梨醇洗涤菌体3次;
f.最后用400μl冰浴1m山梨醇重悬菌体,分装80μl/管。立即使用或冻存于-80℃。
二.携带一拷贝植酸酶基因的质粒ppichka-(phy)1的构建
(1)过夜培养大肠杆菌菌株ppiczαa-phy(参考文献doi:10.1186/s12896-015-0204-2)和ppichka(参考文献doi:10.1186/s12896-015-0204-2),收集细胞,分离、提取质粒ppiczαa-phy和ppichka。
(2)以质粒ppiczαa-phy为模板,使用引物p9,p10扩增来源于柠檬酸杆菌(citrobacteramalonaticus)cgmcc1696的植酸酶基因phy(genbank:abi98040.1)。引物p1和p2由5’到3’端下划线部分分别为kpni和ecori的识别位点。
p9:5′-gctggtaccttacagggaacaggcagggatt-3′;
p10:5′-gctgaattcgaattccagtccgaacctgcgt-3′。
(3)将胶回收纯化后的pcr产物用限制性内切酶kpni和ecori进行双酶切与同样经过kpni和ecori双酶切的质粒ppichka用t4连接酶16℃连接过夜,连接产物化学转化e.colitop10(购自美国invitrogen英杰生命技术有限公司)。经卡那霉素抗性平板筛选阳性转化子,阳性转化子提取质粒经kpni和ecori双酶切鉴定正确后,得到重组质粒ppichka-(phy)1。含有正确重组质粒ppichka-(phy)1的克隆菌株e.colitop10/ppichka-(phy)1加15%甘油于-80℃保存。
三.将重组质粒ppichka-(phy)1使用kpn2i线性化后电转化pichiapastorisgs115-e感受态细胞,并在组氨酸营养缺陷型平板上进行阳性转化子筛选,得到的阳性转化子命名为gs115-e/1c-phy。
实施例3表达六拷贝植酸酶基因的内质网内源sec63蛋白融合表达增强型绿色荧光蛋白egfp的重组菌株gs115-e/6c-phy的构建
一.利用同尾酶bglii和bamhi首尾相连的特性构建含六拷贝植酸酶基因的重组质粒ppichka-(phy)6。
(1)使用bglii和bamhi对质粒ppichka-(phy)1进行双酶切,使用bamhi对质粒ppichka-(phy)1进行单酶切同时使用fastap进行去磷酸化处理,切胶回收两种酶切片段。
(2)将胶回收纯化后的产物用t4连接酶16℃连接过夜,连接产物化学转化e.colitop10(购自美国invitrogen英杰生命技术有限公司)。经卡那霉素抗性平板筛选阳性转化子,阳性转化子提取质粒经bglii和bamhi双酶切鉴定正确后,得到含二拷贝植酸酶基因的重组质粒ppichka-(phy)2。
(3)参考(1)、(2)中的步骤,在重组质粒ppichka-(phy)2中重复插入植酸酶基因表达盒,最终构建得到的含六拷贝植酸酶基因的重组质粒ppichka-(phy)6。
二.将重组质粒ppichka-(phy)6使用kpn2i线性化后电转化pichiapastorisgs115-e感受态细胞,并在组氨酸营养缺陷型平板上进行阳性转化子筛选,得到的阳性转化子命名为gs115-e/6c-phy。
实施例4利用流式细胞仪根据egfp荧光值高低分选植酸酶表达水平不同的重组菌株
(1)将gs115-e/1c-phy和gs115-e/6c-phy接种到同一瓶bmgy培养基,30℃,250rpm培养20h。6000rpm,4℃离心5min收集细胞,然后将收集的细胞重悬到bmmy培养基至起始od600接近0.5,30℃,250rpm震荡培养,每天补加终浓度1%甲醇,培养至96h后取样。样品经6000rpm,4℃离心5min分离菌体和发酵上清液。向收集得到的菌体中加入200μl的重悬缓冲液洗涤三次,之后加入1ml重悬缓冲液重悬菌体,利用分选型流式细胞仪分选出高egfp荧光值酵母细胞群落与低egfp荧光值酵母细胞群落。
(2)将分选到的2种酵母细胞群落分别涂布于ypd平板,30℃培养48h,在高荧光值菌群与低荧光值菌群涂布的平板上各挑选20个单菌落同时接种到bmgy培养基,30℃,250rpm培养20h。6000rpm,4℃离心5min收集细胞,然后将收集的细胞重悬到bmmy培养基至起始od600接近0.5,30℃,250rpm震荡培养,每天补加终浓度1%甲醇并取样。样品经6000rpm,4℃离心5min分离菌体和发酵上清液。检测发酵上清液植酸酶酶活。
植酸酶酶活的测定方法:
发酵液上清液加入适量的醋酸钠缓冲液(0.25mol/l,ph5.50),稀释合适的倍数至终体积为1ml,37℃预热5min。加入2ml植酸钠溶液(0.00761mol/l,ph5.50),混合,37℃反应30min。加入2ml现配的显色/终止液(2.35g/l钒酸铵溶液:100g/l钼酸铵溶液:30%硝酸=1:1:2),室温静止10min后,415nm处测定吸光度。
植酸酶酶活力定义:37℃,ph值为5.5条件下,每分钟水解植酸钠溶液释放1μmol无机磷的酶量为l个酶活单位(u)。
植酸酶活力计算公式:酶活(u/ml)=n×vt×a/(ε×30×ve/103);n:酶液稀释倍数;a:吸光度的变化值;vt:酶反应液的总体积;ve:酶反应液中酶液体积;ε:摩尔吸光系数。
(3)分别从步骤(2)中诱导发酵的20个单菌落中各随机挑选2个单菌落,检测这些菌株中植酸酶基因的拷贝数。
a.菌株基因组的提取:
(1)挑取毕赤酵母菌株单菌落于10mlypd培养基中,30℃、250rpm培养20h;
(2)取含有1~2×108(约2~4od600)个细胞的菌液经8,000×g离心5min收集菌体后,弃上清;
(3)按照yeastdnaisokit说明书提取毕赤酵母基因组dna;
(4)提取完毕后的基因组dna经测定浓度后,置于-20℃保存备用。
b.菌株植酸酶基因拷贝数的测定:
(1)以浓度为10-7、10-6、10-5、10-4、10-3、10-2和10-1的植酸酶基因拷贝数测定标准质粒ppiczαa-phy-g(参考文献doi:10.1186/s12896-015-0204-2)为模板,使用
(2)将所提取的相关菌株的基因组dna利用
在用流式细胞仪分析gs115-e/1c-phy和gs115-e/6c-phy混合培养菌株时可以明显发现混合培养菌株的细胞群落分为两群(图3-a),且低egfp荧光值的细胞群落为gs115-e/1c-phy(图3-b),高egfp荧光值的细胞群落为gs115-e/6c-phy(图3-c)。
对高、低荧光值菌群涂布平板上的单菌落的发酵实验表明高egfp荧光值菌株的植酸酶表达情况显著优于低egfp荧光值菌株的植酸酶表达情况(图4、5)。甲醇诱导120h后,高egfp荧光值菌株的植酸酶酶活平均值达到1030u/ml,相对于低egfp荧光值菌株的植酸酶酶活平均值251u/ml提高了约310%(图4)。而对各菌株phy拷贝数的鉴定试验也验证了高egfp荧光值菌株具有六拷贝的植酸酶基因,而低egfp荧光值菌株具有一拷贝的植酸酶基因(图6)。综合上述结果,说明根据egfp荧光值的高低可以快速,精准地分选出植酸酶表达情况高低不同的菌株。
实施例5根据egfp荧光值高低分选出木聚糖酶表达水平不同的重组菌株(1)将携带来源于耐盐芽孢杆菌(bacillushalodurans)c125的木聚糖酶基因xyn(ncbigeneid:891394)的质粒phka-xyn10(参考文献:林小琼.基于itraq技术的高效表达木聚糖酶重组毕赤酵母细胞的蛋白组学研究[d].华南理工大学,2013.)使用kpn2i线性化后电转化pichiapastorisgs115-e感受态细胞,并在组氨酸营养缺陷型平板上进行阳性单克隆筛选。
(2)随机挑选5个阳性单克隆接种到bmgy培养基,30℃,250rpm培养20h。6000rpm,4℃离心5min收集细胞,然后将收集的细胞重悬到bmmy培养基至起始od600接近0.5,30℃,250rpm震荡培养,每天补加终浓度1%甲醇并取样。样品经6000rpm,4℃离心5min分离菌体和发酵上清液。检测菌体egfp荧光与发酵上清液木聚糖酶酶活。
a.菌体egfp荧光的测定:
往收集得到的菌体中加入200μl的重悬缓冲液洗涤三次后,使用200μl的重悬缓冲液重悬并于酶标仪上检测其荧光强度(激发波长为488nm,发射波长为520nm)。
b.木聚糖酶活的测定方法:
发酵液上清液加入适量的甘氨酸缓冲液(0.5mol/l,ph9.0),稀释合适的倍数至终体积为1ml,将反应液移至于25ml比色管中,加入1ml的10mg/ml的木聚糖溶液,混合,70℃反应30min。马上加入3ml的3,5-二硝基水杨酸溶液,轻轻混匀后,沸水浴10min,然后添加蒸馏水定容至25ml,取反应液于波长540nm下测定其吸收值。
木聚糖酶活力定义:70℃,ph值为9.0条件下,每分钟水解10mg/ml的木聚糖溶液释放1μmol还原糖的酶量为l个酶活单位(u)。
木聚糖酶活力计算公式:酶活(u/ml)=n×vt×a/(ε×30×ve/103);n:酶液稀释倍数;a:吸光度的变化值;vt:酶反应液的总体积;ve:酶反应液中酶液体积;ε:摩尔吸光系数。
甲醇诱导发酵期间,不同单克隆菌株的egfp荧光值与木聚糖酶的表达情况各不相同(图7)。甲醇诱导120h时,单克隆-5的egfp荧光值显著高于单克隆-4的egfp荧光值(图7-a)。而与egfp荧光值高低情况相符,单克隆-5的上清木聚糖酶酶活为44u/ml相比于单克隆-4的上清木聚糖酶酶活16u/ml提高了180%(图7-b)。说明可以根据sec63-egfp的荧光量分选出木聚糖酶的分泌表达量高低不同的菌株。木聚糖酶的分泌表达量测定需要约60min,而检测菌体egfp荧光量只需要约6min的时间,说明根据egfp荧光值的高低可以快速,精准地分选出木聚糖酶表达情况高低不同的菌株。
通常来说,一批发酵实验需要持续约7~10天的时间,并且需要每天花费大量的时间检测重组蛋白的表达水平(如检测植酸酶与木聚糖酶的酶活都需要约60min)以期分选出表达水平不同的菌株,而根据egfp荧光值大小分选重组蛋白表达情况不同的菌株只需要约6min的时间,大大节省了时间与精力。结合实施例4、5中的结果,说明根据sec63-egfp的荧光量大小可以用来快速、简单、准确地分选出重组蛋白表达水平不同的菌株。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
序列表
<110>华南理工大学
<120>一种基于sec63的不同重组蛋白表达水平的毕赤酵母菌株的分选方法
<160>10
<170>siposequencelisting1.0
<210>1
<211>22
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p1
<400>1
gctaataaatgggctggtcgat22
<210>2
<211>22
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p2
<400>2
tttatcttcaccctcatcttcc22
<210>3
<211>37
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p3
<400>3
tacattatacgaacggtagtgatagtgacagatcaaa37
<210>4
<211>22
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p4
<400>4
ggagggaagattcactccaaaa22
<210>5
<211>41
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p5
<400>5
gaagatgagggtgaagataaagtgagcaagggcgaggagct41
<210>6
<211>60
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p6
<400>6
agatctataacttcgtataatgtatgctatacgaacggtattacttgtacagctcgtcca60
<210>7
<211>54
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p7
<400>7
taccgttcgtatagcatacattatacgaagttatagatctaacatccaaagacg54
<210>8
<211>54
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p8
<400>8
taccgttcgtataatgtatgctatacgaagttatgtctccagcttgcaaattaa54
<210>9
<211>31
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p9
<400>9
gctggtaccttacagggaacaggcagggatt31
<210>10
<211>31
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>p10
<400>10
gctgaattcgaattccagtccgaacctgcgt31
- 还没有人留言评论。精彩留言会获得点赞!