一种丁苯酞衍生物及其制备方法和应用与流程
本发明涉一种丁苯酞衍生物及其制备方法和应用,属于药物化学和药物治疗学领域。
背景技术:
脑缺血是由缺血导致的脑部供血受阻而迅速发展的脑功能损伤。伴随着人口快速老龄化,脑缺血的发病率呈持续增长趋势,已成为当今全球第一致残和第二致死的重大疾病,对社会及家庭的负担亦进一步加剧。脑缺血的发病机制非常复杂,是一个多因素、多机制、多环节的恶性级联过程。多年来,围绕着脑缺血及再灌注损伤的相关机制研究包括兴奋性毒性、离子失衡、氧化应激、皮层扩散性去极化、炎症反应等。目前临床常用的抗脑缺血药物主要有抗血小板聚集药、溶栓药、神经保护剂、自由基清除剂等,这些药物虽然可以通过不同机制发挥一定的作用,但治疗靶点单一、毒副作用较大,单独使用难以达到满意的疗效。国内外专家学者对脑缺血的发病机制和防治进行了广泛的研究并取得了长足进展,但至今仍然缺乏切实有效的临床防治措施。因此,如何采取新的策略,研究和开发具有多重作用的新型高效防治脑缺血药物是一项当前全世界医药学家普遍关注的重要课题。
丁苯酞,又称3-丁基-1(3h)-异苯并呋喃酮(3-n-butylphthalide,nbp)是从芹菜籽中分离提纯的一种化合物。经人工合成的nbp消旋体(商品名“恩必普”)于2002年上市,是我国脑血管病领域第一个拥有自主知识产权的国家一类新药,并获9项国内和国际专利。研究表明,nbp具有抗血小板聚集、抗血栓、抑制炎症反应、减轻脑水肿、减缓脑梗死及明显地改善脑微循环等多种生物活性。尽管nbp能作用于脑缺血的多个病理环节,但其总体疗效不高,常与其它药物联合使用;此外,nbp难溶于水,限制了其治疗急性脑缺血的广泛应用。为了获得理想的抗脑缺血药物,研究人员对nbp进行了多方面的结构修饰和改造工作。研究发现,通过对nbp两个对映体进行深入的研究表明,(s)-nbp在抗血栓、改善脑微循环、改善线粒体功能以及神经细胞保护作用等方面较(r)-nbp突出,不仅能用于治疗急性脑缺血,而且可用于脑缺血的二级预防。因此,以单一构型的nbp为母核进行结构改造,获得洁性更好的化合物必将成为当前这一领域药物研发的热点。研究发现nbp开环衍生物2-(α-羟正戊基)苯甲酸(hpba)钾盐,在体内、外均能经酶或化学作用转变成nbp而发挥药效(转化率达90%以上),具有与nbp类似的生物活性和药代动力学特征,且口服后的生物利用度较nbp提高近一倍。因此,hpba是一个具有良好开发前景的抗脑缺血先导物。
硫化氢(hydrogensulfide,h2s)作为一种最新发现的内源性气体信号分子,具有广泛生物学效应。近年来研究表明h2s对于脑缺血-再灌注损伤有积极的作用,其作用机制可能为:h2s能使血管舒张,可通过改善脑血液循环来保护神经细胞;此外,h2s是一种内源性过氧化亚硝酸盐清道夫,可以阻断活性氧自由基(ros)生成的关键途径,从源头上抑制ros的生成,或与ros发生反应,直接清除自由基;h2s具有抗氧化应激的作用,能升高细胞内谷胱甘肽水平,以一种剂量依赖型的方式保护神经细胞免受谷氨酸的氧化毒性作用。因此,h2s在脑缺血损伤中具有重要的作用。
技术实现要素:
本发明公开了一种硫化氢供体型丁苯酞开环衍生物、其制备方法及应用。药理实验证明,该类化合物具有良好的抗血小板聚集和抗血栓活性。因此,它们可用于预防和治疗与血小板聚集相关的疾病,这类疾病包括心肌梗死、心绞痛、心律失常、冠心病、脑缺血、中风、脑梗死或缺血性神经返行性疾病。
本发明的目的是提供一种丁苯酞衍生物,所述丁苯酞衍生物是在丁苯酞开环化学物hpba的羧基上引入了硫化氢分子,其化学结构式如通式i所示:
其中:
r代表氢原子、c1-c8直链或支链烷基、nr1r2;
r1和r2可相同或不同,并各自独立地代表氢原子、c1-c8直链或支链烷基或r1和r2与其所连接的氮原子一起形成5至7元脂肪杂环或芳杂环,该环基可任意地由下述相同或不同的取代基单取代至完全取代,所述取代基包括:c1-c8直链或支链烷基、c1-c8直链或支链烷氧基、羟基或卤素;
式i中一个手性中心*1表示s构型或r构型。
在本发明的一种实施方式中,所述r为氢原子、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、正己基、异己基、二甲胺基、二乙胺基、二丙胺基、二正丁胺基、吡咯基、哌啶基、咪唑基、吗啡啉基、4-羟基哌嗪基、4-甲基哌啶基、n-甲基哌嗪基或n-羟乙基哌嗪基中任意一种。
在本发明的一种实施方式中,所述丁苯酞衍生物为i1-i8任意一种:
在本发明的一种实施方式中,所述丁苯酞衍生物包括化合物的对映体和非对映异构体,以及其与可药用酸的加合盐。
在本发明的一种实施方式中,所述可药用酸包括盐酸、氢溴酸、硫酸、磷酸、乙酸、三氟乙酸、乳酸、丙酮酸、丙二酸、琥珀酸、戊二酸、富马酸、酒石酸、马来酸、柠檬酸、抗坏血酸、甲磺酸、樟脑酸、草酸。
本发明的第二个目的在于提供本上述的丁苯酞衍生物的制备方法,其特征在于,所述方法包括:
(1)茴三硫(adt)在一定温度的碱性条件下脱甲基,得到硫化氢供体adtoh(化合物1);
(2)(s)-或(r)-nbp在溶剂中经皂化、酸化反应制得内酯开环物(s)或(r)-hpba(化合物2),然后与烷基酰氯反应成酯得到酯类化合物(化合物3);
或者与氯乙酰氯反应成酯得到含氯取代的酯类化合物(化合物4),然后与胺类化合物发生sn2取代反应得到含氮化合物(化合物5a-g);
(4)在溶剂中,步骤(2)所述的酯类化合物(化合物3)或步含氮化合物(化合物5a-g)、硫化氢供体adtoh、缩合剂混合,酯化得到目标化合物丁苯酞衍生物。
在本发明的一种实施方式中,所述方法的合成路线如下:
其中,
r’选自氢原子、c1-c8直链支链烷基;r”选自nr1r2;
r”选自nr1r2;r1和r2可相同或不同,并各自独立地代表氢原子、c1-c8直链或支链烷基或r1和r2与其所连接的氮原子一起形成5至7元脂肪杂环或芳杂环,该环基可任意地由下述相同或不同的取代基单取代至五取代,所述取代基包括:c1-c8直链或支链烷基、c1-c8直链或支链烷氧基、羟基或卤素;
式i中一个手性中心*1表示s构型或r构型。
在本发明的一种实施方式中,所述步骤(1)反应条件为碱性条件,反应温度为200-300℃,所述碱为碳酸钾、碳酸钠、碳酸氢钠、氢氧化钠、氢氧化钾或吡啶或三乙胺。
在本发明的一种实施方式中,所述步骤(2)反应条件首先为碱性条件,反应温度为负20℃至回流,所述碱为碳酸钾、碳酸钠、碳酸氢钠、氢氧化钠、氢氧化钾或吡啶或三乙胺;然后为酸性条件,所述酸为盐酸、枸橼酸、抗坏血酸等。
在本发明的一种实施方式中,所述步骤(2)中的溶剂为乙腈、乙酸乙酯、丙酮、二氯甲烷、氯仿、四氢呋喃、n,n-二甲基甲酰胺、二甲亚砜或二氧六环中的一种或多种。
在本发明的一种实施方式中,所述步骤(3)反应条件为碱性条件,反应温度为室温至回流,所述碱为碳酸钾、碳酸钠、碳酸氢钠、氢氧化钠、氢氧化钾或吡啶或三乙胺。
在本发明的一种实施方式中,所述步骤(3)中的溶剂为乙腈、乙酸乙酯、丙酮、二氯甲烷、氯仿、四氢呋喃、n,n-二甲基甲酰胺、二甲亚砜或二氧六环中的一种或多种。
在本发明的一种实施方式中,所述步骤(4)反应条件为碱性条件,反应温度为负20℃至回流,所述碱为三乙胺、吡啶、4-二甲氨基吡啶或n,n-二异丙基甲胺。
在本发明的一种实施方式中,所述步骤(4)中的溶剂为乙腈、乙酸乙酯、丙酮、二氯甲烷、氯仿、四氢呋喃、n,n-二甲基甲酰胺、二甲亚砜或二氧六环中的一种或多种。
在本发明的一种实施方式中,所述步骤(4)缩合剂为n,n’-二环己基碳二亚胺、n-羟基琥珀酰亚胺、1-羟基苯并三唑或1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐。
在本发明的一种实施方式中,所得的中间体化合物或目标化合物均可按照常规分离技术加以纯化得到。
在本发明的一种实施方式中,所述方法还包括将所得的丁苯酞衍生物转化为与可药用酸的加成盐。
在本发明的一种实施方式中,上述丁苯酞衍生物可以单独使用,或者按照规范的制药方法将其与适于药用的载体或稀释剂配合后使用。
本发明的第三个目的在于提供一种药物组合物,所述药物组合物中包含上述的丁苯酞衍生物。
在本发明的一种实施方式中,所述药物组合物中的丁苯酞衍生物包括其旋光异构体、对映体、非对映体、外消旋体或外消旋混合物,或其药学上可接受的盐。
在本发明的一种实施方式中,所述药物组合物的剂型包含注射液、注射用冻干粉针、控释注射剂、脂质体注射剂、混悬剂、植入剂、栓塞剂、胶囊剂、片剂、丸剂和口服液中任意一种。
在本发明的一种实施方式中,所述药物组合物还可以包括药物载体和/或药用辅料。
在本发明的一种实施方式中,所述药物载体包含微囊、微球、纳米粒和脂质体中任意一种。
本发明的第四个目的是提供所述的丁苯酞衍生物在制备预防或治疗与血小板聚集有关的疾病的药物中的应用。
本发明的第五个目的是提供一种预防或治疗心脑缺血性疾病的药物制剂,所述药物制剂包含上述的丁苯酞衍生物或药物组合物。
本发明的第六个目的是提供一种改善心脑循环障碍的药物制剂,所述药物制剂包含上述的丁苯酞衍生物或药物组合物。
本发明的第七个目的是提供一种抗血小板聚集或抗血栓药物制剂,所述药物制剂包含上述的丁苯酞衍生物或药物组合物。
本发明的第八个目的是提供一种抗痴呆药物制剂,所述药物制剂包含上述的丁苯酞衍生物或药物组合物。
在本发明的一种实施方式中,所述药物制剂或药物组合物的给药方式包括口服给药、非胃肠道给药或局部给药。
在本发明的一种实施方式中,所述的非胃肠道给药包括但并不限于静脉注射、肌肉注射、腹腔注射、皮下注射和透皮给药。
本发明的有益效果:
(1)本发明丁苯酞衍生物安全有效,具有较好的抑制血小板聚集和抗血栓活性。其中对于adp诱导的抗血小板聚集,抑制活性ic50可达0.09mm,显著性优于nbp和ticlid;同时,对于aa诱导的抗血小板聚集,抑制活性ic50可达0.10mm。此外,本发明丁苯酞衍生物160mg/kg剂量的存活率可达66.7%,386mg/kg的存活率可达73.3%,对抑制小鼠急性肺血栓的形成作用较nbp或者化合物ii都强。
(2)本发明丁苯酞衍生物化学合成方法简单易行,所需药品价格低廉。
附图说明
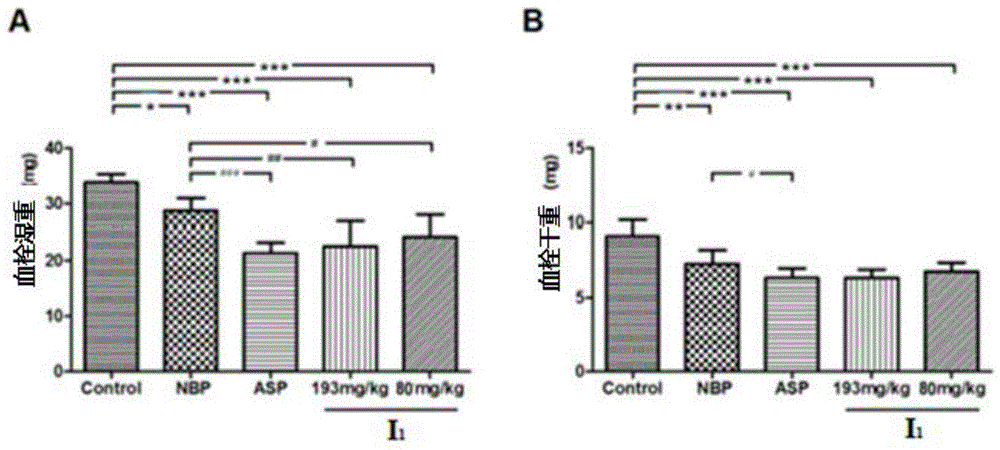
图1为化合物i1对大鼠动静脉旁路血栓的影响。
具体实施方式
为了进一步阐述本发明,以下给出一系列实施例。这些实施例完全是例证性的,它们仅用来对本发明进行具体描述,不应当理解为对本发明的限制。
实施例1化合物i1的制备
将化合物3(1.0mmol)溶于20ml无水二氯甲烷中,搅拌下加入dcc(1.1mmol)和催化量的dmap,室温减半0.5h,再加入化合物1(1.0mmol)室温反应6-8h。反应完毕,过滤,减压浓缩,快速柱层析(pe/etoac=15/1–5/1,v/v)得到桔黄色固体产物366mg,产率80%。
mp94–96℃.ms(esi):m/z459.1[m+h]+.ir(cm-1,kbr):νmax763,1170,1254,1492,1731.1hnmr(300hz,cdcl3):δ0.87(t,3h,ch3,j=6.8hz),1.26–1.50(m,4h,2×ch2),1.83–1.88(m,2h,ch2),2.08(s,3h,oocch3),4.53(t,2h,och2,j=6.0hz),6.65(t,1h,ch,j=6.6hz),7.40–7.44(m,3h,arh),7.45(s,1h,c=ch),7.60–7.65(m,2h,arh),7.73–7.76(m,2h,arh),8.12(d,1h,arh,j=7.8hz).13cnmr(75hz,cdcl3):δ170.4,164.7,153.8,144.7,136.1,133.4,130.6,130.3,129.4,129.4,128.3,128.3,127.4,126.6,123.3,115.2,72.7,36.3,28.0,22.5,21.2,13.9.hrms(esi):m/zcalcdforc23h22o4s3,[m+h]+459.0680;found459.0761.
实施例2化合物i2的制备
将化合物5a(1.0mmol)溶于20ml无水二氯甲烷中,搅拌下加入dcc(1.1mmol)和催化量的dmap,室温减半0.5h,再加入化合物1(1.0mmol)室温反应6-8h。反应完毕,过滤,减压浓缩,快速柱层析(pe/etoac=15/1–5/1,v/v)得到桔黄色油状产物434mg,产率84%。
ms(esi):m/z502.3[m+h]+.ir(cm-1,kbr):νmax764,1169,1276,1490,1739.1hnmr(300hz,cdcl3):δ0.85(t,3h,ch3,j=6.8hz),1.26–1.45(m,4h,2×ch2),1.86–1.90(m,2h,ch2),2.33(s,6h,ch3nch3),3.20–3.21(m,2h,ococh2n),6.65(t,1h,ch,j=6.4hz),7.40–7.45(m,3h,arh),7.46(s,1h,c=ch),7.60–7.62(m,2h,arh),7.73–7.76(m,2h,arh),8.12(d,1h,arh,j=7.8hz).13cnmr(75mhz,cdcl3):δ170.1,164.7,153.8,144.5,136.1,133.3,130.7,130.4,129.4,128.3,127.9,127.1,126.7,123.3,123.1,72.8,45.2,45.2,36.3,28.0,25.6,22.4,13.9.hrms(esi):m/zcalcdforc25h27no4s3,[m+h]+calcd502.1102;found502.1184.
实施例3化合物i3的制备
将化合物5b(1.0mmol)溶于20ml无水二氯甲烷中,搅拌下加入dcc(1.1mmol)和催化量的dmap,室温减半0.5h,再加入化合物1(1.0mmol)室温反应6-8h。反应完毕,过滤,减压浓缩,快速柱层析(pe/etoac=15/1–5/1,v/v)得桔黄色油状产物434mg,产率82%。
ms(esi):m/z530.2[m+h]+.ir(cm-1,kbr):νmax764,1169,1400,1491,1738.1hnmr(300hz,cdcl3):δ0.85(t,3h,ch3,j=6.8hz),0.98(t,6h,2×nch2ch3,j=7.1hz),1.26–1.45(m,4h,2×ch2),1.86–1.96(m,2h,ch2),2.65(q,4h,2×nch2ch3,j=7.1hz),3.32–3.47(m,2h,ococh2n),6.63(t,1h,ch,j=6.5hz),7.40–7.43(m,3h,arh),7.44(s,1h,c=ch),7.60–7.62(m,2h,arh),7.73–7.76(m,2h,arh),8.12(d,1h,arh,j=7.8hz).13cnmr(75mhz,cdcl3):δ170.1,164.7,153.8,144.7,136.1,133.3,130.7,130.4,128.5,128.3,127.9,127.5,127.1,126.7,123.3,123.1,72.6,54.2,47.6,36.4,28.0,22.4,22.4,13.9,12.3.hrms(esi):m/zcalcdforc27h31no4s3,[m+h]+530.1415;found530.1495.
实施例4化合物i4的制备
将化合物5c(1.0mmol)溶于20ml无水二氯甲烷中,搅拌下加入dcc(1.1mmol)和催化量的dmap,室温减半0.5h,再加入化合物1(1.0mmol)室温反应6-8h。反应完毕,过滤,减压浓缩,快速柱层析(pe/etoac=15/1–5/1,v/v)得桔黄色油状产物422mg,产率80%。
ms(esi):m/z528.2[m+h]+.ir(cm-1,kbr):νmax764,1169,1276,1490,1738.1hnmr(300hz,cdcl3):δ0.86(t,3h,ch3,j=6.7hz),1.26–1.28(m,4h,2×nch2ch2),1.32–1.45(m,4h,2×ch2),1.87–1.92(m,2h,ch2),2.62(q,4h,2×nch2,j=7.1hz),3.32–3.45(m,2h,ococh2n),6.62(t,1h,ch,j=6.6hz),7.40–7.43(m,3h,arh),7.44(s,1h,c=ch),7.60–7.61(m,2h,arh),7.73–7.75(m,2h,arh),8.12(d,1h,arh,j=7.8hz).13cnmr(75mhz,cdcl3):δ170.3,164.7,153.8,144.6,136.1,133.3,130.7,129.4,128.7,128.3,127.5,123.3,117.3,72.8,56.9,53.9,36.3,31.9,29.7,28.0,23.8,22.7,22.4,14.1,13.9.hrms(esi):m/zcalcdforc27h29no4s3,[m+h]+530.1259;found528.1341.
实施例5化合物i5的制备
将化合物5d(1.0mmol)溶于20ml无水二氯甲烷中,搅拌下加入dcc(1.1mmol)和催化量的dmap,室温减半0.5h,再加入化合物1(1.0mmol)室温反应6-8h。反应完毕,过滤,减压浓缩,快速柱层析(pe/etoac=15/1–5/1,v/v)得桔黄色油状产物454mg,产率84%。
ms(esi):m/z542.2[m+h]+.ir(cm-1,kbr):νmax764,1169,1276,1490,1739.1hnmr(300hz,cdcl3):δ0.86(t,3h,ch3,j=6.7hz),1.26–1.28(m,4h,2×nch2ch2),1.42–1.43(m,4h,2×ch2),1.57–1.61(m,2h,nch2ch2ch2),1.81–1.91(m,2h,ch2),2.50(q,4h,2×nch2,j=7.1hz),3.16–3.30(m,2h,ococh2n),6.60(t,1h,ch,j=6.6hz),7.40–7.43(m,3h,arh),7.43(s,1h,c=ch),7.60–7.61(m,2h,arh),7.73–7.76(m,2h,arh),8.12(d,1h,arh,j=7.8hz).13cnmr(75mhz,cdcl3):δ170.8,169.1,163.7,152.9,143.8,135.1,132.4,129.7,128.4,127.4,127.0,126.5,125.7,122.1,117.3,71.8,59.4,53.3,36.3,35.4,27.1,24.9,22.9,21.5,14.1,13.0.hrms(esi):m/zcalcdforc28h31no4s3,[m+h]+542.1415;found542.1497.
实施例6化合物i6的制备
将化合物5e(1.0mmol)溶于20ml无水二氯甲烷中,搅拌下加入dcc(1.1mmol)和催化量的dmap,室温减半0.5h,再加入化合物1(1.0mmol)室温反应6-8h。反应完毕,过滤,减压浓缩,快速柱层析(pe/etoac=15/1–5/1,v/v)得到桔黄色油状产物434mg,产率80%。
ms(esi):m/z544.1[m+h]+.ir(cm-1,kbr):νmax764,1169,1400,1491,1739.1hnmr(300hz,cdcl3):δ0.86(t,3h,ch3,j=6.8hz),1.42–1.43(m,4h,2×ch2),1.85–1.92(m,2h,ch2),2.50(q,4h,2×nch2,j=7.0hz),3.18–3.48(m,2h,ococh2n),3.70–3.73(m,4h,2×och2),6.63(t,1h,ch,j=6.5hz),7.40–7.43(m,3h,arh),7.43(s,1h,c=ch),7.60–7.61(m,2h,arh),7.73–7.76(m,2h,arh),8.12(d,1h,arh,j=7.8hz).13cnmr(75mhz,cdcl3):δ169.6,164.7,157.7,154.0,144.5,136.1,133.4,130.7,128.3,127.6,126.6,123.3,116.9,73.0,66.8,59.7,53.3,36.3,35.4,28.0,24.9,22.4,21.5,14.1,13.0.hrms(esi):m/zcalcdforc27h29no5s3,[m+h]+544.1208;found544.1288.
实施例7化合物i7的制备
将化合物5f(1.0mmol)溶于20ml无水二氯甲烷中,搅拌下加入dcc(1.1mmol)和催化量的dmap,室温减半0.5h,再加入化合物1(1.0mmol)室温反应6-8h。反应完毕,过滤,减压浓缩,快速柱层析(pe/etoac=15/1–5/1,v/v)得到桔黄色油状物454mg,产率84%。
ms(esi):m/z557.2[m+h]+.ir(cm-1,kbr):νmax764,1169,1400,1491,1739.1hnmr(300hz,cdcl3):δ0.86(t,3h,ch3,j=6.7hz),1.26–1.28(m,4h,2×nch2ch2),1.42–1.44(m,4h,2×ch2),1.81–1.89(m,2h,ch2),2.33(s,3h,nch3),2.50(m,8h,4×nch2),3.16–3.32(m,2h,ococh2n),6.62(t,1h,ch,j=6.6hz),7.40–7.43(m,3h,arh),7.43(s,1h,c=ch),7.60–7.61(m,2h,arh),7.73–7.76(m,2h,arh),8.12(d,1h,arh,j=7.8hz).13cnmr(75mhz,cdcl3):δ170.8,169.1,163.7,152.9,143.8,135.1,132.4,129.7,128.4,127.4,126.5,126.0,122.1,117.3,71.8,57.3,55.6,46.6,36.3,35.4,27.1,24.9,22.9,21.5,14.1,13.0.hrms(esi):m/zcalcdforc28h32n2o4s3,[m+h]+557.1524;found557.1690.
实施例8化合物i8的制备
将化合物5g(1.0mmol)溶于20ml无水二氯甲烷中,搅拌下加入dcc(1.1mmol)和催化量的dmap,室温减半0.5h,再加入化合物1(1.0mmol)室温反应6-8h。反应完毕,过滤,减压浓缩,快速柱层析(pe/etoac=15/1–5/1,v/v)得到桔黄色油状物454mg,产率84%。
ms(esi):m/z525.3[m+h]+.ir(cm-1,kbr):νmax764,1169,1400,1491,1739.1hnmr(300hz,cdcl3):δ0.86(t,3h,ch3,j=6.7hz),1.42–1.44(m,4h,2×ch2),1.81–1.89(m,2h,ch2),4.24–4.42(m,2h,ococh2n),6.62(t,1h,ch,j=6.6hz),6.77(d,1h,c=ch,j=6.8hz),7.11(d,1h,nch=,j=6.8hz),7.40–7.43(m,3h,arh),7.43(s,1h,c=ch),7.60–7.61(m,2h,arh),7.73–7.76(m,2h,arh),7.83(s,1h,nch=n),8.12(d,1h,arh,j=7.8hz).13cnmr(75mhz,cdcl3):δ170.8,169.1,163.7,152.9,143.8,137.8,132.4,129.7,128.1,127.4,126.5,126.0,122.1,119.1,71.8,57.3,55.6,46.6,36.3,35.4,27.1,24.9,22.9,21.5,14.1,13.0.hrms(esi):m/zcalcdforc26h24n2o4s3,[m+h]+525.0898;found525.1002.
实施例9体外抗血小板聚集活性研究
实验方法:雄性家兔10只,体重2.0~2.5kg。动物于25℃、相对湿度60~75%的条件下饲养1周后用于实验。取家兔2只,用利多卡因局部麻醉,手术分离颈总动脉取血,采取3.8%拘橡酸钠1:9抗凝,以500r/min离心10min,制备富血小板血浆(prp),剩余部分再以3000r/min离心,制备贫血小板血浆(ppp),按比浊法进行血小板聚集实验。测定管中加入prp240μl和不同浓度受试药物30μl,温孵5min,分别以30μl二磷酸腺苷(adp)(终浓度为10μmol/l),30μl凝血酶(终浓度为0.5u/ml)和30μl花生四烯酸(aa)(终浓度为1mmol/l)为诱导剂,观察记录5min内最大聚集率。用生理盐水(ns)作对照,计算各受试化合物的抑制率(%)或ic50值。
抑制率(%)=[对照组5min内最大聚集率-待测样品组5min内最大聚集率]/[对照组5min内最大聚集率]×100%。
测试结果:表1中列出了本发明部分化合物对adp、aa诱导家兔血小板聚集活性数据,阳性对照药为nbp、阿司匹林(asp)及噻氯吡啶(ticlid)。
表1本发明化合物对adp、aa诱导的家兔血小板聚集的抑制活性
以上药理学数据显示,本发明涉及的化合物对不同诱导剂诱导的血小板聚集有较好的抑制作用,且它们的抑制血小板聚集作用均强于nbp和ticlid。化合物i1对于adp诱导的抗血小板聚集,i1的抑制活性(0.09mm)显著性优于nbp(0.73mm)和ticlid(0.36mm);同样,对于aa诱导的抗血小板聚集,i1的抑制活性(0.10mm)也显著性优于nbp(0.58mm),并略优于asp(0.14mm)。
实施例10活性化合物i1对小鼠肺血栓形成的影响
实验方法:选取体重为30~35g的雄性icr小鼠,按体重随机分为12组,每组10只。对照组:i.g.10%dmso;阿司匹林(asp)组:i.g.160mg/kg;nbp组:i.g.160mg/kg;受试化合物i1低、高剂量组(低剂量与阳性药nbp等摩尔量,高剂量与阳性药nbp等重量)。连续给药7天,末次给药2h后,通过尾静脉快速推注含有1mg/mltypei型胶原和44.5μg/ml盐酸肾上腺素的混合诱导剂,剂量为10ml/kg体重。观察并记录5min内小鼠死亡数或15min内偏瘫未恢复的小鼠数。
测试结果:表2中列出了本发明化合物i1对小鼠肺血栓形成的影响,阳性对照药为nbp、阿司匹林(asp)及噻氯吡啶(ticlid)。
表2本发明化合物i1对小鼠肺血栓形成的影响
由表2可见,icr小鼠(每组15只)连续给药7天后,所有给药组对注射胶原-肾上腺素5min内诱导体内血栓形成所致的动物死亡数及15min内偏瘫小鼠未恢复数与空白组相比均有所降低,且存在一定差异。其中:空白对照组(死亡12只,存活率为20.0%),nbp给药组(160mg/kg,死亡7只,存活率为53.3%),asp给药组(160mg/kg,死亡6只,存活率为60.0%),i1等重量给药组(160mg/kg,死亡5只,存活率为66.7%),i1等摩尔量给药组(386mg/kg,死亡4只,存活率为73.3%)。这表明i1对抑制小鼠急性肺血栓的形成作用较nbp强,与asp相当,且存在浓度依赖关系。
实施例11活性化合物i1对大鼠动静脉旁路血栓形成的影响
实验步骤:选取体重为250~300g的sd大鼠,雌雄各半,按体重随机分为6组,每组10只。对照组:i.g.10%dmso;阿司匹林(asp)组:i.g.80mg/kg;nbp组:i.g.80mg/kg;受试化合物i1低、高剂量组(低剂量与阳性药nbp等摩尔量,高剂量与阳性药nbp等重量)。连续给药7天,末次给药2h后,腹腔注射3%戊巴比妥钠溶液(30mg/kg)将大鼠麻醉后,仰位固定,颈正中切口,分离右颈总动脉和左颈外静脉,在聚乙烯管的中段放入一根长6cm的丝线(放入管腔前丝线需先称重),以肝素生理盐水(50u/ml)充满聚乙烯管。将聚乙烯管插入分离出的血管中,使动静脉形成回路。开放血流15min后取出丝线,用滤纸吸去残留的血液后立即称重,总重减去丝线重为血栓湿重。
测试结果:图1中展示出了本发明化合物i1对大鼠动静脉旁路血栓的影响,阳性对照药为nbp、阿司匹林(asp)及噻氯吡啶(ticlid)。
由图1a可见,大鼠连续给药7天后,i1给药等摩尔计量、等重量给药组(21.03±4.24mg,24.07±3.82mg)所形成的血栓湿量均低于空白对照组(33.83±1.48mg)所形成的血栓湿重,且均低于nbp给药组(28.75±2.27mg),但略高于asp给药组(21.40±1.86mg)。血栓干重的结果与之类似。以上结果表明,25a可显著抑制血栓的形成,且明显高于等摩尔剂量的nbp。此外,本实验模型中混合血栓的形成是由于大鼠颈动脉血管内皮损伤,引起一系列活性因子释放,激活血小板和白细胞,引起血小板聚集所产生的。因此,推测i1的抗血栓作用机制很可能与抗血小板聚集有关。
对比例1:
参照实施例9,测定式ii化合物对adp、aa诱导家兔血小板聚集活性数据。测定结果见表3
化合物ii的化学结构式如下所示:
其中ii1:n=2;ii2:n=3;ii3:n=4;ii4:n=5;ii5:n=6;ii6:n=8;ii7:n=12。
表3化合物ii对adp、aa诱导的家兔血小板聚集的抑制活性效果
由表3可见,对于含有碳链连接臂的化合物ii1-ii7,随着碳链长度的增加,对血小板聚集的抑制活性先增强(n=2-6)后减弱(n=6,8,12),其中,化合物ii5(n=6)的抑制作用最为突出。但是,和化合物i1相比,ii5(0.15mm)对adp诱导的血小板聚集的抑制作用不如i1(0.09mm),另外,ii5对aa诱导的血小板聚集的抑制作用也不如i1。
参照实施例10,测定化合物ii5对小鼠肺血栓形成的影响。测定结果见表4。
表4化合物ii对小鼠肺血栓形成的影响
由表4可见,icr小鼠(每组15只)连续给药7天后,所有给药组对注射胶原-肾上腺素5min内诱导体内血栓形成所致的动物死亡数及15min内偏瘫小鼠未恢复数与空白组相比均有所降低,且存在一定差异。化合物ii5等重量给药组(160mg/kg,死亡6只,存活率为60.0%),ii5等摩尔量给药组(386mg/kg,死亡5只,存活率为67.7%)。然而,化合物i1等重量给药组(160mg/kg,死亡5只,存活率为66.7%),i1等摩尔量给药组(386mg/kg,死亡4只,存活率为73.3%),均优于ii5。这表明i1对抑制小鼠急性肺血栓的形成作用较ii5强,且存在浓度依赖关系。
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
- 还没有人留言评论。精彩留言会获得点赞!