一种邻甲氧基苯甲醛的制备方法与流程
本发明涉及有机合成技术领域,特别涉及一种邻甲氧基苯甲醛的制备方法。
背景技术:
邻甲氧基苯甲醛时一种常见的有机合成中间体,其广泛存在于药物,天然产物中,如,拟肾上腺素药物喘咳宁。也可用作香料、医药和荧光增白剂中间体,如生产荧光增白剂CBS、三苯甲烷染料和防蛀剂N。
由于醛基的弱协调性和对氧化剂的敏感性以及弱导向作用导致苯甲醛邻位定位反应很难,其次甲醇中的羟基H,不容易离去,使得在反应中甲氧基很难定位在醛基邻位,且现有的许多邻甲氧基苯甲醛合成方法步骤繁琐,不能一步得到产物,反应温度高,原子利用率低,环保压力大,这些缺点限制了方法的应用。
因此,开发一种制备过程简单,反应时间短,反应条件温和,后处理简单,产率高,底物拓展性强的邻甲氧基苯甲醛的制备方法具有十分重要的意义。
技术实现要素:
有鉴于此,本发明旨在提出一种邻甲氧基苯甲醛的制备方法,以解决现有邻甲氧基苯甲醛合成方法复杂、反应时间长、反应条件苛刻、产品产率和纯度较低、环保压力大的问题。
为达到上述目的,本发明的技术方案是这样实现的:
一种邻甲氧基苯甲醛的制备方法,包括以下步骤:
1)将苯甲醛类衍生物、无水甲醇、醋酸钯、氧化剂、苯胺配体混合在溶剂中,然后,在一定温度条件下进行甲氧基化反应,得到反应液A;
2)将所述反应液A过滤,并将过滤得到的滤液B用萃取剂萃取,得到下层萃取液C;
3)用干燥剂将所述萃取液C干燥除水后过滤,向过滤得到的滤液D中加入硅胶粉旋干所述二氯甲烷,得到混合物E;
4)用层析法分离提纯所述混合物E,得到邻甲氧基苯甲醛。
可选地,所述步骤1)中所述苯甲醛类衍生物、所述无水甲醇、所述醋酸钯、所述氧化剂、所述苯胺配体的摩尔比为1∶(15~20)∶0.1∶2∶0.4。
可选地,所述步骤1)中所述甲氧基化反应的反应温度为40~80℃,反应时间为18~32h。
可选地,所述步骤1)中所述苯甲醛类衍生物的苯甲醛上的取代基为甲基、甲氧基、苯基、氟、氯、溴中的一种,所述苯胺配体为间三氟甲基苯胺、邻三氟甲基苯胺中的一种,所述氧化剂为过硫酸钾、过硫酸钠中的一种,所述溶剂为二氯甲烷、1,2-二氯乙烷、甲苯中的一种。
可选地,所述步骤1)中所述苯甲醛类衍生物的用量与所述溶剂的用量的比值为1mol∶10~15L。
可选地,所述步骤2)中所述萃取剂为饱和碳酸氢钠水溶液和二氯甲烷的混合液;所述饱和碳酸氢钠水溶液和所述二氯甲烷的体积比为1∶(2-3)。
可选地,所述步骤3)中所述干燥剂为无水硫酸钠,且所述萃取液C的用量与所述干燥剂的用量的比值为1L∶25g。
可选地,所述步骤4)中所述层析法包括柱层析法。
可选地,所述柱层析法包括以下步骤:
将硅胶粉用石油醚调成糊状,然后倒入色谱柱中;
向色谱柱中加入石油醚,加压,直至流速恒定,使柱床体积压缩至柱床原始体积的9/10;
采用干法上样将所述混合物E置于色谱柱内,展开并洗脱,待洗脱结束后,点收集目标产物的洗脱液,然后,将收集的所述洗脱液中的溶剂旋干,即得邻甲氧基苯甲醛。
可选地,所述洗脱的洗脱剂为石油醚和乙酸乙酯的混合物;所述石油醚和所述乙酸乙酯的体积比为10∶1。
相对于现有技术,本发明所述的邻甲氧基苯甲醛的制备方法具有以下优势:
1、本发明以简单易得的苯甲醛类衍生物、无水甲醇为起始原料,以醋酸钯为催化剂,并配以氧化剂、溶剂、以及可与苯甲醛类衍生物的醛基缩合形成瞬时导向集团的苯胺配体,使苯甲醛类衍生物、无水甲醇在苯胺配体与苯甲醛类衍生物的醛基缩合形成瞬时导向集团的同时,选择性地在苯甲醛类衍生物的醛基的邻位进行甲氧基化反应,使得所制邻甲氧基苯甲醛具有较高的产率和纯度,其产率可达67%。
2、本发明以醋酸钯为催化剂,并使苯甲醛类衍生物、无水甲醇在苯胺配体与苯甲醛类衍生物的醛基缩合形成瞬时导向集团的同时,选择性地在苯甲醛类衍生物的醛基的邻位进行甲氧基化反应,使得整个制备过程简单,易操作,并使整个反应所需时间缩短,大大提高了生产效率,且有利于降低本发明的反应温度和溶剂消耗量,也大大简化了整个后处理过程,易于工业化生产。另外,本发明当用二氯甲烷做溶剂时,其对人体的伤害较低,侵入机体后,主要由肝内代谢,排泄较快。同时,本发明采用柱层析法提纯,大大减轻对环境的压力同时,有利于进一步提高产品的纯度。
附图说明
构成本发明的一部分的附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
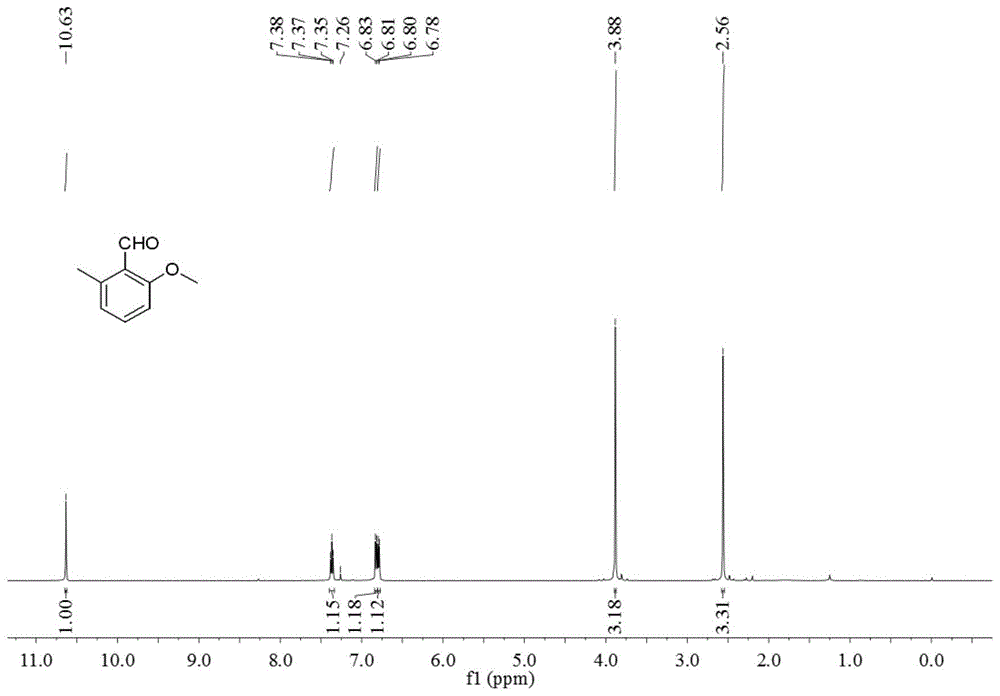
图1为本发明实施例1制备的邻甲氧基苯甲醛的氢谱谱图;
图2为本发明实施例1制备的邻甲氧基苯甲醛的碳谱谱图。
具体实施方式
需要说明的是,在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。
现有邻甲氧基苯甲醛合成方法复杂,有机溶剂消耗量大,产品产率和纯度较低,环保压力大,因此,本发明在现有邻甲氧基苯甲醛合成方法的基础上,以苯甲醛类衍生物、无水甲醇为起始原料,以醋酸钯为催化剂,并配以氧化剂、溶剂、以及可与苯甲醛类衍生物的醛基缩合形成瞬时导向集团的苯胺配体,使苯甲醛类衍生物、无水甲醇在苯胺配体与苯甲醛类衍生物的醛基缩合形成瞬时导向集团的同时,选择性地在苯甲醛类衍生物的醛基的邻位进行甲氧基化反应,提高所制邻甲氧基苯甲醛的产率和纯度,并简化反应步骤,缩短反应时间,降低环保压力,进而提高其推广和应用价值。
本发明的甲氧基化反应的反应过程如下:
其中,R为甲基、甲氧基、苯基、氟、氯、溴中的一种,苯胺配体为间三氟甲基苯胺、邻三氟甲基苯胺中的一种,氧化剂为过硫酸钾、过硫酸钠中的一种,溶剂为二氯甲烷、1,2-二氯乙烷、甲苯中的一种。
下面将结合附图和实施例来详细说明本发明。
实施例1
一种邻甲氧基苯甲醛的制备方法,具体包括以下步骤:
1)将12.0mg(0.1mmol)邻甲基苯甲醛、64.0mg(20.0mmol)无水甲醇(MeOH)、2.25mg(0.01mmol)醋酸钯(Pd(OAc)2)、54.0mg(0.2mmol)过硫酸钾(K2S2O8)、6.44mg(0.04mmol)间三氟甲基苯胺(TDG)混合在1ml二氯甲烷(DCM)中,然后,在60℃的磁力搅拌器上搅拌反应24h,即进行邻甲基苯甲醛中醛基的邻位的甲氧基化反应,得到反应液A,反应过程中,可采用规格为-254的薄层色谱板点板追踪反应是否完成;
2)将反应液A过滤,并向过滤得到的滤液B中加饱和碳酸氢钠水溶液和二氯甲烷萃取,得到下层萃取液C,其中,饱和碳酸氢钠水溶液和二氯甲烷体积比为1∶3;
3)用无水硫酸钠将萃取液C干燥除水后过滤,向过滤得到的滤液D中加入硅胶粉旋干二氯甲烷,得到含有邻甲氧基苯甲醛与硅胶粉的混合物E,其中,萃取液C的用量与无水硫酸钠的用量的比值为1L∶25g,即干燥1L萃取液C所需无水硫酸钠的质量为25g,硅胶粉的质量是滤液D中溶质的质量的30-40倍;
4)用柱层析法分离提纯混合物E,得到10.1mg邻甲氧基苯甲醛(2-甲氧基-6-甲基苯甲醛),经计算,其产率为67%。
在本实施例中,柱层析法分离提纯混合物E具体包括以下步骤:
采用湿法装柱法将硅胶粉用石油醚调成糊状,然后倒入色谱柱中,其中,色谱柱的直径为3.5cm、高度为30cm;
向色谱柱中加入石油醚,用气泵加压,直至流速恒定,使柱床体积压缩至柱床原始体积的9/10;
采用干法上样将所述混合物E上样到色谱柱内,展开并采用洗脱剂洗脱,洗脱过程中通过薄层色谱不断的点板,观察目标产物有没有被洗脱出来,待洗脱结束后,点收集目标产物的洗脱液,然后,将收集的洗脱液合并后,旋干其中的溶剂,即得邻甲氧基苯甲醛,其中,洗脱剂为石油醚和乙酸乙酯的混合物;石油醚和乙酸乙酯的体积比为10∶1。
将10mg实施例1的邻甲氧基苯甲醛溶解在0.5mL的氘代氯仿(CDCl3)中,对其进行核磁共振氢谱和碳谱测试,测试结果分别如图1和图2所示。
由图1可知,本发明实施例1的邻甲氧基苯甲醛的氢谱谱图中含有1H NMR(500MHz,CDCl3)δ10.63(s,1H),7.37(t,J=8.0Hz,1H),6.82(d,J=8.4Hz,1H),6.79(d,J=7.6Hz,1H),3.88(s,3H),2.56(s,3H)的峰,其中,δ10.63(s,1H)为醛基上的H,6.82(d,J=8.4Hz,1H)为位于甲氧基邻位的苯环上的H,6.79(d,J=7.6Hz,1H)为位于甲基临位苯环上的H,7.37(t,J=8.0Hz,1H)为醛基对位苯环上的H,3.88(s,3H)为甲氧基上的H,2.56(s,3H)为甲基H。
由图2可知,本发明实施例1的邻甲氧基苯甲醛的碳谱谱图中含有13C NMR(126MHz,CDCl3)δ192.32,163.17,142.03,134.43,124.09,123.36,109.05,55.77,21.44的峰,其中,δ192.32为醛基上的C,δ163.17,142.03,134.43,124.09,123.36,109.05,为苯环上的C,δ55.77为甲氧基上的C,δ21.44为甲基上的C。
通过对图1和图2的谱图中的峰进行分析可知,本实施例合成的物质为邻甲氧基苯甲醛。
本实施例的甲氧基化反应的反应过程如下:
其中,R为甲基;TDG的结构式为:
苯甲醛类衍生物、无水甲醇、醋酸钯、氧化剂、苯胺配体的摩尔比为1∶20∶0.1∶2∶0.4。当加入0.1mmol的苯甲醛类衍生物时,所加溶剂为1ml。
实施例2
本实施例与实施例1的区别在于:本实施例中与邻甲基苯甲醛中醛基缩合形成瞬时导向基团的苯胺配体为邻三氟甲基苯胺,邻三氟甲基苯胺的用量为6.44mg(0.04mmol),其他制备原料组成以及邻甲氧基苯甲醛的制备过程同实施例1。
本实施例制得的邻甲氧基苯甲醛的量为6.75mg,经计算,其产率为45%。
实施例3
本实施例与实施例1的区别在于:本实施例中氧化剂为过硫酸钠,其用量为47.6mg(0.2mmol),其他制备原料组成以及邻甲氧基苯甲醛的制备过程同实施例1。
本实施例制得的邻甲氧基苯甲醛的量为7.35mg,经计算,其产率为49%。
实施例4
本实施例与实施例1的区别在于:本实施例中溶剂为1,2-二氯乙烷,其用量为1ml,他制备原料组成以及邻甲氧基苯甲醛的制备过程同实施例1。
本实施例制得的邻甲氧基苯甲醛的量为9.45mg,经计算,其产率为63%。
实施例5
本实施例与实施例1的区别在于:本实施例中甲氧基化反应的反应温度为40℃,本实施例中各原料组成以及制备过程同实施例1。
本实施例制得的邻甲氧基苯甲醛的量为5.55mg,经计算,其产率为37%。
实施例6
本实施例与实施例1的区别在于:本实施例中甲氧基化反应的反应温度为80℃,本对比例中各原料组成以及制备过程同实施例1。
本实施例制得的邻甲氧基苯甲醛的量为9.3mg,经计算,其产率为62%。
实施例7
本实施例与实施例1的区别在于:本实施例中溶剂为甲苯,其用量为1ml,甲氧基化反应的反应温度为40℃,其他制备原料组成以及邻甲氧基苯甲醛的制备过程同实施例1。
本实施例制得的邻甲氧基苯甲醛的量为4.5mg,经计算,其产率为30%。
以上仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!