一种胡椒碱衍生物及其制备方法和用途与流程
本发明属于化学药物技术领域,具体涉及一种胡椒碱衍生物及其制备方法和用途。
背景技术:
神经退行性疾病导致脑功能的进行性丧失和临床综合症的重叠。例如,阿尔茨海默病(ad),还有血管性痴呆,额颞叶痴呆(ftd),混合性痴呆和路易体痴呆(lbd)会出现认知缺陷。类似地,肌萎缩侧索硬化(als),亨廷顿舞蹈病(hd),帕金森病(pd)和脊髓小脑性共济失调(sca)会造成运动系统受损。在所有这些疾病中,衰老是一种常见的危险因素。根据2015年联合国关于世界人口老龄化的报告预计在未来35年内增加一倍以上,全世界60岁及以上人口的人数将近21亿。尽管预期寿命有所改善,但阿尔茨海默病(ad)和相关的神经退行性疾病可能成为老年人最可怕的疾病,这些破坏性疾病的流行得不到遏制,会给患者,家庭和社会带来繁重的经济负担。
尽管它们具有不同的临床表现,反映了不同脑区特定神经元和突触的丢失,但神经退行性疾病具有共同的特征和机制。神经变性疾病是一组以神经元结构和/或功能丧失为特征的疾病。在分子水平上,神经退行性疾病具有几种相似性,例如蛋白质的异常沉积(在许多疾病中被错误折叠),线粒体应激导致活性氧(ros)的形成,小胶质细胞活化和神经炎症,蛋白质稳定失调(涉及泛素-蛋白酶体途径和自噬吞噬体途径),程序性细胞死亡(包括细胞凋亡),以及突触可塑性,记忆,学习和其他功能相关的受体失调。目前治疗这些疾病都只能对症治疗,不会减缓或逆转疾病进展。
技术实现要素:
为了解决上述问题,本发明提供了一种胡椒碱衍生物及其制备方法和用途。
本发明提供了一种式(i)所示的化合物、或其盐、或其立体异构体、或其水合物:
其中,
x为o、s或se;
n为0或1;
r1选自
n1为1;
n2、n3、n4为0或1;
r3选自取代或未取代的c1~c8烷基、取代或未取代的c1~c8烷氧基、氰基、卤素、羟基、苯基、
m、x独立为1~8的整数;
r9~r13、r14~r17、r18~r21、r22~r25、r22’~r25’分别独立选自氢、卤素、氰基、三氟甲基或c1~c8的烷基;
r7、r8分别独立选自c1~c5的烷基、c3~c8环烷基;
当n为0或1,r1选自
当n为0,r1选自
当n为1,r1选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c8烷基。
进一步地,所述化合物如式(ii)所示:
其中,
r1选自
n1为1;
n2、n3、n4为0或1;
r3选自取代或未取代的c1~c8烷基、取代或未取代的c1~c8烷氧基、氰基、卤素、羟基、苯基、
m、x独立为1~8的整数;
r9~r13、r14~r17、r18~r21、r22~r25、r22’~r25’分别独立选自氢、卤素、氰基、三氟甲基、或c1~c8的烷基;
r7、r8分别独立选自c1~c5的烷基、c3~c8环烷基;
当r1选自
r1选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c8烷基。
进一步地,所述化合物如式(iii)所示:
其中,
r1选自
n1为1;
n2、n3、n4为0或1;
r3选自取代或未取代的c1~c8烷基、取代或未取代的c1~c8烷氧基、氰基、氯、羟基、苯基、
m、x独立为1~8的整数;
r9~r13、r14~r17、r18~r21、r22~r25、r22’~r25’分别独立选自氢、卤素、氰基、三氟甲基、或c1~c8的烷基;
r7、r8分别独立选自c1~c5的烷基、c3~c8环烷基;
r2选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c8烷基。
进一步地,所述化合物如式(ii-a)所示:
其中,
n1为1;
r3选自c1~c5烷基、c1~c5烷氧基、氰基、苯基、卤素、羟基、
m、x独立为1或2;
r9~r13、r14~r17、r18~r21、r22~r25、r22’~r25’分别独立选自氢、卤素、氰基、三氟甲基或c1~c5的烷基;
r2选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c5烷基。
进一步地,所述化合物如式(ii-b)所示:
其中,
n2为0或1;
r3选自c1~c5烷基、c1~c5烷氧基、氰基、苯基、卤素、羟基、
m、x独立为1或2;
r9~r13、r14~r17、r18~r21、r22~r25、r22’~r25’分别独立选自氢、卤素、氰基、三氟甲基或c1~c5的烷基;
r2选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c5烷基;
或,所述化合物如式(ii-c)所示:
其中,
n3为0或1;
r3选自c1~c5烷基、c1~c5烷氧基、氰基、苯基、卤素、羟基、
m、x独立为1或2;
r9~r13、r14~r17、r18~r21、r22~r25、r22’~r25’分别独立选自氢、卤素、氰基、三氟甲基或c1~c5的烷基;
r2选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c5烷基;
或,所述化合物如式(ii-d)所示:
其中,
n4为0或1;
r3选自c1~c5烷基、c1~c5烷氧基、氰基、苯基、卤素、羟基、
m、x独立为1或2;
r9~r13、r14~r17、r18~r21、r22~r25、r22’~r25’分别独立选自氢、卤素、氰基、三氟甲基或c1~c5的烷基;
r2选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c5烷基;
或,所述化合物如式(ii-e)所示:
其中,
r7选自c1~c5的烷基、c3~c8环烷基;
r2选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c8烷基;
或,所述化合物如式(ii-f)所示:
其中,
r8选自c1~c3的烷基、c3~c5环烷基;
r2选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c8烷基;
或,所述化合物如式(ii-g)所示:
其中,
r2选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c8烷基;
或,所述化合物如式(ii-h)所示:
其中,
r2选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c8烷基。
进一步地,所述化合物如式(iii-a)所示:
其中,
n1为1;
r3选自c1~c5烷基、c1~c5烷氧基、氰基、苯基、氯、羟基、
m、x独立为1或2;
r9~r13、r14~r17、r18~r21、r22~r25、r22’~r25’分别独立选自氢、卤素、氰基、三氟甲基或c1~c5的烷基;
r2选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c5烷基;
或,所述化合物如式(iii-b)所示:
其中,
n2为0或1;
r3选自c1~c5烷基、c1~c5烷氧基、氰基、苯基、卤素、羟基、
m、x独立为1或2;
r9~r13、r14~r17、r18~r21、r22~r25、r22’~r25’分别独立选自氢、卤素、氰基、三氟甲基或c1~c5的烷基;
r2选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c5烷基;
或,所述化合物如式(iii-c)所示:
r2选自
r26~30、r31~34、r35~36、r37~38、r39~42分别独立选自氢、卤素、氰基、三氟甲基或c1~c5烷基。
进一步地,所述化合物为如下化合物之一:
本发明还提供了前述的化合物、或其盐、或其立体异构体、或其水合物在制备治疗神经退行性疾病的药物中的用途。
进一步地,所述神经退行性疾病为阿尔茨海默病、血管性痴呆、额颞叶痴呆、混合性痴呆、路易体痴呆、肌萎缩侧索硬化、亨廷顿舞蹈病、帕金森病、脊髓小脑性共济失调。
本发明还提供了一种药物,它是由前述的化合物、或其盐、或其立体异构体、或其水合物为活性成分,加上药物上可接受的辅料或辅助性成分制备而成的制剂。
本发明中,碳氢基团中碳原子含量的最小值和最大值通过前缀表示,例如,前缀ca~cb烷基表明任何含“a”至“b”个碳原子的烷基。
本发明中,
本发明中,卤素是指氟、氯、溴、碘。
本发明中,室温为25±5℃;过夜为12±2h。
本发明化合物能够有效保护神经细胞,提高神经细胞的存活率,因此,本发明化合物可以有效治疗神经退行性疾病,可以用于制备治疗神经退行性疾病的药物。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
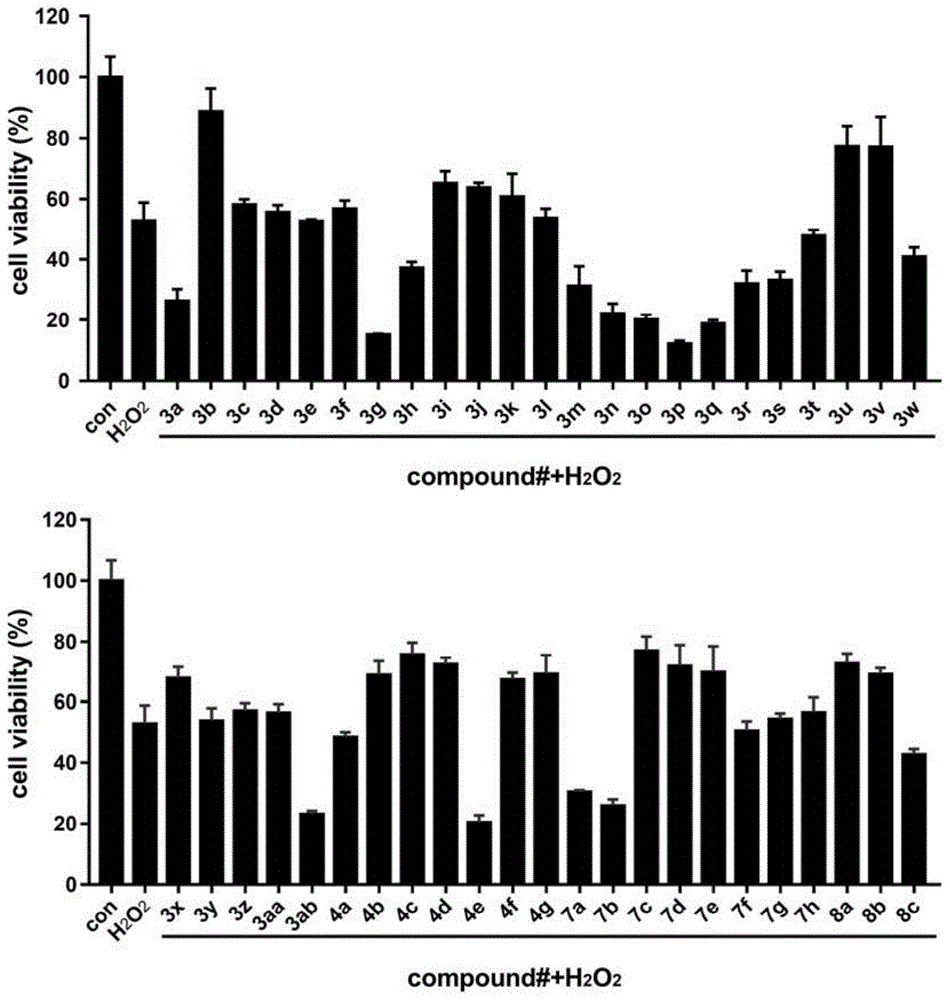
图1为本发明化合物关于pc-12细胞保护活性的平均细胞存活率。
具体实施方式
本发明具体实施方式中使用的原料、设备均为已知产品,通过购买市售产品获得。为方便后面对实例合成路线及方法的描述,现将实施例所用主要原料或试剂的缩写制成下表1。
表1实施例所用主要原料或试剂的缩写
实施例1、(e,e)-1-[4-(1-甲氧基苯-4-基)-1-氧代-2,4-戊二烯基]-哌啶的制备
步骤1:
在0℃下,搅拌哌啶(0.10mol)和et3n(10.12g,0.10mol)的dcm(50ml)溶液,滴加巴豆酰氯(0.10mol),然后将反应缓慢升温至室温并搅拌8小时,通过tlc监测反应。反应完成后,用饱和nahco3水溶液50ml洗涤二氯甲烷层3次,再用水50ml洗二氯甲烷层。无水na2so4干燥,减压浓缩,获得浅黄色粘稠液体2a。1hnmr(400mhz,cdcl3)δ6.58(dq,j=13.9,6.8hz,1h),6.06(d,j=15.0hz,1h),3.31(d,j=34.4hz,4h),1.64(d,j=6.8hz,3h),1.42(dd,j=10.7,5.7hz,2h),1.33(d,j=4.8hz,4h).exactmasscalcdforc9h15no[m+h]+:154.1232;found154.1235.
步骤2:
将中间体2a(1equiv,2.4mmol)用2mldmso溶解,加入1ml饱和naoh溶液,搅拌30分钟,然后加入对甲氧基苯甲醛(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用二氯甲烷萃取,有机相用水洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物3a。1hnmr(400mhz,cdcl3)δ7.43(ddd,j=12.4,6.3,1.7hz,1h),7.40-7.37(m,2h),6.89-6.85(m,2h),6.81-6.77(m,2h),6.43(d,j=14.6hz,1h),3.81(s,3h),3.58(s,4h),1.70-1.63(m,2h),1.58(dt,j=11.0,5.5hz,4h).13cnmr(101mhz,cdcl3)δ165.61,160.12,142.87,138.30,129.38,128.40,125.07,119.72,114.29,55.40,47.04,43.54,26.21,24.76.exactmasscalcdforc17h21no2[m+h]+:272.1572;found272.1576.
实施例2、(e,e)-1-[2-(1-甲氧苯-2-基)-1-氧代-2,4-戊二烯基]-哌啶的制备
将中间体2a(1equiv,2.4mmol)用2mldmso溶解,加入1ml饱和氢氧化钠溶液,搅拌30分钟,然后加入邻甲氧基苯甲醛(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用二氯甲烷萃取,有机相用水洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物3b。1hnmr(400mhz,cdcl3)δ7.42(dd,j=14.5,10.3hz,1h),7.28-7.22(m,1h),7.04(d,j=7.7hz,1h),6.97(s,1h),6.94-6.77(m,3h),6.49(d,j=14.7hz,1h),3.82(s,3h),3.59(s,4h),1.74-1.64(m,2h),1.63-1.51(m,4h).13cnmr(101mhz,cdcl3)δ165.48,159.97,142.33,138.48,137.99,129.80,127.48,121.25,119.77,114.47,112.13,55.38,47.05,43.39,26.80,25.88,24.76.exactmasscalcdforc17h21no2[m+h]+:272.1572;found272.1576.
实施例3、(e,e)-1-[3-(1-甲氧苯-3-基)-1-氧代-2,4-戊二烯基]-哌啶的制备
将中间体2a(1equiv,2.4mmol)用2mldmso溶解,加入1ml饱和氢氧化钠溶液,搅拌30分钟,然后加入间甲氧基苯甲醛(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用二氯甲烷萃取,有机相用水洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物3c。1hnmr(400mhz,cdcl3)δ7.48(dd,j=7.6,1.6hz,1h),7.45(dd,j=14.8,11.2hz,1h),7.28-7.23(m,1h),7.18(d,j=15.7hz,1h),7.00-6.91(m,2h),6.88(d,j=8.3hz,1h),6.45(d,j=14.8hz,1h),3.87(s,3h),3.58(m,4h),1.71-1.63(m,2h),1.59(dt,j=10.9,5.4hz,4h).13cnmr(101mhz,cdcl3)δ165.67,157.45,143.37,133.82,129.74,127.81,127.32,125.61,120.79,120.37,111.18,55.61,47.04,43.35,26.84,25.74,24.79.exactmasscalcdforc17h21no2[m+h]+:272.1572;found272.1576.
实施例4、(e,e)-1-[3-(1-甲氧苯-3-基)-1-氧代-2,4-戊二烯基]-哌啶的制备
将中间体2a(1equiv,2.4mmol)用2mldmso溶解,加入1ml饱和氢氧化钠溶液,搅拌30分钟,然后加入二甲胺基苯甲醛(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用二氯甲烷萃取,有机相用水洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物3d。1hnmr(400mhz,cdcl3)δ7.43(dd,j=14.6,9.9hz,1h),7.34(d,j=8.8hz,2h),6.81-6.72(m,2h),6.67(d,j=8.6hz,2h),6.37(d,j=14.6hz,1h),3.56(d,j=22.9hz,4h),2.98(s,6h),1.71-1.63(m,2h),1.57(dd,j=13.5,10.6hz,4h).exactmasscalcdforc18h24n2o[m+h]+:285.1967;found285.1972.
实施例5、(e,e)-1-[3-(1-甲氧苯-3-基)-1-氧代-2,4-戊二烯基]-哌啶的制备
将中间体2a(1equiv,2.4mmol)用2mldmso溶解,加入1ml饱和氢氧化钠溶液,搅拌30分钟,然后加入二乙胺基苯甲醛(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用二氯甲烷萃取,有机相用水洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物3e。1hnmr(400mhz,cdcl3)δ7.44(dd,j=14.6,10.3hz,1h),7.31(d,j=8.7hz,2h),6.79-6.69(m,2h),6.62(d,j=8.7hz,2h),6.35(d,j=14.6hz,1h),3.66-3.51(m,4h),3.37(q,j=7.0hz,4h),1.65(m,2h),1.62-1.53(m,4h),1.17(t,j=7.1hz,6h).exactmasscalcdforc20h28n2o[m+h]+:313.2280;found313.2285.
实施例6、(e,e)-1-[3-(1-甲氧苯-3-基)-1-氧代-2,4-戊二烯基]-哌啶的制备
将中间体2a(1equiv,2.4mmol)用2mldmso溶解,加入1ml饱和氢氧化钠溶液,搅拌30分钟,然后加入对氰基苯甲醛(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用二氯甲烷萃取,有机相用水洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物3f。1hnmr(400mhz,meod)δ7.90(d,j=8.4hz,2h),7.65(d,j=8.4hz,2h),7.39(dd,j=14.6,10.9hz,1h),7.21(dd,j=15.5,11.0hz,1h),7.00(d,j=15.5hz,1h),6.80(d,j=14.6hz,1h),3.71-3.64(m,4h),1.79-1.71(m,2h),1.67(d,j=22.8hz,4h).exactmasscalcdforc17h18n2o[m+h]+:266.1419;found266.1423.
实施例7、(e,e)-1-[3-(1-甲氧苯-3-基)-1-氧代-2,4-戊二烯基]-哌啶的制备
将中间体2a(1equiv,2.4mmol)用2mldmso溶解,加入1ml饱和氢氧化钠溶液,搅拌30分钟,然后加入4-苯基苯甲醛(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用dcm萃取,有机相用水洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物3g。1hnmr(400mhz,cdcl3)δ7.63-7.56(m,4h),7.52(d,j=8.4hz,2h),7.49-7.40(m,3h),7.36(dt,j=9.3,4.2hz,1h),6.95(dd,j=15.5,10.1hz,1h),6.88(d,j=15.5hz,1h),6.50(d,j=14.7hz,1h),3.60(d,j=40.1hz,4h),1.73-1.64(m,2h),1.60(m,4h).13cnmr(101mhz,cdcl3)δ165.37,142.35,141.28,140.46,138.02,135.53,128.85,127.54,127.41,127.09,126.95,120.94,46.98,43.29,26.77,25.64,24.69.exactmasscalcdforc22h23no[m+na]+:340.1677;found340.1685.
实施例8、(e,e)-1-[3-(1-甲氧苯-3-基)-1-氧代-2,4-戊二烯基]-哌啶的制备
将中间体2a(1equiv,2.4mmol)用2mldmso溶解,加入1ml饱和氢氧化钠溶液,搅拌30分钟,然后加入4-(1-吡咯啉基)苯甲醛(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用dcm萃取,有机相用水洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物3h。1hnmr(400mhz,cdcl3)δ7.44(dd,j=14.6,10.5hz,1h),7.33(d,j=8.7hz,2h),6.78(d,j=15.4hz,1h),6.70(dd,j=15.4,10.5hz,1h),6.52(d,j=8.6hz,2h),6.35(d,j=14.6hz,1h),3.58(m,4h),3.35-3.28(m,4h),2.03-1.98(m,4h),1.69-1.63(m,2h),1.61-1.53(m,4h).13cnmr(101mhz,cdcl3)δ166.01,148.23,143.81,139.70,128.56,122.16,117.61,111.88,47.74,46.96,43.30,26.82,25.56,24.85.exactmasscalcdforc20h26n2o[m+na]+:333.1947;found333.1947.
实施例9-21
其他胡椒碱衍生物的合成方法同实施例2~8,只是更改芳香醛的种类。芳香醛原料、所得化合物结构式以及所得化合物核磁结果如表2所示。
表2制备化合物的原料、所得化合物结构式及化合物核磁结果
实施例22、(e,e)-1-[3-(1-甲氧苯-3-基)-1-氧代-2,4-戊二烯基]-哌啶的制备
步骤1:
在0℃下,搅拌哌啶(0.10mol)和et3n(10.12g,0.10mol)的二氯甲烷(50ml)溶液,滴加巴豆酰氯(0.10mol),然后将反应缓慢升温至室温并搅拌8小时,通过tlc监测反应。反应完成后,用饱和nahco3水溶液50ml洗涤有机层3次,再用h2o50ml洗有机层。无水na2so4干燥,减压浓缩,获得浅黄色粘稠液体2a。(同实施例1的步骤1)
步骤2:
将中间体2a(1equiv,2.4mmol)用2mldmso溶解,加入1ml饱和氢氧化钠溶液,搅拌30分钟,然后加入4-(1-吡咯啉基)苯甲醛(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用dcm萃取,有机相用水洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物p-3i。
将得到的化合物p-3i溶解在2ml三氟乙酸中,室温搅拌1小时,反应完成后加入饱和碳酸氢钠溶液淬灭反应,用10ml乙酸乙酯萃取水相3次,合并有机相,有机相减压浓缩得到淡黄色粉末状固体3i。1hnmr(400mhz,cdcl3)δ7.37(dd,j=9.3,4.5hz,1h),7.33(d,j=8.6hz,2h),6.82(d,j=8.6hz,2h),6.77-6.73(m,2h),6.39(d,j=14.7hz,1h),3.57(s,4h),3.40(s,4h),3.30(s,4h),1.64(m,2h),1.58(m,4h).13cnmr(101mhz,cdcl3)δ166.39,150.08,143.77,138.98,129.46,128.51,125.01,119.13,116.71,46.23,45.27,43.88,43.44,26.47,25.87,24.56.exactmasscalcdforc20h26n2o[m+na]+:348.2052;found348.2056.
实施例23、(2e,4e)-n,n-二乙基-5-(4-(哌嗪-1-基)苯基)戊-2,4-二胺的制备
步骤1:
在0℃下,搅拌二乙胺(0.10mol)和et3n(10.12g,0.10mol)的二氯甲烷(50ml)溶液,滴加巴豆酰氯(0.10mol),然后将反应缓慢升温至室温并搅拌8小时,通过tlc监测反应。反应完成后,用饱和nahco3水溶液50ml洗涤有机层3次,再用h2o50ml洗有机层。无水na2so4干燥,减压浓缩,获得浅黄色粘稠液体2b。
步骤2:
将中间体2b(1equiv,2.4mmol)用2ml二甲基亚砜溶解,加入1ml饱和氢氧化钠溶液,搅拌30分钟,然后加入1-boc-4-(4-甲酰苯基)哌嗪(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用dcm萃取,有机相用水洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物p-3j。
将得到的化合物p-3j溶解在2mltfa中,室温搅拌1小时,反应完成后加入饱和碳酸氢钠溶液淬灭反应,用10ml乙酸乙酯萃取水相3次,合并有机相,有机相减压浓缩得到淡黄色粉末状固体3j。1hnmr(400mhz,cdcl3)δ7.46(ddd,j=14.6,6.1,3.8hz,1h),7.38(d,j=8.6hz,2h),6.87(d,j=8.6hz,2h),6.83-6.75(m,2h),6.34(d,j=14.6hz,1h),3.43(m,8h),3.23(d,j=4.7hz,4h),1.21(m,6h),0.91-0.79(m,1h).13cnmr(101mhz,cdcl3)δ166.17,147.66,143.01,138.55,130.53,128.36,125.00,119.83,116.45,47.73,44.41,42.38,41.15.exactmasscalcdforc19h27n3o[m+na]+:336.2052;found336.2005.
实施例24、(2e,4e)-n-乙基-5-(4-(哌嗪-1-基)苯基)戊-2,4-二胺的制备
步骤1:
在0℃下,搅拌乙胺(0.10mol)和et3n(10.12g,0.10mol)的二氯甲烷(50ml)溶液,滴加巴豆酰氯(0.10mol),然后将反应缓慢升温至室温并搅拌8小时,通过tlc监测反应。反应完成后,用饱和nahco3水溶液50ml洗涤有机层3次,再用h2o50ml洗有机层。无水na2so4干燥,减压浓缩,获得浅黄色粘稠液体2c。
步骤2:
将中间体2c(1equiv,2.4mmol)用2ml二甲基亚砜溶解,加入1ml饱和氢氧化钠溶液,搅拌30分钟,然后加入1-boc-4-(4-甲酰苯基)哌嗪(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的盐酸中和,用二氯甲烷萃取,有机相用水洗涤,无水硫酸钠干燥。减压浓缩。通过硅胶柱层析纯化得到产物p-3k。
将得到的化合物p-3k溶解在2ml三氟乙酸中,室温搅拌1小时,反应完成后加入饱和碳酸氢钠溶液淬灭反应,用10ml乙酸乙酯萃取水相3次,合并有机相,有机相减压浓缩得到淡黄色粉末状固体3k。1hnmr(400mhz,cdcl3)δ7.46(ddd,j=14.6,6.1,3.8hz,1h),7.38(d,j=8.6hz,2h),6.87(d,j=8.6hz,2h),6.83-6.75(m,2h),6.34(d,j=14.6hz,1h),3.43(m,8h),3.23(d,j=4.7hz,4h),1.21(m,6h),0.91-0.79(m,1h).13cnmr(101mhz,cdcl3)δ166.17,147.66,143.01,138.55,130.53,128.36,125.00,119.83,116.45,47.73,44.41,42.38,41.15.exactmasscalcdforc19h27n3o[m+na]+:336.2052;found336.2005.
实施例25、(2e,4e)-5-(苯并[d][1,3]二恶唑-5-基)-n,n-二乙基五甲基-2,4-二烯酰胺的制备
将中间体2b(1equiv,2.4mmol)用2mldmso溶解,加入1ml饱和naoh溶液,搅拌30分钟,然后加入胡椒醛(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用dcm萃取,有机相用h2o洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物3m。1hnmr(400mhz,cdcl3)δ7.44(ddd,j=14.6,7.4,2.6hz,1h),6.99(d,j=1.5hz,1h),6.89(dd,j=8.0,1.5hz,1h),6.77(d,j=8.5hz,2h),6.75(d,j=5.0hz,1h),6.36(d,j=14.6hz,1h),5.97(s,2h),3.53-3.34(m,4h),1.23(t,j=7.1hz,3h),1.17(t,j=6.5hz,3h).13cnmr(101mhz,cdcl3)δ165.88,148.21,148.14,142.50,138.44,131.05,125.36,122.53,120.27,108.50,105.71,101.28,42.22,40.98,15.03,13.26.exactmasscalcdforc16h19no3[m+na]+:296.1263;found296.1265.
实施例26、(2e,4e)-5-(苯并[d][1,3]二恶唑-5-基)-n-乙基戊-2,4-二胺的制备
将中间体2c(1equiv,2.4mmol)用2mldmso溶解,加入1ml饱和naoh溶液,搅拌30分钟,然后加入胡椒醛(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用dcm萃取,有机相用h2o洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物3n。1hnmr(400mhz,dmso)δ8.03(t,j=5.4hz,1h),7.26(d,j=1.5hz,1h),7.15(dd,j=15.0,10.2hz,1h),7.00(dd,j=8.1,1.5hz,1h),6.97-6.88(m,2h),6.85(d,j=15.5hz,1h),6.07(d,j=15.0hz,1h),6.05(s,2h),3.21-3.12(m,2h),1.06(t,j=7.2hz,3h).13cnmr(101mhz,dmso)δ164.89,147.90,147.65,139.05,137.68,130.86,125.27,124.67,122.57,108.40,105.60,101.22,33.40,14.75.exactmasscalcdforc14h15no3[m+na]+:268.0950;found268.0952.
实施例27、(2e,4e)-n,n-二乙基-5-(噻吩-2-基)戊-2,4-二酰胺的制备
将中间体2b(1equiv,2.4mmol)用2mldmso溶解,加入1ml饱和naoh溶液,搅拌30分钟,然后加入2-噻吩甲醛(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用dcm萃取,有机相用h2o洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物3u。1hnmr(400mhz,dmso)δ7.57(d,j=5.1hz,1h),7.28(d,j=3.6hz,1h),7.20-7.15(m,1h),7.15-7.11(m,1h),7.09(dd,j=5.1,3.6hz,1h),6.73(dd,j=15.4,11.1hz,1h),6.06(d,j=15.1hz,1h),3.55-3.34(m,4h),1.24(t,j=7.1hz,3h),1.18(t,j=6.5hz,3h).13cnmr(101mhz,dmso)δ165.06,141.05,138.21,130.47,128.19,127.81,126.68,126.09,125.12,42.12,40.89,15.03,13.16.exactmasscalcdforc11h13nos[m+na]+:258.0929;found258.0928.
实施例28、(2e,4e)-n-乙基-5-(噻吩-2-基)戊-2,4-二酰胺的制备
将中间体2c(1equiv,2.4mmol)用2mldmso溶解,加入1ml饱和naoh溶液,搅拌30分钟,然后加入2-噻吩甲醛(1equiv,2.4mmol)。在室温下搅拌6-8小时。通过tlc监测反应。反应完成后,将反应混合物用5%的hcl中和,用dcm萃取,有机相用h2o洗涤,无水na2so4干燥。减压浓缩。通过硅胶柱层析纯化得到产物3v。1hnmr(400mhz,dmso)δ8.04(t,j=5.3hz,1h),7.55(d,j=5.1hz,1h),7.26(d,j=3.6hz,1h),7.20-7.15(m,1h),7.15-7.11(m,1h),7.09(dd,j=5.1,3.6hz,1h),6.73(dd,j=15.4,11.1hz,1h),6.09(d,j=15.1hz,1h),3.17(qd,j=7.2,5.8hz,2h),1.06(t,j=7.2hz,3h).13cnmr(101mhz,dmso)δ164.76,141.35,138.36,130.66,128.19,127.99,126.79,126.18,125.22,33.42,14.74.exactmasscalcdforc11h13nos[m+na]+:230.0616;found230.0617.
实施例29、(2e,4e)-5-(4-羟苯基)-1-(哌啶-1-基)戊-2,4-二烯-1-酮的制备
将化合物3a溶解于无水dcm中,-78℃预冷30分钟,滴加配好的1mbbr3dcm溶液,-78℃搅拌两小时,缓慢升到室温,常温搅拌4h到完全反应。通过tlc监测反应。反应完成后,将反应液缓慢倒入水中淬灭反应,用饱和的nahco3溶液中和反应液,并用乙酸乙酯萃取,合并有机相,减压浓缩。通过硅胶柱层析纯化得到系列化合物4a。1hnmr(400mhz,cdcl3)δ7.46-7.37(m,1h),7.30(d,j=8.4hz,2h),6.86(d,j=8.4hz,2h),6.74(m,2h),6.40(d,j=14.4hz,1h),3.60(s,4h),1.72-1.63(m,2h),1.60(s,4h).13cnmr(101mhz,cdcl3)δ166.12,157.12,143.73,139.18,128.76,128.63,124.35,118.75,115.93,100.00,27.10,26.35,24.58.exactmasscalcdforc16h19no2[m+na]+:280.1313;found280.1314.
实施例30、(2e,4e)-5-(2-羟苯基)-1-(哌啶-1-基)戊-2,4-二烯-1-酮的制备
将化合物3b溶解于无水dcm中,-78℃预冷30分钟,滴加配好的1mbbr3dcm溶液,-78℃搅拌两小时,缓慢升到室温,常温搅拌4h到完全反应。通过tlc监测反应。反应完成后,将反应液缓慢倒入水中淬灭反应,用饱和的nahco3溶液中和反应液,并用乙酸乙酯萃取,合并有机相,减压浓缩。通过硅胶柱层析纯化得到系列化合物4b。1hnmr(400mhz,dmso)δ9.88(s,1h),7.44(d,j=6.7hz,1h),7.22(ddd,j=14.4,8.4,2.1hz,1h),7.17-7.03(m,3h),6.86(d,j=7.8hz,1h),6.80(t,j=7.4hz,1h),6.70(d,j=14.4hz,1h),3.52(s,4h),1.65-1.55(m,2h),1.48(s,4h).13cnmr(101mhz,dmso)δ164.31,155.49,142.52,133.51,129.53,127.26,127.13,123.04,120.71,119.30,115.94,46.04,42.43,26.48,25.40,24.16.exactmasscalcdforc16h19no2[m+na]+:280.1313;found280.1314.
实施例31、(2e,4e)-5-(3-羟基苯基)-1-(哌啶-1-基)戊-2,4-二烯-1-酮的制备
将化合物3c溶解于无水dcm中,-78℃预冷30分钟,滴加配好的1mbbr3dcm溶液,-78℃搅拌两小时,缓慢升到室温,常温搅拌4h到完全反应。通过tlc监测反应。反应完成后,将反应液缓慢倒入水中淬灭反应,用饱和的nahco3溶液中和反应液,并用乙酸乙酯萃取,合并有机相,减压浓缩。通过硅胶柱层析纯化得到系列化合物4c。1hnmr(400mhz,meod)δ7.33(dd,j=14.7,10.9hz,1h),7.16(t,j=7.9hz,1h),7.03-6.91(m,3h),6.83(d,j=15.6hz,1h),6.73(dd,j=8.0,2.0hz,1h),6.66(d,j=14.7hz,1h),3.67-3.56(m,4h),1.69(dd,j=10.5,5.5hz,2h),1.59(s,4h).13cnmr(101mhz,meod)δ167.62,158.88,144.26,140.43,139.20,130.77,128.02,121.54,119.77,116.93,114.43,48.14,44.54,27.86,26.87,25.53.exactmasscalcdforc16h19no2[m+na]+:280.1313;found280.1314.
实施例32、(2e,4e)-n,n-二乙基-5-(4-羟苯基)戊-2,4-二烯酰胺的制备
将化合物3d溶解于无水dcm中,-78℃预冷30分钟,滴加配好的1mbbr3dcm溶液,-78℃搅拌两小时,缓慢升到室温,常温搅拌4h到完全反应。通过tlc监测反应。反应完成后,将反应液缓慢倒入水中淬灭反应,用饱和的nahco3溶液中和反应液,并用乙酸乙酯萃取,合并有机相,减压浓缩。通过硅胶柱层析纯化得到系列化合物4d。1hnmr(400mhz,dmso)δ8.02(dd,j=15.9,11.2hz,1h),7.30(d,j=8.5hz,2h),6.80(d,j=8.5hz,2h),6.64(d,j=15.9hz,1h),6.45(t,j=11.2hz,1h),5.59(d,j=11.2hz,1h),2.75(q,j=7.2hz,4h),1.11(t,j=7.2hz,6h).13cnmr(101mhz,dmso)δ169.84,158.33,138.98,136.85,128.14,127.55,123.84,123.09,115.77,41.74,12.45.exactmasscalcdforc15h17no2[m+na]+:268.1313;found268.1316.
实施例33、(2e,4e)-n-乙基-5-(4-羟苯基)戊-2,4-二胺的制备
将化合物3e溶解于无水dcm中,-78℃预冷30分钟,滴加配好的1mbbr3dcm溶液,-78℃搅拌两小时,缓慢升到室温,常温搅拌4h到完全反应。通过tlc监测反应。反应完成后,将反应液缓慢倒入水中淬灭反应,用饱和的nahco3溶液中和反应液,并用乙酸乙酯萃取,合并有机相,减压浓缩。通过硅胶柱层析纯化得到系列化合物4e。1hnmr(400mhz,dmso)δ8.11(dd,j=15.9,11.1hz,1h),7.29(d,j=8.4hz,2h),6.84(d,j=8.4hz,2h),6.58(d,j=15.9hz,1h),6.35(t,j=11.1hz,1h),5.59(d,j=11.1hz,1h),2.79(q,j=7.2hz,2h),1.13(t,j=7.2hz,3h).13cnmr(101mhz,dmso)δ170.69,158.54,137.07,135.54,127.90,127.55,126.30,123.50,115.82,34.16,13.63.exactmasscalcdforc13h15no2[m+na]+:240.1000;found240.1001.
实施例34、(e)-1-(哌啶-1-基)-3-(4-(吡咯烷基-1-基)苯基)丙-2-烯-1-酮的制备
步骤1:
用15ml吡啶和1.5ml哌啶溶解相应的芳香醛(1.3mmol),加入丙二酸(0.3g,2.86mmol),加热回流8h,用tlc检测反应。反应完成后,在冰浴下加盐酸调ph至酸性,生成沉淀,抽滤,滤饼用冰水洗涤。用乙醇重结晶,得到相应的酸6a。
步骤2:
将6a(1mmol)溶解于2ml的n,n-二甲基甲酰胺(dmf),加入2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(hatu)(1.5mmol),然后加入哌啶(1.2mmol)。反应在室温下搅拌4小时,用tlc检测反应。反应完成后,加入10ml水淬灭反应,用乙酸乙酯2ml萃取3次,合并有机相,用无水硫酸钠干燥。减压浓缩。通过硅胶柱层析纯化得到产物7a。1hnmr(400mhz,cdcl3)δ7.61(d,j=15.2hz,1h),7.41(d,j=8.7hz,2h),6.67(d,j=15.2hz,1h),6.52(d,j=8.7hz,2h),3.62(s,4h),3.32(t,j=6.6hz,4h),2.04-1.97(m,4h),1.71-1.63(m,2h),1.60(d,j=4.6hz,4h).13cnmr(101mhz,cdcl3)δ166.30,148.76,143.11,129.37,122.73,111.55,111.49,47.55,46.85,43.37,26.72,25.46,24.78.exactmasscalcdforc18h24n2o[m+na]+:307.1786;found307.1784.
实施例35、(e)-n,n-二乙基-3-(4-(吡咯烷-1-基)苯基)丙烯酰胺的制备
将6a(1mmol)溶解于2ml的n,n-二甲基甲酰胺(dmf),加入2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(hatu)(1.5mmol),然后加入二乙胺(1.2mmol)。反应在室温下搅拌4小时,用tlc检测反应。反应完成后,加入10ml水淬灭反应,用乙酸乙酯2ml萃取3次,合并有机相,用无水硫酸钠干燥。减压浓缩。通过硅胶柱层析纯化得到产物7b。1hnmr(400mhz,cdcl3)δ7.64(d,j=15.2hz,1h),7.40(d,j=8.7hz,2h),6.58(d,j=15.2hz,1h),6.51(d,j=8.7hz,2h),3.55-3.39(m,4h),3.30(t,j=6.6hz,4h),2.10-1.92(m,4h),1.32-1.10(m,6h).13cnmr(101mhz,cdcl3)δ166.73,148.88,143.15,129.51,122.77,111.77,111.64,47.63,42.31,41.11,25.54,15.13,13.43.exactmasscalcdforc17h24n2o[m+na]+:295.1786;found295.1792.
实施例36、(e)-3-(苯并[d][1,3]二氧杂-5-基)-1-(哌啶-1-基)丙-2-烯-1-酮的制备
步骤1:
用15ml吡啶和1.5ml哌啶溶解相应的芳香醛(1.3mmol),加入丙二酸(0.3g,2.86mmol),加热回流8h,用tlc检测反应。反应完成后,在冰浴下加盐酸调ph至酸性,生成沉淀,抽滤,滤饼用冰水洗涤。用乙醇重结晶,得到相应的酸6b。
步骤2:
将6b(1mmol)溶解于2ml的n,n-二甲基甲酰胺(dmf),加入2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(hatu)(1.5mmol),然后加入哌啶(1.2mmol)。反应在室温下搅拌4小时,用tlc检测反应。反应完成后,加入10ml水淬灭反应,用乙酸乙酯2ml萃取3次,合并有机相,用无水硫酸钠干燥。减压浓缩。通过硅胶柱层析纯化得到产物7c。1hnmr(400mhz,cdcl3)δ7.56(d,j=15.3hz,1h),7.03(d,j=1.4hz,1h),6.99(dd,j=8.0,1.4hz,1h),6.79(d,j=8.0hz,1h),6.73(d,j=15.3hz,1h),5.98(s,2h),3.61(s,4h),1.71-1.64(m,2h),1.64-1.56(m,4h).13cnmr(101mhz,cdcl3)δ165.62,148.98,148.34,142.13,130.10,123.74,115.86,108.63,106.50,101.53,26.22,24.81.exactmasscalcdforc15h17no3[m+na]+:282.1106;found282.1105.
实施例37、(e)-3-(苯并[d][1,3]二氧杂-5-基)-n,n-二乙基丙烯酰胺的制备
将6b(1mmol)溶解于2ml的n,n-二甲基甲酰胺(dmf),加入2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(hatu)(1.5mmol),然后加入二乙胺(1.2mmol)。反应在室温下搅拌4小时,用tlc检测反应。反应完成后,加入10ml水淬灭反应,用乙酸乙酯2ml萃取3次,合并有机相,用无水硫酸钠干燥。减压浓缩。通过硅胶柱层析纯化得到产物7d。1hnmr(400mhz,cdcl3)δ7.62(d,j=15.3hz,1h),7.03(d,j=1.6hz,1h),7.00(dd,j=8.0,1.6hz,1h),6.80(d,j=8.0hz,1h),6.65(d,j=15.3hz,1h),5.99(s,2h),3.47(s,4h),1.25(t,j=6.5hz,3h),1.18(t,j=6.5hz,3h).13cnmr(101mhz,cdcl3)δ166.01,149.00,148.32,142.18,130.08,123.83,115.97,108.65,106.51,101.53,42.41,41.22,15.22,13.39.exactmasscalcdforc14h17no3[m+na]+:270.1106;found270.1107.0
实施例38、(e)-3-(苯并[d][1,3]二氧杂-5-基)-n-乙基丙烯酰胺的制备
将6b(1mmol)溶解于2ml的n,n-二甲基甲酰胺(dmf),加入2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(hatu)(1.5mmol),然后加入乙胺(1.2mmol)。反应在室温下搅拌4小时,用tlc检测反应。反应完成后,加入10ml水淬灭反应,用乙酸乙酯2ml萃取3次,合并有机相,用无水硫酸钠干燥。减压浓缩。通过硅胶柱层析纯化得到产物7e。1hnmr(400mhz,cdcl3)δ7.52(d,j=15.5hz,1h),6.98(d,j=1.5hz,1h),6.96(dd,j=8.0,1.5hz,1h),6.78(d,j=8.0hz,1h),6.21(d,j=15.5hz,1h),5.70(s,1h),3.41(qd,j=7.3,5.9hz,2h),1.20(t,j=7.3hz,3h).13cnmr(101mhz,cdcl3)δ166.08,149.11,148.34,140.65,129.45,123.88,118.99,108.65,106.45,101.54,34.73,15.05.exactmasscalcdforc12h13no3[m+na]+:242.0793;found242.0787.
实施例39、(e)-1-(哌啶-1-基)-3-(噻吩-2-基)丙-2-烯-1-酮的制备
步骤1:
用15ml吡啶和1.5ml哌啶溶解相应的芳香醛(1.3mmol),加入丙二酸(0.3g,2.86mmol),加热回流8h,用tlc检测反应。反应完成后,在冰浴下加盐酸调ph至酸性,生成沉淀,抽滤,滤饼用冰水洗涤。用乙醇重结晶,得到相应的酸6c。
步骤2:
将6c(1mmol)溶解于2ml的n,n-二甲基甲酰胺(dmf),加入2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(hatu)(1.5mmol),然后加入哌啶(1.2mmol)。反应在室温下搅拌4小时,用tlc检测反应。反应完成后,加入10ml水淬灭反应,用乙酸乙酯2ml萃取3次,合并有机相,用无水硫酸钠干燥。减压浓缩。通过硅胶柱层析纯化得到产物7f。1hnmr(400mhz,cdcl3)δ7.78(d,j=14.9hz,1h),7.30(d,j=5.0hz,1h),7.20(d,j=3.5hz,1h),7.03(dd,j=5.0,3.5hz,1h),6.72(d,j=14.9hz,1h),3.62(s,4h),1.68(d,j=4.2hz,2h),1.62(s,4h).exactmasscalcdforc12h15nos[m+na]+:244.0772;found244.0775.
实施例40、(e)-n,n-二乙基-3-(噻吩-2-基)丙烯酰胺的制备
将6c(1mmol)溶解于2ml的n,n-二甲基甲酰胺(dmf),加入2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(hatu)(1.5mmol),然后加入二乙胺(1.2mmol)。反应在室温下搅拌4小时,用tlc检测反应。反应完成后,加入10ml水淬灭反应,用乙酸乙酯2ml萃取3次,合并有机相,用无水硫酸钠干燥。减压浓缩。通过硅胶柱层析纯化得到产物7g。1hnmr(400mhz,cdcl3)δ7.81(d,j=15.1hz,1h),7.29(d,j=5.1hz,1h),7.20(d,j=3.5hz,1h),7.02(dd,j=5.1,3.5hz,1h),6.62(d,j=15.1hz,1h),3.55-3.37(m,4h),1.21(s,6h).13cnmr(101mhz,cdcl3)δ205.56,165.53,140.77,135.17,130.10,128.08,127.02,116.75,42.41,41.23,15.21,13.35.exactmasscalcdforc11h15nos[m+na]+:232.0772;found232.0774.
实施例41、(e)-n-乙基-3-(噻吩-2-基)丙烯酰胺的制备
将6c(1mmol)溶解于2ml的n,n-二甲基甲酰胺(dmf),加入2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(hatu)(1.5mmol),然后加入乙胺(1.2mmol)。反应在室温下搅拌4小时,用tlc检测反应。反应完成后,加入10ml水淬灭反应,用乙酸乙酯2ml萃取3次,合并有机相,用无水硫酸钠干燥。减压浓缩。通过硅胶柱层析纯化得到产物7h。1hnmr(400mhz,cdcl3)δ7.73(d,j=15.3hz,1h),7.29(d,j=5.1hz,1h),7.19(d,j=3.4hz,1h),7.02(dd,j=5.1,3.7hz,1h),6.19(d,j=15.3hz,1h),5.67(s,1h),3.48-3.36(m,2h),1.20(t,j=7.3hz,3h).13cnmr(101mhz,cdcl3)δ165.68,140.18,133.67,130.24,128.10,127.23,119.88,34.75,15.02.exactmasscalcdforc9h11nos[m+na]+:204.0459;found204.0453.
实施例42、(e)-3-(4-(哌嗪-1-基)苯基)-1-(哌啶-1-基)丙-2-烯-1-酮的制备
步骤1:
用15ml吡啶和1.5ml哌啶溶解相应的芳香醛(1.3mmol),加入丙二酸(0.3g,2.86mmol),加热回流8h,用tlc检测反应。反应完成后,在冰浴下加盐酸调ph至酸性,生成沉淀,抽滤,滤饼用冰水洗涤。用乙醇重结晶,得到相应的酸6d。
步骤2:
将6d(1mmol)溶解于2ml的n,n-二甲基甲酰胺(dmf),加入2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(hatu)(1.5mmol),然后加入哌啶(1.2mmol)。反应在室温下搅拌4小时,用tlc检测反应。反应完成后,加入10ml水淬灭反应,用乙酸乙酯2ml萃取3次,合并有机相,用无水硫酸钠干燥。减压浓缩。通过硅胶柱层析纯化得到产物p-8i。
步骤3:
将得到的化合物p-8a溶解在2ml三氟乙酸中,搅拌1小时,反应完成后加入饱和碳酸氢钠溶液淬灭反应,并用10ml乙酸乙酯萃取三次,合并有机相,减压浓缩得到淡黄色粉末状固体8a。1hnmr(400mhz,cdcl3)δ7.56(d,j=15.4hz,1h),7.44(d,j=8.7hz,2h),6.86(d,j=8.7hz,2h),6.77(d,j=15.4hz,1h),3.61(d,j=19.9hz,4h),3.49-3.36(m,4h),3.34-3.16(m,4h),1.72-1.64(m,2h),1.60(d,j=4.1hz,4h).13cnmr(101mhz,cdcl3)δ165.71,150.99,141.74,129.11,127.96,116.23,115.19,47.03,46.82,43.77,43.38,26.76,25.64,24.66.exactmasscalcdforc18h25n3o[m+h]+:300.2076;found300.2076.
实施例43、(e)-n,n-二乙基-3-(4-(哌嗪-1-基)苯基)丙烯酰胺的制备
将6d(1mmol)溶解于2ml的n,n-二甲基甲酰胺(dmf),加入2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(hatu)(1.5mmol),然后加入二乙胺(1.2mmol)。反应在室温下搅拌4小时,用tlc检测反应。反应完成后,加入10ml水淬灭反应,用乙酸乙酯2ml萃取3次,合并有机相,用无水硫酸钠干燥。减压浓缩。通过硅胶柱层析纯化得到产物p-8b。将得到的化合物p-8b溶解在2ml三氟乙酸中,搅拌1小时,反应完成后加入饱和碳酸氢钠溶液淬灭反应,并用10ml乙酸乙酯萃取三次,合并有机相,减压浓缩得到淡黄色粉末状固体8b。1hnmr(400mhz,cdcl3)δ7.64(d,j=15.3hz,1h),7.44(d,j=8.7hz,2h),6.88(d,j=8.7hz,2h),6.67(d,j=15.3hz,1h),3.48(d,j=2.6hz,4h),3.35-3.19(m,4h),3.08(dd,j=6.0,4.0hz,4h),1.26(s,6h).13cnmr(101mhz,cdcl3)δ166.21,152.17,142.17,129.08,126.67,115.38,114.36,48.94,45.56,42.26,41.09,29.70,15.06.exactmasscalcdforc17h25n3o[m+h]+:288.2076;found288.2077.
实施例44、(e)-n-乙基-3-(4-(哌嗪-1-基)苯基)丙烯酰胺的制备
将6d(1mmol)溶解于2ml的n,n-二甲基甲酰胺(dmf),加入2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(hatu)(1.5mmol),然后加入乙胺(1.2mmol)。反应在室温下搅拌4小时,用tlc检测反应。反应完成后,加入10ml水淬灭反应,用乙酸乙酯2ml萃取3次,合并有机相,用无水硫酸钠干燥。减压浓缩。通过硅胶柱层析纯化得到产物p-8c。将得到的化合物p-8c溶解在2ml三氟乙酸中,搅拌1小时,反应完成后加入饱和碳酸氢钠溶液淬灭反应,并用10ml乙酸乙酯萃取三次,合并有机相,减压浓缩得到淡黄色粉末状固体8c。1hnmr(400mhz,cdcl3)δ7.54(d,j=15.5hz,1h),7.40(d,j=8.8hz,2h),6.86(d,j=8.8hz,2h),6.21(d,j=15.5hz,1h),5.55(s,1h),3.46-3.35(m,2h),3.27-3.16(m,4h),3.07-2.97(m,4h),1.20(t,j=7.3hz,3h).13cnmr(101mhz,cdcl3)δ166.54,152.62,140.82,129.20,125.80,117.32,115.29,49.43,46.05,34.68,15.11.exactmasscalcdforc15h21n3o[m+na]+:260.1763;found260.1762.
以下通过具体的试验例证明本发明的有益效果
试验例1、本发明化合物的药理试验
本发明还提供上述化合物的药理活性筛选实验,即体外生化水平pc-12细胞保护实验。pc-12细胞来源于rattusnorvegicus(褐家鼠)肾上腺嗜铬细胞瘤,是一种交感神经系统的肿瘤细胞,为儿茶酚胺能细胞,常用于神经退行性疾病药物基于表型的体外药理活性筛选,并有大量学者利用该细胞制作体外疾病细胞模型来研究不同发病因素在神经系统疾病发病过程中的作用,同时也被用来做抗神经系统退行性疾病的药物开发研究。通过双氧水诱导的pc-12细胞损伤,可以模拟神经细胞受到的氧化应激发生的反应;采用小分子药物进行干预,通过其抑制双氧水诱导的pc-12细胞的损伤情况,可以初步判定药物对神经细胞的保护作用。
材料:h2o2:科伦化工;mtt,碧云天生物技术有限公司;血清,草原绿野生物科技有限公司;培养基,hyclone;
方法:h2o2损伤模型的制备:将pc-12细胞(1×104细胞/孔)接种在96孔板中培养24小时,然后用12.5μm化合物处理24小时,再用含有650μmh2o2新鲜培养基替换,24小时后,更换新鲜培养基,每孔加入20μl的mtt(5mg/ml),通过mtt测定法测定细胞存活率。不用化合物处理,仅使用h2o2造模为模型对照组;并设立空白对照组(正常培养pc-12细胞)。
1.化合物的配制
1)用100%的dmso将受试化合物配置成浓度为10mmol/l的溶液。
2)用rpmi1640培养基将h2o2配制成浓度为10mmol/l的溶液。
2.读出并记录每孔的原始数据,并对原始数据进行相应的转换。
细胞存活率=化合物转换值/空白值*100,其中,空白值为dmso控制组数据。
图1和表3均为本发明化合物关于pc-12细胞保护活性的平均细胞存活率。
表3本发明化合物对pc-12细胞存活率的影响
由图1和表3可知:部分化合物(3a、3g、3h、3m-3t、3w、3ab、4e、7a和7b等)具有较大的毒性,使得pc-12细胞损伤更严重,对神经细胞没有保护作用。但其余化合物显示出不同程度的神经细胞保护作用,其中化合物3b、3i~3k、3u、3v、3x、4b~4d、4f、4g、7c-7e、8a和8b,在12.5μm浓度作用下,可使pc-12细胞存活率达到60~90%,效果良好;而化合物3b对神经细胞的保护效果最优,在12.5μm浓度作用下,可使pc-12细胞存活率达到88.82%。
综上,本发明化合物能够有效保护神经细胞,提高神经细胞的存活率,因此,本发明化合物可以有效治疗神经退行性疾病,可以用于制备治疗神经退行性疾病的药物。
- 还没有人留言评论。精彩留言会获得点赞!