第四代EGFR酪氨酸激酶抑制剂的制作方法
本发明涉及具有egfr酪氨酸激酶抑制活性的化合物及含有该化合物的egfr酪氨酸激酶抑制剂。
背景技术:
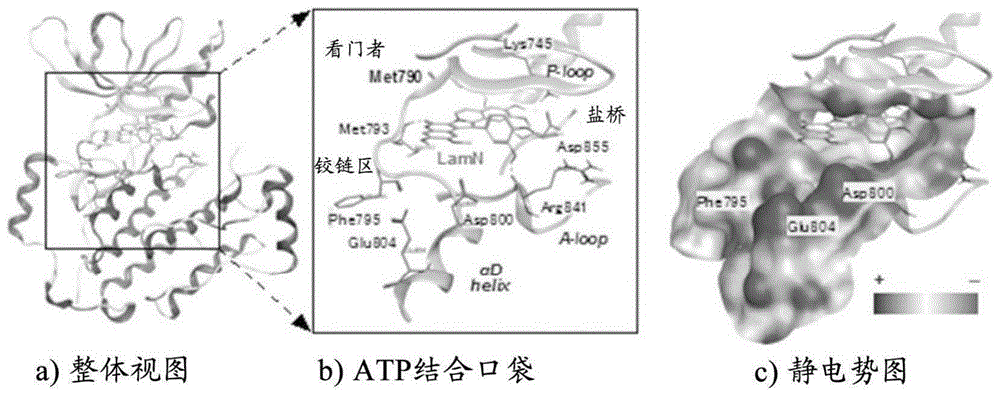
:肺癌的80%为非小细胞肺癌,该非小细胞肺癌的20~30%中确认到了表皮生长因子受体(epidermalgrowthfactorreceptor:egfr)的基因突变。主要的突变是egfr激酶结构域的l858r突变或exon19缺失。由于该突变,从egfr至核的细胞增殖信号传递不断地活性化,细胞发生癌变。对于具有这样的活性化突变egfr(l858r或exon19缺失)的非小细胞肺癌,gefitinib(1)、erlotinib(2)这样的酪氨酸激酶抑制剂(第一代egfr-tki)显示出显著的抗肿瘤效果。另一方面,持续使用这些药剂时,大多数情况下1年左右egfr发生二次耐药性突变(t790m),癌会复发(非专利文献1)。为了应对该耐药性突变,开发了将迈克尔受体导入gefitinib(1)的骨架而成的不可逆抑制剂afatinib(3)(第二代egfr-tki)(非专利文献2)。然而,该药剂不仅强烈抑制t790m二次耐药性突变egfr,也会强烈抑制正常细胞中存在的野生型egfr(wt),因此不能充分提高药剂的血药浓度,对于具有耐药性突变egfr的非小细胞肺癌没有显示出足够的治疗效果(非专利文献3)。化学式1另一方面,作为对t790m二次耐药性突变egfr具有选择性抑制活性的第三代egfr-tki,最近开发了具有嘧啶骨架的不可逆抑制剂wz4002(4)(非专利文献4)、rociletinib(co-1686)(5)(非专利文献5)、osimertinib(azd9291)(6)(非专利文献6)。其中,对于astrazeneca公司开发的6,作为突破性治疗药由美国fda认定为快速审查的对象,于2015年11月批准为egfr-t790m阳性突变非小细胞肺癌治疗药(非专利文献7)。日本国内也于2016年5月起可以进行6的临床使用。化学式2然而,在6的临床实验阶段,报告了对第三代egfr-tki也显示耐药性的新的c797s突变型三次耐药性egfr(非专利文献8)。以t790m二次耐药性突变egfr作为靶标的第三代egfr-tki均为不可逆抑制剂,通过egfr激酶结构域中存在的cys797的侧链硫醇基与抑制剂的丙烯酰胺部位形成共价键,显示出强烈的抗肿瘤活性。因此,从cys797向具有反应性低的侧链羟基的ser797的突变对于不可逆抑制剂来说是致命的。实际上,对于具有c797s突变型三次耐药性egfr的非小细胞肺癌,不仅是不可逆抑制剂,现有的egfr-tki全部无效(非专利文献9)。可知,今后随着第三代egfr-tki的临床使用,具有c797s突变型三次耐药性egfr的非小细胞肺癌会明显地表现出来,可以认为开发对该突变体有效的第四代egfr-tki是紧要的课题(非专利文献10)。现有技术文献非专利文献非专利文献1:v.sharma,etal.epidermalgrowthfactorreceptormutationsinlungcancer,naturereviews,7,169-181(2007)非专利文献2:d.li,etal.bibw2992,anirreversibleegfr/her2inhibitorhighlyeffectiveinpreclinicallungcancermodels,oncogene,27,4702-4711(2008)非专利文献3:y.kim,etal.theegfrt790mmutationinacquiredresistancetoanirreversiblesecond-generationegfrinhibitors,molecularcancertherapeutics,11,784-791(2012)非专利文献4:w.zhou,etal.novelmutant-selectiveegfrkinaseinhibitorsagainstegfrt790m,nature,462,1070-1074(2009)非专利文献5:a.o.walter,etal.discoveryofamutant-selectivecovalentinhibitorofegfrthatovercomest790m-mediatedresistanceinnsclc,cancerdiscovery,1404-1415(2013)非专利文献6:d.a.e.cross,etal.azd9291,anirreversibleegfrtki,overcomest790m-mediatedresistancetoegfrinhibitorsinlungcancer,cancerdiscovery,1047-1061(2014)非专利文献7:fdanewsrelease,fdaapprovesnewpilltotreatcertainpatientswithnon-smallcelllungcancer,november13,2015非专利文献8:k.s.thress,etal.acquiredegfrc797smutationmediatesresistancetoazd9291innon-smallcelllungcancerharboringegfrt790m,naturemedicine,21,560-564(2015)非专利文献9:d.ercan,etal.egfrmutationsandresistancetoirreversiblepyrimidine-basedinhibitors,clinicalcancerresearch,21,3913-3923(2015)非专利文献10:y.jia,etal.overcomingegfr(t790m)andegfr(c797s)resistancewithmutant-selectiveallostericinhibitors,nature,534,129-132(2016)技术实现要素:发明要解决的课题本发明的目的在于提供对c797s突变型耐性egfr(特别是c797s突变型三次耐药性egfr)有效的第四代egfr-tki。解决课题的方法本发明人等为了解决上述课题而进行了深入研究,结果发现,下式(i)所示的化合物对c797s突变型耐性egfr(特别是c797s突变型三次耐药性egfr)具有特异性的酪氨酸激酶抑制活性,从而完成了本发明。具体而言,作为以海洋天然产物片螺素(lamellarin)作为结构模体的抗癌药开发研究的一环,本发明人等实施了片螺素类对癌相关激酶的活性评价。其结果是,发现了片螺素n(7)对耐药性突变egfr(l858r/t790m)显示出抑制活性。片螺素n(7)是可逆性抑制剂,由于活性表达不依赖于与cys797形成共价键,因此可以认为是用于创制对c797s突变有效的egfr-tki的命中化合物(hitcompound)。这里,为了提高命中化合物的活性,进行了片螺素n(7)对egfr(l858r/t790m)激酶结构域的对接模拟(图1)。其结果是可以预想,沿激酶的atp结合口袋的开口部取向的片螺素n(7)的a环部取代基与其附近存在的phe795、asp800、glu804的相互作用增强会引起活性提高。因此,合成将各种取代基导入片螺素n(7)的a环部的氧官能团部分(oh、ome)而成的新型片螺素n衍生物,并进行了评价。另外,对于将片螺素n(7)的b环部内酯环取代为内酰胺环而成的氮杂片螺素(azalamellarin)n(8),也进行了同样的结构研究。化学式2即,本发明如下所述。[1]式(i)所示的化合物或其盐(本说明书中,有时简称为“化合物(i)”),化学式4[式中,x表示o或nh,r1及r2各自独立地表示氢原子或任选被取代的烃基;r3及r4各自独立地表示氢原子、卤原子或任选被取代的烃基,r5及r6各自独立地表示任选被取代的烃基。]其中,式(i)所示的化合物不包括以下的化合物:化学式5[2]一种c797s突变型耐药性egfr特异性酪氨酸激酶抑制剂,其含有式(i)所示的化合物或其盐,化学式5[式中,x表示o或nh,r1及r2各自独立地表示氢原子或任选被取代的烃基;r3及r4各自独立地表示氢原子、卤原子或任选被取代的烃基,r5及r6各自独立地表示任选被取代的烃基。]。发明效果本发明的化合物对c797s突变型耐药性egfr(特别是c797s突变型三次耐药性egfr)具有特异性的酪氨酸激酶抑制活性,作为c797s突变型耐药性egfr(特别是c797s突变型三次耐药性egfr)特异性酪氨酸激酶抑制剂、具有耐药性突变egfr的非小细胞肺癌等的预防和/或治疗剂等是有用的。附图说明图1示出了片螺素n(7)对egfr(l858r/t790m)激酶结构域的对接模拟。图2示出了利用试验例4中的化合物26d进行的egfr信号抑制。图3示出了试验例5中的化合物26d在小鼠异种移植模型中的抗肿瘤效果。图4示出了试验例5中的化合物26d给药期间中裸鼠体重的变化。具体实施方式以下,对本发明进行详细说明。以下,对本说明书中使用的各取代基的定义进行详细说明。在没有特别说明的情况下,各取代基具有以下的定义。在本说明书中,作为“卤原子”,可以列举例如:氟、氯、溴、碘。在本说明书中,作为“烃基”,可以列举例如:c1-6烷基、c2-6烯基、c2-6炔基、c3-10环烷基、c3-10环烯基、c6-14芳基、c7-16芳烷基。在本说明书中,作为“c1-6烷基”,可以列举例如:甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、戊基、异戊基、新戊基、1-乙基丙基、己基、异己基、1,1-二甲基丁基、2,2-二甲基丁基、3,3-二甲基丁基、2-乙基丁基。在本说明书中,作为“c2-6烯基”,可以列举例如:乙烯基、1-丙烯基、2-丙烯基、2-甲基-1-丙烯基、1-丁烯基、2-丁烯基、3-丁烯基、3-甲基-2-丁烯基、1-戊烯基、2-戊烯基、3-戊烯基、4-戊烯基、4-甲基-3-戊烯基、1-己烯基、3-己烯基、5-己烯基。在本说明书中,作为“c2-6炔基”,可以列举例如:乙炔基、1-丙炔基、2-丙炔基、1-丁炔基、2-丁炔基、3-丁炔基、1-戊炔基、2-戊炔基、3-戊炔基、4-戊炔基、1-己炔基、2-己炔基、3-己炔基、4-己炔基、5-己炔基、4-甲基-2-戊炔基。在本说明书中,作为“c3-10环烷基”,可以列举例如:环丙基、环丁基、环戊基、环己基、环庚基、环辛基、双环[2.2.1]庚基、双环[2.2.2]辛基、双环[3.2.1]辛基、金刚烷基。在本说明书中,作为“c3-10环烯基”,可以列举例如:环丙烯基、环丁烯基、环戊烯基、环己烯基、环庚烯基、环辛烯基。在本说明书中,作为“c6-14芳基”,可以列举例如:苯基、1-萘基、2-萘基、1-蒽基、2-蒽基、9-蒽基。在本说明书中,作为“c7-16芳烷基”,可以列举例如:苄基、苯乙基、萘基甲基、萘基丙基。在本说明书中,作为“任选被取代的烃基”的取代基,可以列举例如:卤原子、羟基、氰基、硝基、羧基、任选被c1-6烷基单取代或二取代的氨基、胍基。取代基的数量例如为1~5个,优选为、1~3个。在存在多个取代基的情况下,各取代基可以相同,也可以不同。以下,对式(i)中的各符号的定义进行详细说明。x表示o或nh。r1及r2各自独立地表示氢原子或任选被取代的烃基。作为r1及r2所示的“任选被取代的烃基”的“烃基”,优选为c1-6烷基(例如,甲基、乙基、正丙基)。r1及r2优选各自独立地表示氢原子或任选被取代的c1-6烷基(例如,甲基、乙基、正丙基);更优选各自独立地表示氢原子、或任选被以下的1~3个取代基取代的c1-6烷基(例如,甲基、乙基、正丙基),所述取代基选自任选被c1-6烷基单取代或二取代的氨基(例如,二甲基氨基)及胍基;进一步优选各自独立地表示氢原子、或任选被氨基取代的c1-6烷基(例如,甲基、乙基、正丙基),所述氨基为被c1-6烷基二取代的氨基(例如,二甲基氨基)。r3及r4各自独立地表示氢原子、卤原子或任选被取代的烃基。作为r3及r4所示的“任选被取代的烃基”的“烃基”,优选为c1-6烷基(例如,甲基)。r3及r4优选各自独立地表示氢原子、卤原子或任选被取代的c1-6烷基(例如,甲基),更优选各自独立地表示氢原子或卤原子,进一步优选分别为氢原子。r5及r6各自独立地表示任选被取代的烃基。作为r5及r5所示的“任选被取代的烃基”的“烃基”,优选为c1-6烷基(例如,甲基)。r5及r6优选各自独立地表示任选被取代的c1-6烷基(例如,甲基),更优选各自独立地表示c1-6烷基(例如,甲基)。作为化合物(i)的优选例,可以举出以下的化合物。[化合物i-1]化合物(i)x为o或nh;r1及r2各自独立地表示氢原子或任选被取代的c1-6烷基(例如,甲基、乙基、正丙基);r3及r4各自独立地表示氢原子、卤原子或任选被取代的c1-6烷基(例如,甲基);r5及r6各自独立地表示任选被取代的c1-6烷基(例如,甲基)。[化合物i-2]化合物(i)x为o或nh;r1及r2各自独立地表示氢原子、或任选被以下的1~3个取代基取代的c1-6烷基(例如,甲基、乙基、正丙基),所述取代基选自任选被c1-6烷基单取代或二取代的氨基(例如,二甲基氨基)及胍基;r3及r4各自独立地表示氢原子、卤原子或c1-6烷基(例如,甲基);r5及r6各自独立地表示c1-6烷基(例如,甲基)。[化合物i-3]化合物(i)x为o或nh;r1及r2各自独立地表示氢原子、或任选被以下的1~3个取代基取代的c1-6烷基(例如,甲基、乙基、正丙基),所述取代基选自任选被c1-6烷基单取代或二取代的氨基(例如,二甲基氨基)及胍基;r3及r4各自独立地表示氢原子或卤原子;r5及r6各自独立地表示c1-6烷基(例如,甲基)。[化合物i-4]化合物(i)x为o或nh;r1及r2各自独立地表示氢原子、或任选被氨基取代的c1-6烷基(例如,甲基、乙基、正丙基),氨基是被c1-6烷基二取代的氨基(例如,二甲基氨基);r3及r4分别为氢原子;r5及r6各自独立地表示c1-6烷基(例如,甲基)。在化合物(i)为盐的情况下,作为这样的盐,可以列举例如:与无机碱形成的盐、与有机碱形成的盐、与无机酸形成的盐、与有机酸形成的盐、与碱性或酸性氨基酸形成的盐等。作为与无机碱形成的盐的优选例,可以列举:钠盐、钾盐等碱金属盐;钙盐、镁盐等碱土金属盐;铝盐;铵盐。作为与有机碱形成的盐的优选例,可以列举:与三甲胺、三乙胺、吡啶、甲基吡啶、乙醇胺、二乙醇胺、三乙醇胺、氨丁三醇[三(羟甲基)甲胺]、叔丁胺、环己胺、苄胺、二环己胺、n,n-二苄基乙二胺形成的盐。作为与无机酸形成的盐的优选例,可以列举:与氯化氢、溴化氢、硝酸、硫酸、磷酸形成的盐。作为与有机酸形成的盐的优选例,可以列举:与甲酸、酢酸、三氟乙酸、邻苯二甲酸、富马酸、草酸、酒石酸、马来酸、柠檬酸、丁二酸、苹果酸、甲磺酸、苯磺酸、对甲苯磺酸形成的盐。作为与碱性氨基酸形成的盐的优选例,可以列举:与精氨酸、赖氨酸、鸟氨酸形成的盐。作为与酸性氨基酸形成的盐的优选例,可以列举:与天冬氨酸、谷氨酸形成的盐。在这些盐中,优选为药学上可以允许的盐。作为药学上可以允许的盐,在化合物内具有碱性官能团的情况下,可以列举例如:与盐酸、氢溴酸、硝酸、硫酸、磷酸等无机酸形成的盐;或与乙酸、邻苯二甲酸、富马酸、草酸、酒石酸、马来酸、柠檬酸、丁二酸、甲磺酸、对甲苯磺酸等有机酸形成的盐。另外,在化合物内具有酸性官能团的情况下,可以列举:碱金属盐(例如,钠盐、钾盐等)、碱土金属盐(例如,钙盐、镁盐、钡盐等)等无机盐;铵盐等。另外,化合物(i)可以用同位素(例、3h、13c、14c、18f、35s、125i)等进行了标记。另外,化合物(i)可以是水合物,也可以是非水合物,可以是非溶剂化物,也可以是溶剂化物。此外,将1h转化为2h(d)的氘转化体也包含于化合物(i)。在化合物(i)具有手性中心的情况下,可以存在对映体或非对映体等异构体。这样的异构体及它们的混合物均包含在本发明的范围内。另外,有时生成因构象或互变异构引起的异构体,这样的异构体或其混合物也包含于本发明的化合物(i)。另外,有时生成阻转异构体(具体而言,在式(i)中,r3及r4中的任一者或两者为除氢原子以外的情况),这样的异构体或其混合物也包含于本发明的化合物(i)。接下来,对本发明的化合物(i)的制造方法进行说明。本发明的化合物(i)例如可以通过j.org.chem.2014,79,529-537中记载的片螺素类的模块合成法或基于此的方法等来合成。作为一例,将r5及r6为甲基的化合物(i)的合成方案示于以下。化学式7(式中,pg1及pg2表示羟基的保护基,其它符号如上述所定义。)可以以能够由市售的吡咯合成的化合物9作为起始物质,以2步合成三环化合物10。通过使本化合物10与芳基硼酸11或芳基硼酸酯12进行铃木-宫浦偶联,可以合成片螺素衍生物13及氮杂片螺素衍生物14。进而,将片螺素衍生物13及氮杂片螺素衍生物14的a环部氧上的保护基(pg1、pg2)选择性地脱保护,进一步使得到的酚衍生物官能化,由此可以合成本发明的化合物(i)(片螺素n衍生物(x=o)及氮杂片螺素n衍生物(x=nh)。本发明的化合物(i)对于c797s突变型耐药性egfr(特别是c797s突变型三次耐药性egfr)具有特异性的酪氨酸激酶抑制活性,可以用作c797s突变型耐药性egfr(特别是c797s突变型三次耐药性egfr)特异性酪氨酸激酶抑制剂、具有耐药性突变egfr的非小细胞肺癌等的预防和/或治疗剂等。本发明的化合物(i)对于c797s突变型耐药性egfr(特别是c797s突变型三次耐药性egfr)显示出特异性的酪氨酸激酶抑制活性,且本发明的化合物(i)在药效显现、药代动力学(例如,吸收性、分布、代谢、排泄)、溶解性(例如,水溶性)、与其它医药品的相互作用(例如,药物代谢酶抑制作用)、安全性(例如,急性毒性、慢性毒性、遗传毒性、生殖毒性、心脏毒性、致癌性、中枢毒性)、稳定性(例如,化学稳定性、对酶的稳定性)方面也优异,因此可以用作医药品。因此,对于哺乳动物(例如,小鼠、大鼠、仓鼠、兔、猫、犬、牛、羊、猴、人),本发明的化合物(i)可以用于对c797s突变型耐药性egfr(特别是c797s突变型三次耐药性egfr)特异性的酪氨酸激酶抑制作用。本发明的化合物(i)可以直接或与药理学上允许的载体配合制成医药品而对哺乳动物(优选为人)口服或非口服给药。以下,对含有本发明的化合物(i)而成的医药(有时简称为“本发明的医药”)进行详细说明。作为本发明的医药的剂型,可以列举例如:片剂(例如,糖衣片、薄膜包衣片、舌下片、口含片、口腔速崩片)、丸剂、颗粒剂、散剂、胶囊剂(例如,软胶囊剂、微胶囊剂)、糖浆剂、乳剂、悬浮剂、膜剂(例如,口腔崩解膜、口腔粘膜粘贴膜)等口服制剂。另外,作为本发明的医药的剂型,可以列举例如:注射剂、输液剂、透皮剂(例如,离子电渗透皮剂)、栓剂、软膏剂、滴鼻剂、肺部给药剂、滴眼剂等非口服制剂。另外,本发明的医药可以为速释制剂、缓释制剂(例如,缓释微胶囊)等控释制剂。本发明的医药可以通过制剂
技术领域:
通常使用的公知的制造方法(例如,日本药局方中记载的方法)来制造。另外,本发明的医药可以根据需要适当、适量地含有制剂领域通常使用的赋形剂、粘合剂、崩解剂、润滑剂、甜味剂、表面活性剂、助悬剂、乳化剂、着色剂、防腐剂、芳香剂、矫味剂、稳定剂、增稠剂等添加剂。作为上述的药理学上允许的载体,可以列举这些添加剂。例如,片剂可以使用赋形剂、粘合剂、崩解剂、润滑剂等制造,丸剂及颗粒剂可以使用赋形剂、粘合剂、崩解剂来制造。另外,散剂及胶囊剂可以使用赋形剂等制造,糖浆剂可以使用甜味剂等制造,乳剂或悬浮剂可以使用助悬剂、表面活性剂、乳化剂等制造。作为赋形剂的例子,可以列举:乳糖、蔗糖、葡萄糖、淀粉、蔗糖、微晶纤维素、甘草粉末、甘露醇、碳酸氢钠、磷酸钙、硫酸钙。作为粘合剂的例子,可以列举:5~10重量%淀粉糊液、10~20重量%阿拉伯胶液或明胶液、1~5重量%黄蓍胶液、羧甲基纤维素液、藻酸钠液、甘油。作为崩解剂的例子,可以列举:淀粉、碳酸钙。作为润滑剂的例子,可以列举:硬脂酸镁、硬脂酸、硬脂酸钙、纯化的滑石。作为甜味剂的例子,可以列举:葡萄糖、果糖、转化糖、山梨糖醇、木糖醇、甘油、单糖浆。作为表面活性剂的例子,可以列举:月桂基硫酸钠、聚山梨醇酯80、脱水山梨糖醇单脂肪酸酯、聚乙二醇硬脂酸酯40。作为助悬剂的例子,可以列举:阿拉伯胶、藻酸钠、羧甲基纤维素钠、甲基纤维素、膨润土。作为乳化剂的例子,可以列举:阿拉伯胶、黄蓍胶、明胶、聚山梨醇酯80。例如,在本发明的医药为片剂的情况下,该片剂可以通过按照本身公知的方法,在本发明的化合物(i)中添加例如赋形剂(例如,乳糖、蔗糖、淀粉)、崩解剂(例如,淀粉、碳酸钙)、粘合剂(例如,淀粉、阿拉伯胶、羧甲基纤维素、聚乙烯吡咯烷酮、羟丙基纤维素)或润滑剂(例如,滑石、硬脂酸镁、聚乙二醇6000)并进行压缩成形,接着根据需要,为了掩盖气味、肠溶性或持续性而用本身公知的方法进行包衣来制造。作为包衣所用的包衣剂,可以使用例如,羟丙基甲基纤维素、乙基纤维素、羟甲基纤维素、羟丙基纤维素、聚氧乙二醇、吐温80、普朗尼克(pluronic)f68、邻苯二甲酸乙酸纤维素、羟丙基甲基纤维素邻苯二甲酸酯、乙酸羟甲基纤维素琥珀酸酯、尤特奇(eudragit,rohm公司制、德国、甲基丙烯酸-丙烯酸共聚物)及色素(例如,铁丹、二氧化钛)。作为上述注射剂,除静脉注射剂以外,还包括皮下注射剂、皮内注射剂、肌肉注射剂、腹腔内注射剂、输液注射剂等。所述注射剂可以通过本身公知的方法、即将本发明的化合物(i)溶解、悬浮或乳化于无菌的水性液体或油性液体来制备。作为水性液体,可以列举:生理盐水、包含葡萄糖、其它佐剂的等渗液(例如,d-山梨糖醇、d-甘露醇、氯化钠)等。该水性液体可以包含适当的增溶剂、例如醇(例如,乙醇)、多元醇(例如,丙二醇、聚乙二醇)、非离子型表面活性剂(例如,聚山梨醇酯80、hco-50)。作为油性液体,可以列举:芝麻油、大豆油等。该油性液体可以包含适当的增溶剂。作为该增溶剂,可以列举:苯甲酸苄酯、苯甲醇等。另外,该注射剂中可以配合缓冲剂(例如,磷酸缓冲液、乙酸钠缓冲液)、无痛化剂(例如,苯扎氯铵、盐酸普鲁卡因)、稳定剂(例如,人血清白蛋白、聚乙二醇)、防腐剂(例如,苯甲醇、苯酚)等。制备的注射液通常可以填充于安瓿。本发明的医药中的本发明的化合物(i)的含量根据制剂的形态而不同,通常相对于全部制剂为约0.01~约100重量%,优选为约2~约85重量%,进一步优选为约5~约70重量%。本发明的医药中的添加剂的含量根据制剂的形态而不同,通常相对于全部制剂为约1~约99.9重量%,优选为约10~约90重量%。本发明的化合物(i)可以稳定且低毒性地安全使用。本发明的化合物(i)的1天给药量根据患者的状态、体重、化合物的种类、给药途径等而不同,例如,在为了治疗癌而对患者口服给药的情况下,成人(体重约60kg)每1天的给药量以本发明的化合物(i)计为约1~约1000mg,优选为约3~约300mg,进一步优选为约10~约200mg,可以将它们分成1次或2~3次给药。在非口服给药本发明的化合物(i)的情况下,通常以液体制剂(例如,注射剂)的形式给药。本发明的化合物(i)的1次给药量根据给药对象、对象脏器、症状、给药方法等而不同,例如,优选将每1kg体重通常为约0.01~约100mg、优选为约0.01~约50mg、更优选为约0.01~约20mg的本发明的化合物(i)通过静脉注射进行给药。实施例以下,列举实施例对本发明进行更具体说明,但本发明并不受以下实施例的限制,可以在能够符合上述/下述的主旨的范围内适当进行变更来实施,这些均包含于本发明的技术范围内。在本说明书中,以下的方案中使用的缩写符号表示以下含义。-me:甲基-et:乙基-ome:甲氧基-oi-pr:异丙氧基-co2me:甲氧羰基-nme2:二甲基氨基-mom:甲氧基甲基-omom:甲氧基甲氧基-obn:苄氧基-boc:叔丁氧羰基-nhboc:叔丁氧羰基氨基[片螺素n衍生物的合成法]化学式8化合物9可以通过j.org.chem.2014,79,529-537中记载的方法或以此为基准的方法来合成。工序(a):化合物15的合成在氩气氛围下,将由化合物9(2.66g,5.00mmol)、2-溴-1,1-二甲氧基乙烷(3.72ml,31.5mmol)、碳酸铯(11.7g,35.8mmol)、及干燥二甲基甲酰胺(60ml)形成的混合物在110℃下搅拌了16小时。冷却后,加入水,用己烷/乙酸乙酯混合溶剂(1∶1)进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥后,减压蒸馏除去溶剂。用硅胶快速色谱(己烷∶乙酸乙酯=3∶1)纯化残渣,以白色的半固体(2.83g)的形式得到了化合物15。收率91%。1hnmr(400mhz,cdcl3):δ1.21(d,j=6.1hz,6h),1.35(d,j=6.1hz,6h),3.23(s,6h),3.67(s,3h),3.81(s,3h),3.93(s,3h),4.27(sep,j=6.1hz,1h),4.41-4.46(m,3h),4.50(sep,j=6.1hz,1h),6.66(s,1h),6.72-6.76(m,3h),6.76(d,j=1.7hz,1h),6.79(d,j=8.3hz,1h).hrdartmsm/z.calcdforc30h38brno8(m+):619.1781.found:619.1782.工序(b):化合物10的合成在氩气氛围下,在0℃下向化合物15(1.84g,2.97mmol)的二氯甲烷(110ml)溶液中加入三氟甲磺酸三甲基硅酯(270μl,1.49mmol)。在0℃下搅拌1.5小时后,加入饱和碳酸氢钠水溶液,将混合物升温至室温,然后在该温度下搅拌30分钟。加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥,减压蒸馏除去二氯甲烷。用硅胶快速色谱(己烷∶乙酸乙酯=4∶1)纯化残渣,以白色固体(1.50g)的形式得到了化合物10。收率91%。熔点165-166℃(二乙基醚/己烷混合溶剂)。1hnmr(400mhz,cdcl3):δ1.35(d,j=6.1hz,3h),1.38(d,j=6.1hz,3h),1.42(d,j=6.1hz,6h),3.41(s,3h),3.93(s,3h),3.98(s,3h),4.53(sep,j=6.1hz,1h),4.66(sep,j=6.1hz,1h),6.93(d,j=7.6hz,1h),6.95(d,j=1.9hz,1h),6.99(dd,j=1.9and8.2hz,1h),7.02(s,1h),7.05(d,j=8.2hz,1h),7.07(s,1h),9.28(d,j=7.6hz,1h).hrdartmsm/z.calcdforc28h31brno6[(m+h)+]:556.13347.found:556.13474.化学式9工序(a):化合物11a的合成在氩气氛围下,在-78℃下,向市售的、或者通过本身公知的方法或以其为基准的方法合成的化合物17a(13.8g,39.1mmol)的四氢呋喃(120ml)溶液滴加叔丁基锂戊烷溶液(1.35m,60.9ml,82.2mmol),在该温度下搅拌1小时。在-78℃下滴加了硼酸三甲酯(6.54ml,58.7mmol)后,在该温度下搅拌1小时,升温至室温,在该温度下搅拌1小时。在反应溶液中加入饱和氯化铵水溶液,减压蒸馏除去溶剂。向残渣中一点一点地加入乙酸,使其为ph=3。然后,用二氯甲烷进行萃取,用水、饱和碳酸氢钠水溶液及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥。减压蒸馏除去二氯甲烷后,用己烷清洗残渣,以淡黄色固体(12.3g)的形式得到了化合物11a。收率99%。化学式10工序(a):化合物19的合成在氩气氛围下,向市售的、或者通过本身公知的方法或以其为基准的方法合成的化合物18(5.01g,36.2mmol)的干燥二甲基甲酰胺(30ml)溶液依次加入碳酸钾(5.00g,36.2mmol)、碘化钾(6.02g,36.3mmol)及2-溴丙烷(4.42ml,47.1mmol)。将混合物在40℃下搅拌21小时。冷却后,加入2m盐酸,用乙酸乙酯进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥后,减压蒸馏除去乙酸乙酯。用硅胶柱色谱(己烷∶乙酸乙酯=5∶1~1∶1)纯化残渣,以白色固体(4.25g)的形式得到了化合物19。收率65%。1hnmr(400mhz,cdcl3):δ1.42(d,j=6.1hz,6h),4.74(sep,j=6.1hz,1h),5.94(brs,1h),6.96(d,j=8.2hz,1h),7.41(dd,j=2.0and8.0hz,1h),7.45(d,j=2.0hz,1h),9.83(s,1h).工序(b):化合物20的合成在氩气氛围下,向化合物19(3.89g,21.6mmol)的干燥丙酮(40ml)溶液依次加入碳酸钾(3.87g,36.2mmol)及溴化苄(3.33ml,28.0mmol)。将混合物加热回流21小时。冷却后,加入2m盐酸,减压蒸馏除去溶剂。向残渣中加入水,用乙酸乙酯进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥后,减压蒸馏除去乙酸乙酯。用硅胶柱色谱(己烷∶乙酸乙酯=5∶1)纯化残渣,以白色固体(5.71g)的形式得到了化合物20。收率98%。1hnmr(400mhz,cdcl3):δ1.41(d,j=6.0hz,6h),4.68(sep,j=6.0hz,1h),5.17(s,2h),7.00(d,j=8.2hz,1h),7.29-7.47(m,7h),9.80(s,1h).工序(c):化合物21的合成在氩气氛围下,在0℃下向化合物20(1.02g,3.79mmol)的二氯甲烷(12ml)溶液加入间氯过氧苯甲酸(含有水约30%,1.31g)。在0℃下搅拌18小时后,加入饱和碳酸氢钠水溶液,升温至室温,用二氯甲烷进行萃取。用水清洗萃取液,并用无水硫酸钠干燥后,减压蒸馏除去乙酸乙酯。在氩气氛围下,将残渣、碳酸钾(1.27g,4.28mmol)、及甲醇(13ml)的混合物在室温下搅拌30分钟,然后减压蒸馏除去甲醇。向残渣中加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥后,减压蒸馏除去二氯甲烷。用硅胶柱色谱(己烷∶乙酸乙酯=3∶1~1∶1~乙酸乙酯)纯化残渣,以黄色油状物(785mg)的形式得到了化合物21。收率80%。1hnmr(400mhz,cdcl3):δ1.29(d,j=6.0hz,6h),4.34(sep,j=6.0hz,1h),5.01(s,2h),5.13(brs,1h),6.31(dd,j=2.8and8.6hz,1h),6.45(d,j=2.8hz,1h),6.80(d,j=8.6hz,1h),7.27-7.41(m,5h).工序(d):化合物22的合成在氩气氛围下,在0℃下向化合物21(785mg,3.04mmol)及n,n-二异丙基乙胺(1.06ml,6.08mmol)的二氯甲烷(11.5ml)溶液加入氯二甲醚(0.35ml,4.56mmol),在该温度下搅拌1小时。升温至室温,在该温度下搅拌18小时,然后加入28%氨水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥后,减压蒸馏除去二氯甲烷。用硅胶柱色谱(己烷∶乙酸乙酯=5∶1~1∶1~乙酸乙酯)纯化残渣,以淡黄色油状物(856mg)的形式得到了化合物22。收率93%。1hnmr(400mhz,cdcl3):δ1.31(d,j=6.2hz,6h),3.45(s,3h),4.38(sep,j=6.1hz,1h),5.08(s,2h),5.09(s,2h),6.57(dd,j=2.9,8.7hz,1h),6.68(d,j=2.8hz,1h),6.86(d,j=8.7hz,1h),7.28-7.45(m,5h).工序(e):化合物17b的合成在氩气氛围下,在0℃下向化合物22(201mg,0.663mmol)的干燥二甲基甲酰胺(3.3ml)溶液加入n-溴代琥珀酰亚胺(119mg,0.668mmol)。在0℃下搅拌2小时,然后加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥,减压蒸馏除去二氯甲烷。用硅胶柱色谱(己烷∶乙酸乙酯=10∶1)纯化残渣,以淡黄色油状物(232mg)的形式得到了化合物17b。收率92%。1hnmr(400mhz,cdcl3):δ1.32(d,j=6.1hz,6h),3.48(s,3h),4.40(sep,j=6.1hz,1h),5.09(s,2h),5.10(s,2h),6.83(s,1h),7.10(s,1h),7.28-7.43(m,5h).工序(f):化合物11b的合成在氩气氛围下,在-100℃下向化合物17b(574mg,1.51mmol)的四氢呋喃(9.5ml)溶液滴加正丁基锂己烷溶液(1.50m,1.1ml,1.66mmol),并在该温度下搅拌20分钟。在-100℃下滴加硼酸三甲酯(0.30ml,2.67mmol)后,升温至室温,在该温度下搅拌1小时。向反应溶液中加入饱和氯化铵水溶液,减压蒸馏除去溶剂。向残渣中一点一点地加入乙酸,使其为ph=3。然后,用二氯甲烷进行萃取,用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥。减压蒸馏除去二氯甲烷后,用己烷清洗残渣,以白色固体(379mg)的形式得到了化合物11b。收率73%。1hnmr(400mhz,cdcl3):δ1.32(d,j=6.2hz,6h),3.46(s,3h),4.45(sep,1h,6.1hz),5.15(s,2h),5.16(s,2h),6.32(brs,2h),6.81(s,1h),7.40(s,1h),7.29-7.45(m,5h).化学式11工序(a):化合物17c的合成在氩气氛围下,在0℃下向化合物23(1.20mg,3.43mmol)的干燥二甲基甲酰胺(20ml)溶液加入n-溴代琥珀酰亚胺(0.64g,3.60mmol)。在0℃下搅拌2小时后,加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥,减压蒸馏除去二氯甲烷。用硅胶柱色谱(己烷∶乙酸乙酯=7∶1)纯化残渣,以白色固体(1.23g)的形式得到了化合物17c。收率83%。工序(b):化合物11c的合成在氩气氛围下,在-78℃下向化合物17c(1.22g,2.87mmol)的四氢呋喃(50ml)溶液滴加正丁基锂己烷溶液(1.54m,2.05ml,3.16mmol),在该温度下搅拌1小时。在-78℃下滴加硼酸三甲酯(0.48ml,4.31mmol)后,升温至室温,在该温度下搅拌1小时。向反应溶液中加入饱和氯化铵水溶液,减压蒸馏除去溶剂。向残渣中一点一点地加入乙酸,使其为ph=3。然后,用二氯甲烷进行萃取,用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥。减压蒸馏除去二氯甲烷后,用己烷清洗残渣,以淡黄色固体(905mg)的形式得到了化合物11c。收率89%。1hnmr(500mhz,cdcl3):δ3.46(s,3h),5.12(s,2h),5.16(s,2h),5.18(s,2h),5.93(s,2h),6.83(s,1h),7.25-7.37(s,6h),7.42-7.46(m,5h).化学式12工序(a):化合物16a的合成在氩气氛围下,将由化合物10(290mg,0.521mmol)、化合物11a(497mg,1.56mmol)、双(二亚苄基丙酮)钯(0)(30.0mg,52.1μmol)、1,1’-双(二苯基膦)二茂铁(28.9mg,52.1μmol)、na2co3(365mg,3.44mmol)、1,2-二甲氧基乙烷(12ml)、及脱气水(1.0ml)形成的混合物加热回流24小时。冷却后,减压蒸馏除去溶剂。向残渣中加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥,减压蒸馏除去二氯甲烷。用硅胶快速色谱(己烷∶乙酸乙酯=3∶1)纯化残渣,以淡褐色固体(302mg)的形式得到了化合物16a。收率77%。1hnmr(400mhz,cdcl3):δ0.98-1.33(m,6h),1.40-1.48(m,6h),3.22(brs,3h),3.43(brs,3h),3.61(brs,6h),3.84(brs,3h),4.20-4.43(m,1h),4.63-4.95(m,3h),5.06-5.16(m,2h),6.48-7.50(m,13h),9.27(d,j=7.5hz,1h).hrdartmsm/z.calcdforc44h48no10[(m+h)+]:750.32782.found:750.32788.工序(a):化合物16b的合成在氩气氛围下,将由化合物10(186mg,0.334mmol)、化合物11b(173mg,0.500mmol)、双(二亚苄基丙酮)钯(0)(19.6mg,34.1μmol)、1,1’-双(二苯基膦)二茂铁(18.3mg,33.0μmol)、na2co3(233mg,2.20mmol)、1,2-二甲氧基乙烷(15ml)、及脱气水(1ml)形成的混合物加热回流24小时。冷却后,减压蒸馏除去溶剂。向残渣中加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥,减压蒸馏除去二氯甲烷。用硅胶快速色谱(己烷∶乙酸乙酯=2∶1)纯化残渣,以淡褐色固体(55.3mg)的形式得到了化合物16b。收率94%。1hnmr(500mhz,cdcl3):δ1.02-1.31(m,12h),1.42(d,j=6.1hz,6h),3.23(brs,3h),3.42(s,3h),3.60(s,3h),3.82(s,3h),4.14-4.42(m,2h),4.66(sep,j=6.1hz,1h),4.69-4.93(m,2h),5.02-5.13(m,2h),6.57(brs,0.5h),6.66(brs,0.5h),6.75-6.90(m,3.5h),6.91(d,j=7.6hz,1h),6.96-7.02(m,0.5h),7.04(s,1h),7.20-7.33(m,2h),7.34-7.39(m,2h),7.42-7.47(m,2h),9.28(d,j=7.6hz,1h).hrdartmsm/z.calcdforc46h52no10[(m+h)+]:778.35912.found:778.35688.工序(a):化合物16c的合成在氩气氛围下,将由化合物10(1.23g,2.21mmol)、化合物11c(1.31mg,3.32mmol)、双(二亚苄基丙酮)钯(0)(123mg,0.213mmol)、1,1’-双(二苯基膦)二茂铁(132mg,0.237mmol)、na2co3(1.55g,14.6mmol)、1,2-二甲氧基乙烷(80ml)、及脱气水(6ml)形成的混合物加热回流24小时。冷却后,减压蒸馏除去溶剂。向残渣中加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥,减压蒸馏除去二氯甲烷。用硅胶快速色谱(己烷∶乙酸乙酯=5∶1)纯化残渣,以淡黄色固体(1.83g)的形式得到了化合物16c。收率100%。1hnmr(500mhz,cdcl3):δ1.03-1.30(m,6h),1.42(d,j=6.1hz,6h),3.23(brs,3h),3.42(s,3h),3.56(brs,3h),3.82(s,3h),4.23-4.40(m,1h),4.66(sep,j=6.1hz,1h),4.79-4.93(m,4h),5.04-5.14(m,2h),6.63(brs,0.5h),6.71-6.90(m,4.5h),6.91(d,j=7.6hz,1h),7.03(s,1h),7.20-7.38(m,9h),7.40-7.45(m,2h),9.27(d,j=7.6hz,1h).hrdartmsm/z.calcdforc50h52no10[(m+h)+]:826.35912.found:826.36040.化学式13工序(a):化合物13a的合成在氩气氛围下,用封管将由化合物16a(30.0mg,40.0μmol)、对甲苯磺酸一水合物(30.4mg,0.160mmol)、甲醇(1.5ml)形成的混合物在65℃下加热搅拌18小时。冷却后,减压蒸馏除去溶剂。向残渣中加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥,减压蒸馏除去二氯甲烷。用硅胶柱色谱(己烷∶乙酸乙酯=2∶1)纯化残渣,以淡黄色固体(25.0mg)的形式得到了化合物13a。收率93%。1hnmr(400mhz,cdcl3):δ1.35(d,j=6.1hz,3h),1.37(d,j=6.1hz,3h),1.43(d,j=6.1hz,6h),3.45(s,3h),3.49(s,3h),3.96(s,3h),4.55(sep,j=6.1hz,1h),4.69(sep,j=6.1hz,1h),5.17(s,2h),6.76(s,1h),6.93(s,1h),7.01(d,j=7.4hz,1h),7.09(s,1h),7.14(d,j=1.8hz,1h),7.15(d,j=8.2hz,1h),7.17(s,1h),7.20(dd,j=1.8and8.2hz,1h),7.27-7.33(m,1h),7.33-7.40(m,2h),7.40-7.45(m,2h),9.19(d,j=7.4hz,1h).hrdartmsm/z.calcdforc41h40no8[(m+h)+]:674.27539.found:674.27820.工序(a):化合物13b的合成在氩气氛围下,将由化合物16b(202mg,0.260mmol)、对甲苯磺酸一水合物(198mg,1.04mmol)、甲醇(10ml)形成的混合物加热回流18小时。冷却后,减压蒸馏除去溶剂。向残渣中加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥,减压蒸馏除去二氯甲烷。用硅胶柱色谱(己烷∶乙酸乙酯=2∶1)纯化残渣,以淡黄色固体(161mg)的形式得到了化合物13b。收率88%。1hnmr(400mhz,cdcl3):δ1.19(d,j=6.1hz,3h),1.20(d,j=6.1hz,3h),1.35(d,j=6.1hz,3h),1.36(d,j=6.1hz,3h),1.43(d,j=6.1hz,6h),3.44(s,3h),3.98(s,3h),4.02(sep,j=6.1hz,1h),4.54(sep,j=6.1hz,1h),4.69(sep,j=6.1hz,1h),5.15(s,2h),6.77(s,1h),6.94(s,1h),7.00(d,j=7.4hz,1h),7.08(s,1h),7.11-7.16(m,3h),7.17(s,1h),7.27-7.33(m,1h),7.33-7.39(m,2h),7.40-7.45(m,2h),9.18(d,j=7.4hz,1h).hrdartmsm/z.calcdforc43h44no8[(m+h)+]:702.30669.found:702.30518.工序(a):化合物13c的合成在氩气氛围下,将由化合物16c(1.77g,2.14mmol)、对甲苯磺酸一水合物(1.63g,8.57mmol)、甲醇(60ml)形成的混合物加热回流18小时。冷却后,减压蒸馏除去溶剂。向残渣中加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥,减压蒸馏除去二氯甲烷。将残渣用二氯甲烷-己烷进行重结晶,以白色固体(1.35g)的形式得到了化合物13c。收率84%。1hnmr(400mhz,cdcl3):δ1.34(d,j=6.1hz,3h),1.37(d,j=6.1hz,3h),1.43(d,j=6.1hz,3h),1.43(d,j=6.1hz,3h),3.44(s,3h),3.97(s,3h),4.54(sep,j=6.1hz,1h),4.69(sep,j=6.1hz,1h),4.76(d,j=12.4hz,1h),4.78(d,j=12.4hz,1h),5.19(s,2h),6.91(s,1h),6.98(s,1h),7.00(d,j=7.4hz,1h),7.08(s,1h),7.10(s,1h),7.12(s,1h),7.13-7.16(m,2h),7.23-7.39(m,8h),7.43-7.46(m,2h),9.20(d,j=7.4hz,1h).hrdartmsm/z.calcdforc47h44no8[(m+h)+]:750.30669.found:750.30535.化学式14工序(a):化合物24a的合成在氩气氛围下,向由化合物13a(405mg,0.601mmol)、乙酸乙酯(15ml)及乙醇(15ml)形成的溶液依次加入钯碳(pd:10%,80.8mg)及甲酸铵(1.14g,18.0mmol),加热回流1小时。冷却后,用硅藻土对反应混合物进行过滤。将滤液浓缩,用硅胶柱色谱(己烷∶乙酸乙酯=1∶1)纯化残渣,以淡黄色固体(328mg)的形式得到了化合物24a。收率93%。1hnmr(400mhz,cdcl3):δ1.36(d,j=6.0hz,3h),1.37(d,j=6.0hz,3h),1.43(d,j=6.0hz,6h),3.45(s,3h),3.51(s,3h),3.97(s,3h),4.57(sep,j=6.0hz,1h),4.70(sep,j=6.0hz,1h),5.95(brs,1h),6.71(s,1h),6.99(s,1h),6.99(d,j=7.3hz,1h),7.08(s,1h),7.15(d,j=8.1hz,1h),7.15(d,j=1.7hz,1h),7.16(s,1h),7.19(dd,j=1.7and8.1hz,1h),9.18(d,j=7.3hz,1h).hrdartmsm/z.calcdforc34h34no8[(m+h)+]:584.22844.found:584.22588.工序(a):化合物24b的合成在氩气氛围下,向由化合物13b(84.9mg,0.121mmol)、乙酸乙酯(5ml)及乙醇(5ml)形成的溶液依次加入钯碳(pd:10%,17mg)及甲酸铵(233mg,3.70mmol),加热回流30分钟。冷却后,用硅藻土对反应混合物进行过滤。将滤液浓缩,用硅胶柱色谱(丙酮)纯化残渣,以淡黄色固体(71.2mg)的形式得到了化合物24b。收率97%。1hnmr(400mhz,cdcl3):δ1.19(d,j=6.0hz,6h),1.35(d,j=6.0hz,3h),1.36(d,j=6.0hz,3h),1.44(d,j=6.0hz,6h),3.45(s,3h),3.98(s,3h),4.01(sep,j=6.0hz,1h),4.55(sep,j=6.0hz,1h),4.70(sep,j=6.0hz,1h),5.92(s,1h),6.68(s,1h),6.99(d,j=7.3hz,1h),6.99(s,1h),7.09(s,1h),7.12-7.17(m,3h),7.17(s,1h),9.18(d,j=7.3hz,1h).hrdartmsm/z.calcdforc36h38no8[(m+h)+]:612.25974.found:612.26035.工序(a):化合物24c的合成在氩气氛围下,向由化合物13c(200mg,0.267mmol)、乙酸乙酯(10ml)及乙醇(10ml)形成的溶液依次加入钯碳(pd:10%,40mg)及甲酸铵(505mg,8.01mmol),加热回流1小时。冷却后,用硅藻土对反应混合物进行过滤。将滤液浓缩,用硅胶柱色谱(丙酮)纯化残渣,以淡黄色固体(146mg)的形式得到了化合物24c。收率96%。1hnmr(500mhz,dmso-d6):δ1.21(d,j=6.0hz,3h),1.25(d,j=6.0hz,3h),1.31(d,j=6.0hz,6h),3.33(s,3h),3.89(s,3h),4.58(sep,j=6.0hz,1h),4.74(sep,j=6.0hz,1h),6.74(s,1h),6.84(s,1h),6.90(s,1h),7.08-7.12(m,2h),7.26(d,j=7.3hz,1h),7.27(d,j=8.4hz,1h),7.41(s,1h),9.04(brs,1h),9.08(d,j=7.3hz,1h),9.76(brs,1h).hrdartmsm/z.calcdforc33h32no8[(m+h)+]:570.21279.found:570.21008.化学式15工序(a):化合物25a的合成在氩气氛围下,向化合物24a(150mg,0.257mmol)的干燥丙酮(7ml)溶液依次加入3-(二甲基氨基)丙基氯盐酸盐(48.7mg,0.308mmol)、碳酸钾(178mg,1.29mmol),然后将混合物加热回流24小时。冷却后,加入饱和碳酸氢钠水溶液,在减压下浓缩混合物。向残渣中加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥后,减压蒸馏除去二氯甲烷。用chromatorexnh-dm1020柱色谱(己烷∶乙酸乙酯=3∶2)纯化残渣,以淡黄色固体(120mg)的形式得到了化合物25a。收率70%。1hnmr(400mhz,cdcl3):δ1.36(d,j=6.1hz,3h),1.36(d,j=6.1hz,3h),1.43(d,j=6.1hz,6h),2.01(quin,j=7.0hz,2h),2.25(s,6h),2.44(t,j=7.0hz,2h),3.45(s,3h),3.47(s,3h),3.97(s,3h),4.07(t,j=7.0hz,2h),4.56(sep,j=6.1hz,1h),4.69(sep,j=6.1hz,1h),6.74(s,1h),6.94(s,1h),7.00(d,j=7.4hz,1h),7.09(s,1h),7.16(d,j=1.8hz,1h),7.16(d,j=8.2hz,1h),7.18(s,1h),7.21(dd,j=1.8and8.2hz,1h),9.18(d,j=7.4hz,1h).hrdartmsm/z.calcdforc39h45n2o8[(m+h)+]:669.31759.found:669.31472.化学式16工序(a):化合物25b的合成在氩气氛围下,向化合物24b(30.0mg,49.0μmol)的干燥丙酮(2ml)溶液依次加入3-(二甲基氨基)丙基氯盐酸盐(9.3mg,58.8μmol)、碳酸钾(40.6mg,0.294mmol),然后将混合物加热回流21小时。冷却后,加入饱和碳酸氢钠水溶液,在减压下浓缩混合物。向残渣中加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥后,减压蒸馏除去二氯甲烷。用硅胶柱色谱(己烷∶乙酸乙酯=1∶1~二氯甲烷∶甲醇=1∶1)纯化残渣,以淡黄色固体(11.6mg)的形式得到了化合物25b。收率34%。1hnmr(400mhz,cdcl3):δ1.17(d,j=6.1hz,3h),1.17(d,j=6.1hz,3h),1.34(d,j=6.1hz,3h),1.35(d,j=6.1hz,3h),1.43(d,j=6.1hz,6h),2.00(quin,j=6.8hz,2h),2.25(s,6h),2.47(t,j=7.3hz,2h),3.44(s,3h),3.97(s,3h),3.97(sep,j=6.1hz,1h),4.07(t,j=6.4hz,2h),4.53(sep,j=6.1hz,1h),4.70(sep,j=6.1hz,1h),6.76(s,1h),6.96(s,1h),7.02(d,j=7.4hz,1h),7.10(s,1h),7.11(s,1h),7.14(s,2h),7.17(s,1h),9.22(d,j=7.4hz,1h).hrdartmsm/z.calcdforc41h49n2o8[(m+h)+]:697.34889.found:697.35000.化学式17工序(a):化合物25c的合成在氩气氛围下,向化合物24c(40.3mg,70.8μmol)的干燥丙酮(10ml)溶液依次加入2-(二甲基氨基)乙基氯盐酸盐(51.0mg,0.354mmol)、碘化钠(10.0mg,66.7μmol)、碳酸钾(147mg,1.06mmol),然后将混合物加热回流10小时。暂时冷却后,追加2-(二甲基氨基)乙基氯盐酸盐(51.0mg,0.354mmol)及碳酸钾(147mg,1.06mmol),进一步将混合物加热回流12小时。冷却后,用二氯甲烷稀释,用硅藻土过滤。将滤液在减压下浓缩后,用chromatorexnh-dm1020柱色谱(己烷∶乙酸乙酯=1∶2~乙酸乙酯)纯化残渣,以淡黄色固体(27.0mg)的形式得到了化合物25c。收率54%。1hnmr(500mhz,cdcl3):δ1.34(d,j=6.1hz,3h),1.34(d,j=6.1hz,3h),1.43(d,j=6.1hz,6h),2.28(s,6h),2.34(s,6h),2.55-2.65(m,2h),2.78(t,j=5.8hz,2h),3.44(s,3h),3.67(t,j=5.8hz,2h),3.96(s,3h),4.12(t,j=5.8hz,2h),4.53(sep,j=6.1hz,1h),4.70(sep,j=6.1hz,1h),6.76(s,1h),6.96(s,1h),7.02(d,j=7.4hz,1h),7.10(s,1h),7.11(d,j=1.8hz,1h),7.15(d,j=8.1hz,1h),7.17(s,1h),7.18(dd,j=1.8and8.1hz,1h),9.22(d,j=7.4hz,1h).hrdartmsm/z.calcdforc41h50n3o8[(m+h)+]:712.35979.found:712.36040.化学式18工序(a):化合物25d的合成在氩气氛围下,向化合物24c(146mg,0.256mmol)的干燥丙酮(25ml)溶液依次加入3-(二甲基氨基)丙基氯盐酸盐(194mg,1.23mmol)、碳酸钾(708mg,5.12mmol),然后将混合物加热回流48小时。冷却后,加入饱和碳酸氢钠水溶液,在减压下浓缩混合物。向残渣中加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥后,减压蒸馏除去二氯甲烷。将残渣用二氯甲烷-己烷进行重结晶,以淡褐色固体(134mg)的形式得到了化合物25d。收率71%。1hnmr(400mhz,cdcl3):δ1.34(d,j=6.1hz,3h),1.35(d,j=6.1hz,3h),1.43(d,j=6.1hz,6h),1.81(quin,j=6.9hz,2h),2.00(quin,j=6.8hz,2h),2.23(s,6h),2.25(s,6h),2.31-2.41(m,2h),2.47(t,j=7.3hz,2h),3.44(s,3h),3.62(t,j=6.3hz,2h),3.97(s,3h),4.08(t,j=6.5hz,2h),4.53(sep,j=6.1hz,1h),4.70(sep,j=6.1hz,1h),6.75(s,1h),6.96(s,1h),7.01(d,j=7.3hz,1h),7.09(s,1h),7.11(d,j=1.8hz,1h),7.14(d,j=8.1hz,1h),7.16(s,1h),7.18(dd,j=1.8and8.1hz,1h),9.22(d,j=7.3hz,1h).hrdartmsm/z.calcdforc43h54n3o8[(m+h)+]:740.39109.found:740.39380.化学式19工序(b):化合物26a的合成在氩气氛围下,在室温下向化合物25a(60.0mg,89.7μmol)的干燥二氯甲烷(4.0ml)溶液滴加氯化铝-硝基苯溶液(1.0m,540μl,0.540mmol),在该温度下搅拌18小时。在反应溶液中加入由碳酸氢钠(200mg,2.38mmol)、罗谢尔盐(440mg,1.56mmol)及水(7.0ml)形成的混合溶液,剧烈搅拌1小时后,减压蒸馏除去二氯甲烷及水。向残渣中加入水,在减压下共沸除去硝基苯。向残渣中加入水,使其充分悬浮后,抽滤滤取固体。在减压干燥后,以淡灰色固体(47.5mg)的形式得到了脱保护体。收率91%。1hnmr(500mhz,dmso-d6):δ1.85(quin,j=6.8hz,2h),2.14(s,6h),2.34(t,j=7.1hz,2h),3.39(s,3h),3.40(s,3h),3.87(s,3h),4.04(t,j=6.5hz,2h),6.78(s,1h),7.00-7.04(m,2h),7.08(s,1h),7.19(s,1h),7.21(s,1h),7.22(d,j=7.3hz,1h),7.24(d,j=8.7hz,1h),9.00(d,j=7.3hz,1h),9.39(brs,1h).向脱保护体(20.0mg,34.2μmol)的二氯甲烷(1.0ml)悬浮液加入三氟乙酸(1.0ml),在室温下搅拌30分钟。减压蒸馏除去溶剂后,用柱色谱(sephadexlh-20,含有0.1%v/v三氟乙酸的甲醇)纯化残渣,以褐色固体(21.6mg)的形式得到了化合物26a。收率90%。1hnmr(500mhz,dmso-d6):δ2.13(quin,j=6.9hz,2h),2.82(s,3h),2.83(s,3h),3.21(quin,j=5.1hz,2h),3.40(s,3h),3.40(s,3h),3.88(s,3h),4.13(t,j=6.1hz,2h),6.81(s,1h),7.00-7.04(m,2h),7.17(s,1h),7.19(s,1h),7.21(s,1h),7.25(d,j=7.3hz,1h),7.25(d,j=9.2hz,1h),9.01(d,j=7.3hz,1h),9.43(brs,2h),9.98(brs,1h).hrdartmsm/z.calcdforc33h33n2o8[(m-cf3coo)+]:585.22369.found:585.22208.化学式20工序(b):化合物26b的合成在氩气氛围下,在室温下向化合物25b(20.0mg,28.7μmol)的干燥二氯甲烷(5.0ml)溶液滴加氯化铝-硝基苯溶液(1.0m,218μl,0.218mmol),并在该温度下搅拌48小时。在反应溶液中加入由碳酸氢钠(55.0mg,0.654mmol)、罗谢尔盐(185mg,0.654mmol)及水(3.3ml)形成的混合溶液,剧烈搅拌1小时后,减压蒸馏除去二氯甲烷及水。向残渣中加入水,在减压下共沸除去硝基苯后,进行减压干燥。向残渣加入二氯甲烷(2.0ml)及三氟乙酸(2.0ml),然后减压蒸馏除去溶剂。用柱色谱(sephadexlh-20,含有0.1%v/v三氟乙酸的水~含有0.1%v/v三氟乙酸的水∶甲醇=1∶1~含有0.1%v/v三氟乙酸的甲醇)纯化残渣,以褐色固体(19.3mg)的形式得到了化合物26b。收率98%。1hnmr(500mhz,dmso-d6):δ2.12(quin,j=5.5hz,2h),2.82(s,6h),3.28(t,j=7.8hz,2h),3.37(s,3h),3.90(s,3h),4.12(t,j=5.9hz,2h),6.81(s,1h),6.92(s,1h),6.94(d,j=2.0hz,1h),6.95(dd,j=2.0and8.0hz,1h),7.11(s,1h),7.18(s,1h),7.21(d,j=7.4hz,1h),7.23(d,j=8.0hz,1h),9.03(d,j=7.4hz,1h),9.04(brs,1h),9.41(brs,1h),9.53(brs,1h),9.98(brs,1h).hrdartmsm/z.calcdforc32h31n2o8[(m-cf3coo)+]:571.20804.found:571.20608.化学式21工序(b):化合物26c的合成在氩气氛围下,在室温下向化合物25c(13.4mg,18.8μmol)的干燥二氯甲烷(1.0ml)溶液滴加氯化铝-硝基苯溶液(1.0m,120μl,0.120mmol),并在该温度下搅拌48小时。在反应溶液中加入由碳酸氢钠(30.3mg,0.361mmol)、罗谢尔盐(102mg,0.361mmol)及水(0.7ml)形成的混合溶液,剧烈搅拌1小时后,减压蒸馏除去二氯甲烷及水。向残渣中加入水,在减压下共沸除去硝基苯。向残渣中加入水,使其充分悬浮后,抽滤滤取固体。在得到的固体的二氯甲烷(1.0ml)悬浮液中加入三氟乙酸(1.0ml),减压蒸馏除去溶剂。用柱色谱(sephadexlh-20,含有0.1%v/v三氟乙酸的甲醇)纯化残渣,以褐色固体(15.0mg)的形式得到了化合物26c。收率93%。1hnmr(500mhz,dmso-d6):δ2.82(s,6h),2.88(s,6h),3.40(s,3h),3.54(t,j=4.8hz,2h),3.90(s,3h),3.90-3.98(m,2h),4.44(t,j=5.0hz,2h),6.87(s,1h),7.01(d,j=2.1hz,1h),7.03(dd,j=2.1and8.1hz,1h),7.14(s,1h),7.21(s,1h),7.25(d,j=8.1hz,1h),7.27(d,j=7.4hz,1h),7.35(s,1h),9.02(d,j=7.4hz,1h),9.48(brs,1h),10.04(brs,3h).hrdartmsm/z.calcdforc35h38n3o8[(m-2cf3coo-h)+]:628.26589.found:628.26746.化学式22工序(b):化合物26d的合成在氩气氛围下,在室温下向化合物25d(90.2mg,0.122mmol)的干燥二氯甲烷(19.5ml)溶液滴加氯化铝-硝基苯溶液(1.0m,780μl,0.780mmol),并在该温度下搅拌84.5小时。在反应溶液中加入由碳酸氢钠(166mg,1.97mmol)、罗谢尔盐(557mg,1.97mmol)及水(3.9ml)形成的混合溶液,剧烈搅拌1小时后,减压蒸馏除去二氯甲烷及水。向残渣中加入水,在减压下共沸除去硝基苯。向残渣中加入水,使其充分悬浮后,抽滤滤取固体。在减压干燥后,以褐色固体(80.0mg)的形式得到了脱保护体。收率100%。1hnmr(500mhz,dmso-d6):δ1.63-1.70(m,2h),1.82-1.89(m,2h),2.14(s,6h),2.17(s,6h),2.23-2.30(m,2h),2.36-2.43(m,2h),3.39(s,3h),3.53-3.60(m,2h),3.88(s,3h),4.01-4.08(m,2h),6.76(s,1h),6.97-7.02(m,2h),7.07(s,1h),7.15(s,1h),7.19(s,1h),7.20-7.24(m,2h),8.99(d,j=7.3hz,1h).向脱保护体(78.3mg,0.106mmol)的二氯甲烷(3.0ml)悬浮液加入三氟乙酸(3.0ml),在室温下搅拌30分钟。减压蒸馏除去溶剂后,用柱色谱(sephadexlh-20,含有0.1%v/v三氟乙酸的甲醇)纯化残渣,以褐色固体(94.2mg)的形式得到了化合物26d。收率100%。1hnmr(500mhz,dmso-d6):δ1.92-2.00(m,2h),2.09-2.17(m,2h),2.81(s,6h),2.83(s,6h),3.06-3.13(m,2h),3.21(t,j=7.2hz,2h),3.40(s,3h),3.59-3.70(m,2h),3.90(s,3h),4.13(t,j=6.2hz,2h),6.79(s,1h),7.01(d,j=2.0hz,1h),7.02(dd,j=2.0and8.0hz,1h),7.17(s,1h),7.19(s,1h),7.21(s,1h),7.25(d,j=7.4hz,1h),7.26(d,j=8.0hz,1h),9.00(d,j=7.4hz,1h),9.50(brs,1h),9.84(brs,1h),9.87(brs,1h),10.05(brs,1h).hrdartmsm/z.calcdforc37h42n3o8[(m-2cf3coo-h)+]:656.29719.found:656.29442.[氮杂片螺素n衍生物的合成法]化学式23化学式24工序(a):化合物29a的合成在氩气氛围下,在室温下向市售的、或者通过本身公知的方法或以其为基准的方法合成的化合物28a(4.33g,28.3mmol)的干燥四氢呋喃(120ml)溶液滴加二碳酸二叔丁酯(7.20ml,31.2mmol),加热回流2小时。冷却后,减压蒸馏除去溶剂。将残渣用乙醚进行重结晶,以淡褐色固体(6.68g)的形式得到了化合物29a。收率93%。1hnmr(400mhz,cdcl3):δ1.51(s,9h),3.84(s,3h),3.87(s,3h),6.47(brs,1h),6.72(dd,j=2.2and8.6hz,1h),6.78(d,j=8.6hz,1h),7.18(br,1h).hrdartmsm/z.calcdforc13h20no4[(m+h)+]:254.1392.found:254.1399.工序(a):化合物29b的合成按照化合物28a的合成中记载的方法,使市售的、或者通过本身公知的方法或以其为基准的方法合成的化合物28b(6.87g,30.0mmol)发生反应。用硅胶快速色谱(己烷∶乙酸乙酯=4∶1)纯化后,以淡褐色固体(3.10g)的形式得到了化合物29b。收率31%。1hnmr(400mhz,cdcl3):δ1.50(s,9h),3.83(s,3h),5.12(s,2h),6.37(brs,1h),6.81(s,2h),7.12(brs,1h),7.27-7.32(m,1h),7.33-7.39(m,2h),7.42-7.47(m,2h).hrdartmsm/z.calcdforc19h23brno4(m+):329.1627.found:329.1639.工序(b):化合物30a的合成在氩气氛围下,在0℃下向化合物29a(333mg,1.31mmol)的干燥四氢呋喃(5ml)溶液加入n-溴代琥珀酰亚胺(254mg,1.43mmol)。在0℃下搅拌1小时,然后加入水,升温至室温。用二氯甲烷进行萃取后,用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥,减压蒸馏除去二氯甲烷。用硅胶柱色谱(己烷∶乙酸乙酯=10∶1)纯化残渣,以淡黄色固体(377mg)的形式得到了化合物30a。收率87%。1hnmr(300mhz,cdcl3):δ1.53(s,9h),3.84(s,3h),3.91(s,3h),6.79(brs,1h),6.97(s,1h),7.81(brs,1h).hrdartmsm/z.calcdforc13h18brno4(m+):331.0419.found:331.0425.工序(b):化合物30b的合成按照化合物30a的合成中记载的方法使化合物29b(3.00g,9.11mmol)发生反应。用硅胶快速色谱(己烷∶乙酸乙酯=3∶1)纯化后,以淡黄色固体(3.38g)的形式得到了化合物30b。收率91%。1hnmr(400mhz,cdcl3):δ1.53(s,9h),3.82(s,3h),5.13(s,2h),6.75(brs,1h),6.99(s,1h),7.28-7.33(m,1h),7.34-7.39(m,2h),7.45-7.50(m,2h),7.90(brs,1h).hrdartmsm/z.calcdforc19h22brno4(m+):407.0732.found:409.0743.工序(c):化合物12a的合成在氩气氛围下,在室温下向由化合物30a(377mg,1.13mmol)、[1,1’-双(二苯基膦)二茂铁]二氯化钯(ii)二氯甲烷加合物(46.9mg,57.4μmol)及1,4-二烷(6.0ml)形成的溶液依次加入三乙胺(640μl,4.54mmol)及频哪醇硼烷(490μl,3.40mmol),加热回流2.5小时。冷却后,加入饱和氯化铵水溶液,蒸馏除去溶剂。向残渣中加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥。减压蒸馏除去二氯甲烷。用硅胶柱色谱(己烷∶乙酸乙酯=50∶1)纯化残渣,以淡黄色固体(282mg)的形式得到了化合物12a。收率65%。1hnmr(400mhz,cdcl3):δ1.35(s,12h),1.52(s,9h),3.88(s,3h),3.94(s,3h),7.15(s,1h),7.92(brs,1h),8.67(brs,1h).hrdartmsm/z.calcdforc19h30bno6(m+):379.2166.found:379.2169.工序(c):化合物12b的合成按照化合物12a的合成中记载的方法使化合物30b(3.38g,8.28mmol)发生反应。用硅胶快速色谱(己烷∶乙酸乙酯=5∶1)纯化后,以淡黄色固体(2.72g)的形式得到了化合物12b。收率72%。1hnmr(300mhz,cdcl3):δ1.35(s,12h),1.52(s,9h),3.86(s,3h),5.19(s,2h),7.18(s,1h),7.27-7.32(m,1h),7.33-7.39(m,2h),7.47-7.51(m,2h),8.02(brs,1h),8.63(brs,1h).hrdartmsm/z.calcdforc25h34bno6(m+):455.2479.found:455.2485.化学式25工序(a):化合物27a的合成在氩气氛围下,用封管将由化合物10(100mg,0.180mmol)、化合物12a(114mg,0.301mmol)、pd(pph3)4(23.2mg,20.1μmol)、na2co3(141mg,1.32mmol)、1,2-二甲氧基乙烷(5.0ml)、及脱气水(0.5ml)构成的混合物在85℃下加热搅拌24小时。冷却后,减压蒸馏除去溶剂。向残渣中加入水,用二氯甲烷进行萃取。用水及饱和食盐水依次清洗萃取液,用无水硫酸钠干燥,减压蒸馏除去二氯甲烷。用硅胶柱色谱(己烷∶乙酸乙酯=3∶1~2∶1~1∶1~乙酸乙酯)纯化残渣,以淡黄色半固体(115mg)的形式得到了化合物27a。收率88%。1hnmr(500mhz,dmso-d6):δ1.02(d,j=6.1hz,1.72h),1.09(d,j=6.1hz,1.72h),1.11(d,j=6.1hz,1.28h),1.16(d,j=6.1hz,1.28h),1.30(d,j=6.1hz,6h),1.37(s,9h),3.29(s,1.28h),3.29(s,1.72h),3.48(s,1.72h),3.53(s,1.28h),3.53(s,1.72h),3.56(s,1.28h),3.69(s,1.72h),3.70(s,1.28h),3.71(s,1.28h),3.74(s,1.72h),4.26(sep,j=6.1hz,0.57h),4.43(sep,j=6.1hz,0.43h),4.70(sep,j=6.1hz,1h),6.52(s,0.57h),6.71(s,0.43h),6.85-7.17(m,4h),7.18(d,j=7.6hz,0.57h),7.18(d,j=7.6hz,0.43h),7.34(s,1h),7.35(brs,0.43h),7.44(brs,0.57h),7.60(brs,0.43h),7.85(brs,0.57h),9.20(d,j=7.6hz,0.57h),9.23(d,j=7.6hz,0.43h).hrdartmsm/z.calcdforc41h49n2o10[(m+h)+]:729.3387.found:729.3358.工序(a):化合物27b的合成按照化合物27a的合成中记载的方法使化合物10(100mg,0.180mmol)及化合物12b(137mg,0.302mmol)发生反应。用硅胶柱色谱(己烷∶乙酸乙酯=3∶1~2∶1~1∶1~1∶2)纯化后,以淡黄色半固体(131mg)的形式得到了化合物27b。收率91%。1hnmr(500mhz,dmso-d6):δ1.02(d,j=6.1hz,1.67h),1.10(d,j=6.1hz,1.67h),1.13(d,j=6.1hz,1.33h),1.17(d,j=6.1hz,1.33h),1.30(d,j=6.1hz,6h),1.37(s,9h),3.29(s,1.33h),3.30(s,1.67h),3.49(s,1.67h),3.53(s,1.33h),3.54(s,1.67h),3.57(s,1.33h),3.72(s,1.33h),3.74(s,1.67h),4.28(sep,j=6.1hz,0.56h),4.44(sep,j=6.1hz,0.44h),4.71(sep,j=6.1hz,1h),4.92-5.01(m,2h),6.56(s,0.56h),6.75(s,0.44h),6.86-7.10(m,4h),7.18(d,j=7.6hz,0.56h),7.19(d,j=7.6hz,0.44h),7.23-7.47(m,7h),7.64(brs,0.44h),7.89(brs,0.56h),9.21(d,j=7.6hz,0.56h),9.23(d,j=7.6hz,0.44h).hrdartmsm/z.calcdforc47h53n2o10[(m+h)+]:805.3700.found:805.3715.化学式26工序(a):化合物14a的合成在氩气氛围下,在封管中向化合物27a(50.0mg,68.6μmol)及四氢呋喃(3.0ml)溶液滴加四丁基氟化铵四氢呋喃溶液(1.0m,343μl,0.343mmol),然后在85℃下加热搅拌19小时。冷却后,加入水,蒸馏除去溶剂。对析出的固体进行抽滤,用水及甲醇依次清洗回收的固体,进行真空干燥,由此,以淡褐色固体(33.6mg)的形式得到了化合物14a。收率82%。1hnmr(500mhz,cdcl3):δ1.35(d,j=6.1hz,6h),1.43(d,j=6.1hz,6h),3.45(s,3h),3.47(s,3h),3.96(s,3h),3.99(s,3h),4.54(sep,j=6.1hz,1h),4.69(sep,j=6.1hz,1h),6.90(s,1h),6.94(d,j=7.4hz,1h),6.94(s,1h),7.10(s,1h),7.16(d,j=8.1hz,1h),7.17(d,j=1.7hz,1h),7.20(s,1h),7.22(dd,j=1.7and8.1hz,1h),9.59(d,j=7.4hz,1h),10.588(s,1h).hrdartmsm/z.calcdforc35h37n2o7[(m+h)+]:597.2601.found:597.2612.工序(a):化合物14b的合成按照化合物14a的合成中记载的方法使化合物27b(100mg,0.180mmol)发生反应。其结果是以淡褐色固体(78.2mg)的形式得到了化合物14b。收率85%。1hnmr(500mhz,cdcl3):δ1.34(d,j=6.1hz,3h),1.34(d,j=6.1hz,3h),1.43(d,j=6.1hz,3h),1.44(d,j=6.1hz,3h),3.42(s,3h),3.46(s,3h),3.93(s,3h),4.53(sep,j=6.1hz,1h),4.68(sep,j=6.1hz,1h),5.22(s,2h),6.76-6.88(m,2h),7.00-7.30(m,9h),7.43-7.47(m,2h),9.44(brs,1h),11.05(brs,1h).hrdartmsm/z.calcdforc41h41n2o7[(m+h)+]:673.2914.found:673.2939.化学式27工序(a):化合物8a的合成在氩气氛围下,在-78℃下向化合物14a(20.0mg,33.5μmol)的干燥二氯甲烷(12ml)溶液滴加三氯化硼庚烷溶液(1.0m,235μl,0.235mmol),并在该温度下搅拌30分钟。然后,升温至室温,并在该温度下搅拌3小时。向反应溶液加入饱和碳酸氢钠水溶液,减压蒸馏除去溶剂。对析出的固体进行抽滤,用水清洗回收的固体,进行真空干燥。用硅胶柱色谱(丙酮)纯化得到的固体,以淡褐色固体(14.2mg)的形式得到了化合物8a。收率71%。1hnmr(500mhz,dmso-d6):δ3.34(s,3h),3.39(s,3h),3.78(s,3h),3.86(s,3h),6.87(s,1h),6.98-7.02(m,3h),7.04(d,j=7.4hz,1h),7.14(s,1h),7.16(s,1h),7.23(d,j=8.7hz,1h),9.34(s,1h),9.38(d,j=7.4hz,1h),9.73(brs,1h),11.34(s,1h).hrfabmsm/z.calcdforc29h25n2o7[(m+h)+]:513.1662.found:513.1676.工序(a):化合物8的合成按照化合物8a的合成中记载的方法使化合物14b(30.0mg,44.6μmol)及三氯化硼庚烷溶液(1.0m,445μl,0.445mmol)发生反应。用硅胶柱色谱(丙酮)纯化得到的粗产物的固体,以淡褐色固体(18.6mg)的形式得到了化合物8。收率84%。1hnmr(500mhz,cdcl3):δ3.36(s,3h),3.39(s,3h),3.86(s,3h),6.84(s,1h),6.89(s,1h),6.98-7.03(m,3h),7.13(s,1h),7.15(s,1h),7.23(d,j=8.2hz,1h),9.33(s,1h),9.37(d,j=7.4hz,1h),9.49(s,1h),9.71(s,1h),11.27(s,1h).hrdartmsm/z.calcdforc28h23n2o7[(m+h)+]:499.1505.found:499.1502.化学式28工序(a):化合物31的合成在氩气氛围下,向由化合物14b(300mg,0.446mmol)、乙酸乙酯(45ml)及乙醇(45ml)形成的溶液依次加入钯碳(pd:10%,60.0mg)及甲酸铵(844mg,13.4mmol),加热回流30分钟。冷却后,用硅藻土对反应混合物进行过滤。将滤液浓缩,用硅胶柱色谱(丙酮)纯化残渣,以淡黄色固体(258mg)的形式得到了化合物31。收率99%。1hnmr(300mhz,cdcl3):δ1.35(d,j=6.1hz,3h),1.36(d,j=6.1hz,3h),1.43(d,j=6.1hz,6h),3.44(s,3h),3.50(s,3h),3.97(s,3h),4.55(sep,j=6.1hz,1h),4.69(sep,j=6.1hz,1h),5.84(brs,1h),6.87(s,2h),6.94(d,j=7.4hz,1h),7.09(s,1h),7.14-7.24(m,4h),9.34(brs,1h),9.53(d,j=7.4hz,1h).化学式29工序(a):化合物32的合成在氩气氛围下,向化合物31(70.0mg,0.120mmol)的干燥丙酮(15ml)溶液依次加入3-(二甲基氨基)丙基氯盐酸盐(22.8mg,0.144mmol)、碳酸钾(83.0mg,0.601mmol),然后将混合物加热回流18小时。暂时冷却后,追加3-(二甲基氨基)丙基氯盐酸盐(22.8mg,0.144mmol)及碳酸钾(83.0mg,0.601mmol),进一步将混合物加热回流5小时。冷却后,用丙酮稀释,用硅藻土过滤。将滤液在减压下浓缩后,用chromatorexdiol柱色谱(乙酸乙酯~二氯甲烷∶甲醇=10∶1)及chromatorex纯化残渣,以淡黄色固体(39.7mg)的形式得到了化合物32。收率50%。1hnmr(500mhz,cdcl3):δ1.34(d,j=6.1hz,6h),1.43(d,j=6.1hz,6h),2.08(quin,j=6.9hz,2h),2.28(s,6h),2.51(t,j=7.2hz,2h),3.45(s,6h),3.96(s,3h),4.17(t,j=6.7hz,2h),4.54(sep,j=6.1hz,1h),4.68(sep,j=6.1hz,1h),6.88(s,1h),6.93(d,j=7.4hz,1h),7.00(s,1h),7.09(s,1h),7.14(d,j=8.2hz,1h),7.16(d,j=1.7hz,1h),7.19(s,1h),7.21(dd,j=1.7and8.2hz,1h),9.59(d,j=7.4hz,1h),10.58(brs,1h).化学式30工序(b):化合物33的合成在氩气氛围下,在室温下向化合物32(15.2mg,22.8μmol)的干燥二氯甲烷(5.0ml)溶液滴加氯化铝-硝基苯溶液(1.0m,145μl,0.145mmol),并在该温度下搅拌72小时。向反应溶液加入由碳酸氢钠(36.7mg,0.437mmol)、罗谢尔盐(123mg,0.437mmol)及水(2.1ml)形成的混合溶液,剧烈搅拌1小时后,减压蒸馏除去二氯甲烷及水。向残渣中加入水,在减压下共沸除去硝基苯,然后进行减压干燥。向残渣加入二氯甲烷(1.0ml)及三氟乙酸(1.0ml)后,减压蒸馏除去溶剂。用柱色谱(sephadexlh-20,含有0.1%v/v三氟乙酸的水~含有0.1%v/v三氟乙酸的水∶甲醇=1∶1~含有0.1%v/v三氟乙酸的甲醇)进行纯化,褐色固体(15.8mg)的形式得到了化合物33。收率100%。1hnmr(500mhz,dmso-d6):δ2.11-2.18(m,2h),2.82(s,3h),2.83(s,3h),3.20-3.26(m,2h),3.36(s,3h),3.39(s,3h),3.86(s,3h),4.05(t,j=5.9hz,2h),6.90(s,1h),6.98-7.02(m,3h),7.05(d,j=7.5hz,1h),7.14(s,1h),7.16(s,1h),7.23(d,j=8.7hz,1h),9.37(d,j=7.5hz,1h),9.60(brs,1h),9.75(brs,2h),11.37(brs,1h).试验例1使用了导入egfr基因的baf3细胞的增殖抑制活性的评价对于药剂在酶水平上的活性而言,由于该药剂的细胞膜通透性、脱靶抑制等等,未必与细胞水平的活性直接相关。因此,作为激酶抑制剂在细胞水平上的评价方法,进行了确立了有效性的使用了转导baf3细胞的egfr抑制活性评价。baf3细胞(小鼠前淋巴细胞)仅能够在il-3(interleukin-3)存在下增殖。另一方面,转导了egfr基因的baf3细胞即使不存在il-3,也可以依赖于egfr进行增殖。向其中加入egfr抑制剂时,baf3细胞变得无法增殖,但加入il-3时,即使存在egfr抑制剂,也能够增殖。利用这样的性质,可以评价egfr抑制剂的活性、特异性。此次,进行了片螺素类(化合物26a、26b、26c、26d)及氮杂片螺素类(化合物8、8a、33)对于转导了双重突变egfr基因的baf3(l858r/t790m)、baf3(delexon19/t790m)及转导了三重突变egfr基因的baf3(l858r/t790m/c797s)、baf3(delexon19/t790m/c797s)的增殖抑制活性评价。作为阳性对照,使用了第一代抑制剂吉非替尼、第二代抑制剂阿法替尼及第三代抑制剂奥希替尼。具体的试验方法如下所述。使用neon电穿孔系统(thermofisherscientific公司)对baf3细胞2×106细胞导入2μg的各种pbabe-egfr质粒,用包含10ng/mlil-3、100u/ml青霉素、100μg/ml链霉素、以及10%胎牛血清的rpmi-1640培养基在5%二氧化碳/95%空气氛围中于37℃进行培养。2天后,添加1μg/ml嘌呤霉素,培养了4天。用pbs清洗细胞后,通过不含有il-3的培养基进行培养,使用了不依赖il-3进行增殖的细胞。将该细胞以3000细胞/孔接种于96孔微孔板(thermofisherscientific公司),并加入各种浓度的试验化合物,在150μl的培养基中培养4天。在孔中添加15μl的5mg/mlmtt(噻唑蓝溴化四唑)溶液,在37℃下温育4小时,然后添加100μl的20%sds,进一步温育过夜。使用酶标仪(beckmancoulter公司)测定570nm的吸光度,计算出50%细胞增殖抑制浓度(ic50)。将结果示于下表。[表1]使用了导入egfr(l858r/t790m)基因的baf3细胞的增殖抑制活性[表2]使用了导入egfr(delexon19/t790m)基因的baf3细胞的增殖抑制活性[表3]使用了导入egfr(t790m/l858r/c797s)基因的baf3细胞的增殖抑制活性[表4]使用了导入egfr(delexon19/t790m/c797s)基因的baf3细胞的增殖抑制活性通过试验例1可知以下结果。(1)对于具有双重突变egfr的baf3(l858r/t790m),片螺素类显示出与阿法替尼相同程度的活性,而且氮杂片螺素类显示出与奥希替尼相当的活性。然而,两者均治疗浓度范围(在该情况下,定义为-il3与+il3之差)狭窄,未确认到相对于阿法替尼、奥希替尼的优势。(2)对于具有双重突变egfr的baf3(delexon19/t790m),片螺素类显示出与阿法替尼相同程度的活性。然而,两者均治疗浓度范围狭窄,未确认到相对于阿法替尼、奥希替尼的优势。(3)另一方面,对于具有三重突变egfr的baf3(l858r/t790m/c797s)、baf3(delexon19/t790m/c797s),阿法替尼、奥希替尼的活性大幅减弱(特别是奥希替尼)。然而,在作为可逆性抑制剂的片螺素类、氮杂片螺素类的情况下,没有确认到这样的减弱,反而确认到了活性的提高。其活性超过了阿法替尼、奥希替尼的活性。而且,在各化合物中确认到明显的治疗浓度范围扩大,明确地表明egfr抑制与细胞增殖抑制相关。试验例2化合物26d对导入野生型egfr基因的baf3细胞的增殖抑制活性的评价使用表达不具有活性型突变的野生型egfr的baf3细胞研究了化合物26d对具有活性型突变的egfr的选择性。使用neon电穿孔系统(thermofisherscientific公司)对baf3细胞2×106细胞导入2μg的pbabe-egfr野生型,用包含10ng/mlil-3、100u/ml青霉素、100μg/ml链霉素、以及10%胎牛血清的rpmi-1640培养基在5%二氧化碳/95%空气氛围中、37℃下进行培养。2天后,添加1μg/ml嘌呤霉素,对耐药性细胞进行选择。用pbs将该细胞清洗2次后,以30000细胞/孔接种于96孔微孔板(thermofisherscientific公司),加入10ng/mlegf或il-3及各种浓度的化合物26d,在150μl的培养基中培养4天。在孔中添加15μl的5mg/mlmtt(噻唑蓝溴化四唑)溶液,在37℃下温育4小时,然后添加100μl的20%sds,进一步温育过夜。用酶标仪(beckmancoulter公司)测定570nm的吸光度,计算出50%细胞增殖抑制浓度(ic50)。将结果使用下表。[表5]化合物26d对导入野生型egfr基因的baf3细胞的增殖抑制活性对于具有活性型突变的baf3(delexon19/t790m/c797s),在表达野生型egfr的baf3细胞的情况下,化合物26d的效果低,为与il-3存在下的增殖抑制活性相同程度。因此,化合物26d对于具有三重突变的egfr显示出选择性。试验例3使用了导入egfr基因的肺腺癌细胞的增殖抑制活性的评价研究了化合物26d对肺腺癌细胞株的增殖抑制活性。使用viafect转染试剂(promega公司)将2μg的pbabe-egfr导入egfr基因中缺失外显子19(delexon19)的肺腺癌细胞株pc-9,用包含100u/ml青霉素、100μg/ml链霉素、以及10%胎牛血清的rpmi-1640培养基在5%二氧化碳/95%空气氛围中、37℃下进行培养。2天后,添加2μg/ml嘌呤霉素,对耐药性细胞进行选择。将这些细胞及表达野生型egfr的a549细胞分别以3000细胞/孔、7500细胞/孔接种于96孔微孔板(thermofisherscientific公司),加入各种浓度的试验化合物,在150μl的培养基中培养4天。在孔中添加15μl的5mg/mlmtt(噻唑蓝溴化四唑)溶液,在37℃下温育4小时后,添加100μl的20%sds,进一步温育过夜。使用酶标仪(beckmancoulter公司)测定570nm的吸光度,计算出50%细胞增殖抑制浓度(ic50)。将结果示于下表。[表6]化合物26d对导入egfr基因的肺腺癌pc-9细胞、egfr野生型a549的增殖抑制活性ic50(nm)pc-9658.5delexon19/t790m493.9l858r/t790m719.0delexon19/t790m/c797s739.9l858r/t790m/c797s595.7a5492861.7向pc-9导入具有活性型突变的egfr基因,研究了化合物26d对肺腺癌细胞株的增殖抑制活性,结果是,无论有无耐药性突变(t790m、c797s突变),化合物26d均以基本相同程度的浓度抑制了增殖。对于表达不具有突变的野生型egfr的肺腺癌细胞株a549,以高于具有活性型突变的pc-9细胞的浓度抑制了增殖。因此,化合物26d对于具有活性型突变的egfr显示出选择性。试验例4化合物26d对egfr信号的抑制的评价egfr酪氨酸激酶抑制剂抑制egfr的自磷酸化及下游的信号。因此,对于化合物26d是否在细胞水平上抑制egfr(delexon19/t790m/c797s)的自磷酸化及下游的信号进行了研究。以100000细胞/孔将肺腺癌细胞pc-9及分别表达egfr(delexon19/t790m)、egfr(delexon19/t790m/c797s)的pc-9细胞接种于6孔板,用包含100μg/ml链霉素及10%胎牛血清的rpmi-1640培养基在5%二氧化碳/95%空气氛围中、37℃下进行培养。次日,添加奥希替尼或化合物26d,培养4小时。用冰冷的pbs清洗后,悬浮于含有5mm钒酸钠、5mm氟化钠、proteaseinhibitorcocktail(roche公司)的100μl的ripa缓冲液(20mmtris-hclph7.5、150mmnacl、1%np40、1%脱氧胆酸钠、0.1%sds),在冰上温育30分钟,以15000转离心30分钟,将上清作为细胞提取液。利用sds聚丙烯酰胺凝胶对细胞提取液进行分离,转印至pvdf膜。pvdf膜用5%脱脂乳封闭30分钟,使用抗磷酸化egfr抗体、抗egfr抗体、抗磷酸化akt抗体、抗akt抗体、抗磷酸化erk抗体(cellsignalingtechnologies公司)、抗erk抗体(santacruz)、抗肌动蛋白抗体(sigma公司)、辣根过氧化物酶结合抗小鼠igg、辣根过氧化物酶结合抗兔igg进行了蛋白质印迹试验(westernblot)。检测使用了eclprimedetectionreagent、las3000(gehealthcare公司)。将结果示于图2。化合物26d抑制了pc-9细胞的egfr的磷酸化、存在于egfr信号的下游的erk的磷酸化。另一方面,未观察到egfr信号的下游的akt的磷酸化的抑制。无论有无耐药性突变(t790m、c797s突变),这些蛋白质的磷酸化以基本相同程度的浓度被抑制。因此,化合物26d显示出抑制现有的药剂耐药性突变细胞的egfr信号的结果。试验例5化合物26d在小鼠异种移植模型中的抗肿瘤效果的评价为调查化合物26d在动物水平上的治疗效果,使用小鼠异种移植模型进行了研究。将表达egfr(delexon19/t790m/c797s)的pc-9制备为5×107细胞,与基质胶(matrigel)(becton,dickinson公司)以1∶1混合。将其100μl注射于balb/c裸鼠(charlesriverjapan公司)的皮下。从观察到形成肿瘤起,每天腹腔内给药0.2mg的化合物26d,测定肿瘤直径。通过长轴(mm)×短轴(mm)2/2计算出肿瘤体积。将结果示于图3及4。化合物26d抑制了移植至裸鼠皮下的表达egfr(delexon19/t790m/c797s)基因的pc-9细胞的肿瘤的增大。因此,化合物26d显示出在动物水平上抑制对吉非替尼、奥希替尼呈现耐药性的人肺癌移植肿瘤的增大的结果。由于在给药期间中未观察到体重减少,因此可以认为化合物26d不具有明显的毒性。如上所述可知,按照基于结构的药剂设计(structure-baseddrugdesign)创制出的本发明的化合物在细胞水平上强烈地抑制egfr(l858r/t790m/c797s)、egfr(delexon19/t790m/c797s)。与已批准的现有egfr-tki(吉非替尼、阿法替尼、奥希替尼)相比,其活性非常强,可以认为作为用于目前尚无治疗方法的具有三重突变egfr的非小细胞肺癌治疗药(第四代egfr-tki)开发的先导化合物是有用的。工业实用性本发明的化合物对c797s突变型耐药性egfr(特别是c797s突变型三次耐药性egfr)具有特异性的酪氨酸激酶抑制活性,作为c797s突变型耐药性egfr(特别是c797s突变型三次耐药性egfr)酪氨酸激酶抑制剂、具有耐药性突变egfr的非小细胞肺癌等的预防和/或治疗剂等是有用的。本申请以在日本提出申请的日本特愿2017-064866为基础,本说明书包含其全部内容。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种快速检测BCR/ABL基因融合的探针组、试剂盒及方法与流程
- 酪氨酸激酶抑制剂的制造方法与工艺
- 芳香族酰胺类衍生物、其制备方法及其在医药上的应用与制造工艺
- 包含5α‑还原酶抑制剂的药物组合物的制造方法与工艺
- 作为脾酪氨酸激酶抑制剂的二氮杂螺烷酮取代的噁唑衍生物的制造方法与工艺
- 包含热弹性感觉剂和TRPA1受体抑制剂的皮肤接合剃刮助剂的制造方法与工艺
- 检测EGFR耐药性突变的方法和组合物与制造工艺
- 5‑溴‑7‑氮杂吲哚啉‑2‑酮类化合物及其制备方法与制造工艺
- 3‑[(3‑{[4‑(4‑吗啉基甲基)‑1H‑吡咯‑2‑基]亚甲基}‑2‑氧代‑2,3‑二氢‑1H‑吲哚‑5‑基)甲基]‑1,3‑噻唑烷‑2,4‑二酮和EGFR TYR激酶抑制剂的组合的制造方法与工艺
- 一种抗癌药物的晶体化合物及其制备方法与制造工艺