靶向敲除人MC1R基因的sgRNA及其构建的细胞株的制作方法
本发明属于基因工程
技术领域:
,具体涉及一种靶向敲除人mc1r基因的sgrna及其构建的细胞株,尤其是构建的人mc1r基因敲除的a375细胞株。
背景技术:
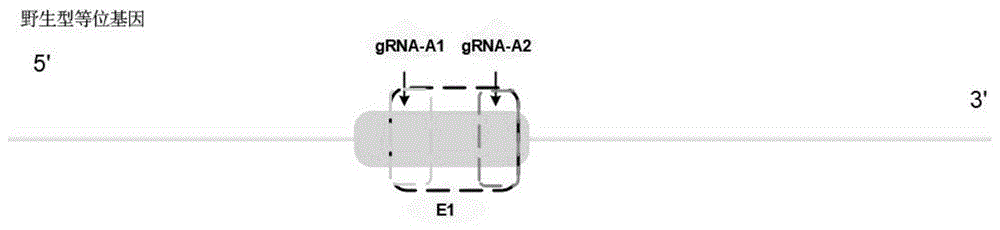
:mc1r基因在哺乳动物中由extension位点编码,为g蛋白偶联受体-黑素皮质素受体(melanocortinreceptors,mcrs)家族成员之一,只有一个编码区,其编码的蛋白质有7个跨膜结构域,主要表达于动物的黑色素细胞,为g蛋白偶联受体中最小的一个。mc1r蛋白是美白通路上研究较为透彻,也比较重要的受体之一。它是控制动物黑色素合成的重要基因,在黑素细胞、肾上腺皮质等多种细胞,以及神经系统和免疫系统中表现出不同的功能。crispr-cas9基因编辑技术作为第三代基因编辑技术,相对于前两代基因编辑技术具有构建系统简单、精准度高、成本低、并且能同时对多个位点进行定点编辑等优势,已成为目前研究的热点。它主要通过一段短的引导rna(guiderna)来识别特定的dna序列,然后由其引导的cas9蛋白定位到该特定的dna序列上进行切割,从而起到基因编辑的作用。现有实验技术中,与crispr-cas9系统最为相似的是sirna靶向的基因沉默技术,其主要缺点是在rna水平的干扰,效率低下,不适用于长期抑制研究。对于黑素生成研究尚无mc1r基因缺失细胞模型,因此建立一株mc1r基因缺失的细胞株,对于研究黑素生成及皮肤相关疾病治疗等有着十分重要的意义。技术实现要素:基于现有技术存在的缺陷,本发明的第一目的在于提供一种用于靶向敲除人mc1r基因的sgrna;本发明的第二目的在于提供一种用于靶向敲除人mc1r基因的grna表达载体;本发明的第三目的在于提供grna表达载体的构建方法;本发明的第四目的在于提供扩增上述sgrna的引物;本发明的第五目的在于提供一种用于敲除人mc1r基因的试剂盒;本发明的第六目的在于提供一种crispr-cas9系统;本发明的第七目的在于提供一种用于构建人mc1r基因敲除的细胞株的试剂盒;本发明的第八目的在于提供一种人mc1r基因敲除的细胞株的构建方法;本发明的第九目的在于提供一种人mc1r基因敲除的a375细胞株;本发明的第十目的在于提供上述人mc1r基因敲除的a375细胞株作为细胞模型在研究肤色调控机制和相关皮肤疾病中的应用。本发明的目的通过以下技术手段得以实现:一方面,本发明提供一种用于靶向敲除人mc1r基因的sgrna,其包括sgrna1和sgrna2,所述sgrna1和所述sgrna2的核苷酸序列如下:a、sgrna1:agaggtgtcgaaatgtcctgggg(seqidno:1)b、sgrna2:agacggagtgtcccaggagtggg(seqidno:2)。本发明的上述sgrna是发明人采用crispr在线设计工具(http://crispr.mit.edu/),输入mc1r基因序列(id:4157)。由于mc1r仅有一个外显子,本发明的目的是尽可能敲除或敲低mc1r基因,使其表达量降低,从而使能与其结合的效应因子如a-msh无法结合而阻断下游黑素生成通路,因此在设计生成的众多sgrna序列中,除了考虑得分率高外,还需综合评估敲除或敲低位点能否破坏mc1r蛋白编码,确定a-msh可能的结合位点是否包含在敲除序列中,同时评估脱靶风险,经过筛选最终获得了本发明上述两条sgrna寡核苷酸序列。采用上述两条sgrna能够精确指导cas9蛋白靶向切割mc1r基因。另一方面,本发明还提供一种用于靶向敲除人mc1r基因的grna表达载体,该grna表达载体包括包含上述sgrna1的dna序列的grna表达载体和包含上述sgrna2的dna序列的grna表达载体。再一方面,本发明还提供上述grna表达载体的构建方法,其包括:分别扩增上述sgrna1和sgrna2、退火获得各自的双链dna片段,通过dna连接酶将各自的双链dna片段分别连接到质粒载体上获得各自的grna表达载体。再一方面,本发明还提供一种扩增上述sgrna的引物,其引物序列包括:扩增sgrna1的引物序列:crispr-f1:5’-caccgagaggtgtcgaaatgtcctgggg-3’(seqidno:3)crispr-r1:5’-cccccaggacatttcgacacctctcaaa-3’(seqidno:4);扩增sgrna2的引物序列:crispr-f2:5’-caccgagacggagtgtcccaggagtggg-3’(seqidno:5)crispr-r2:5’-ccccactcctgggacactccgtctcaaa-3’(seqidno:6)。再一方面,本发明还提供一种用于敲除人mc1r基因的试剂盒,该试剂盒包括上述的引物序列。再一方面,本发明还提供一种crispr-cas9系统,该crispr-cas9系统包括上述grna表达载体和cas9质粒。再一方面,本发明还提供一种用于构建人mc1r基因敲除的细胞株的试剂盒,该试剂盒包括上述sgrna1构建的grna表达载体体外转录的mrna和上述sgrna2构建的grna表达载体体外转录的mrna。再一方面,本发明还提供一种人mc1r基因敲除的细胞株的构建方法,其包括如下步骤:将上述sgrna1构建的grna表达载体体外转录的mrna、上述sgrna2构建的grna表达载体体外转录的mrna和cas9质粒体外转录的mrna一并电转入宿主细胞中,然后进行单克隆培养并进行pcr鉴定;将pcr鉴定正确的阳性克隆进行扩增冻存,完成敲除细胞株的构建。上述的构建方法中,优选地,所述宿主细胞包括肿瘤细胞株。上述的构建方法中,优选地,所述肿瘤细胞株包括黑色素瘤细胞株。上述的构建方法中,优选地,所述黑色素瘤细胞株包括a375细胞株。上述的构建方法中,优选地,所述sgrna1、所述sgrna2分别构建的grna表达载体体外转录采用mmessagemmachinetmt7ultratranscriptionkit试剂盒。上述的构建方法中,优选地,所述cas9质粒体外转录采用megashortscripttmt7transcriptionkit试剂盒。上述的构建方法中,优选地,在进行电转入宿主细胞中时,所述sgrna1构建的grna表达载体体外转录的mrna和所述sgrna2构建的grna表达载体体外转录的mrna的总质量与cas9质粒转录所得的mrna质量比为1:(1~3)。上述的构建方法中,优选地,所述sgrna1构建的grna表达载体体外转录的mrna和所述sgrna2构建的grna表达载体体外转录的mrna的质量比为1:1。再一方面,本发明还提供一种人mc1r基因敲除的a375细胞株。再一方面,本发明还提供上述人mc1r基因敲除的a375细胞株作为细胞模型在研究肤色调控机制和相关皮肤疾病中的应用。本发明的有益效果:(1)本发明提供靶向敲低mc1r基因表达的sgrna能有效靶向mc1r基因,精确指导cas9蛋白靶向切割mc1r基因,将其构建入crispr-cas9系统中,转染入细胞可得到低表达的细胞株;本发明体外转录mrna采用的是mmessagemmachinetmt7ultratranscriptionkit试剂盒和megashortscripttmt7transcriptionkit试剂盒,其相对于现有三组分cas9系统,即在体外直接合成crrna和tracrrna,与cas9质粒共转细胞的系统具有构建方法简单、成本低、可操作性强的优势。(2)体外将构建成功的grna表达载体和cas9质粒转录成mrna,然后电转细胞,转化效率高,同时极大提高了敲除基因的成功率。(3)目前,用于降低蛋白表达常使用rna干扰技术,其主要缺点是在rna水平的干扰效率低下,不适用于长期抑制研究;而使用crispr-cas9系统可以构建出稳定低表达蛋白的细胞;在科研研究黑素生成与相关疾病中所涉及的分子机制中有很大的优势。(4)本发明构建的人mc1r基因敲除的a375细胞株作为细胞模型在研究肤色调控机制和相关皮肤疾病中具有重要的应用前景。附图说明图1为本发明实施例3中单克隆pcr鉴定扩增区域示意图;其中grna1、grna2及e1(mc1r编码区)分别在箭头所示位置,三个虚线框分别表示扩增的三个区域。图2为本发明实施例3中单克隆pcr扩增区域1鉴定结果;理论扩增条带大小分别为wt(野生型):600bp;杂合子:600bp;纯合子:0bp。其中1g8、3a2、3a8为3个不同单克隆,m为marker,wt为野生型即未敲除mc1r组,water为阴性对照组。图3为本发明实施例3中单克隆pcr扩增区域2鉴定结果;理论扩增条带大小分别为wt:570bp;杂合子:570bp;纯合子:0bp。其中1g8、3a2、3a8为3个不同单克隆,m为marker,wt为野生型即未敲除mc1r组,water为阴性对照组。图4为本发明实施例3中单克隆pcr扩增区域3鉴定结果;理论扩增条带大小分别为wt:2169bp;杂合子:2169/~587bp;纯合子:~587bp。其中1g8、3a2、3a8为3个不同单克隆,m为marker,wt为野生型即未敲除mc1r组,water为阴性对照组。图5为本发明实施例3中1g8克隆基因组测序结果。图6为本发明实施例4中1g8克隆细胞形态(100x)。图7为本发明实施例4中1g8细胞株与野生a375细胞株10-7ma-msh刺激下黑素生成水平检测对比图。具体实施方式为了对本发明的技术特征、目的和有益效果有更加清楚的理解,现对本发明的技术方案进行以下详细说明,但不能理解为对本发明的可实施范围的限定。文中未注明具体条件的实验方法,通常按照《分子克隆实验指南》中所述常规条件,或试剂制造厂商所建议的条件实施。除非另行定义,文中所使用的所以专业和科学用语与本领域熟练人员所熟悉的意义相同。实施例1用于靶向敲除人mc1r基因的sgrna筛选采用crispr在线设计工具(http://crispr.mit.edu/),输入mc1r基因序列(id:4157)。由于mc1r仅有一个外显子,本发明的目的是尽可能敲除或敲低mc1r基因,使其表达量降低,从而使能与其结合的效应因子如a-msh无法结合而阻断下游黑素生成通路,因此在设计生成的众多sgrna序列中,除了考虑得分率高外,还需综合评估敲除或敲低位点能否破坏mc1r蛋白编码,确定a-msh可能的结合位点是否包含在敲除序列中,同时评估脱靶风险,经过付出创造性劳动的筛选最终获得了本发明如下两条sgrna寡核苷酸序列,交由上海生工合成序列。sgrna1:agaggtgtcgaaatgtcctgggg(seqidno:1)sgrna2:agacggagtgtcccaggagtggg(seqidno:2)采用本发明上述两条sgrna能够精确指导cas9蛋白靶向切割mc1r基因。实施例2用于靶向敲除人mc1r基因的表达载体的构建将实施例1中的sgrna分别扩增退火获得双链dna片段,具体操作如下:用如下引物扩增sgrna(sgrna1或sgrna2)片段,其中crispr-f1和crispr-r1扩增sgrna1,crispr-f2和crispr-r2扩增sgrna2。crispr-f1:5’-caccgagaggtgtcgaaatgtcctgggg-3’(seqidno:3)crispr-r1:5’-cccccaggacatttcgacacctctcaaa-3’(seqidno:4)。crispr-f2:5’-caccgagacggagtgtcccaggagtggg-3’(seqidno:5)crispr-r2:5’-ccccactcctgggacactccgtctcaaa-3’(seqidno:6)。引物稀释,将crispr-f1、crispr-r1、crispr-f2、crispr-r2引物用灭菌ddh2o稀释到终浓度为10μm,pcr反应体系如下表1所示:表1:组成x1genomicdna1.5μl正向引物(10μm)1.0μl反向引物(10μm)1.0μlp112taqdna聚合酶(vazyme)12.5μlddh2o9.0μl总计25.0μl将上述表1试剂混匀后,上pcr仪,pcr反应条件如表2所示:表2:通过dna连接酶分别将两个sgrna的双链dna片段连接到各自的质粒载体lenticrisprv2上获得各自的grna表达载体。实施例3人mc1r基因敲除的细胞株的构建1、获得各自mrna:将实施例2的grna表达载体和cas9质粒(赛业(广州)生物科技有限公司提供)分别体外转录获得各自的mrna;具体转录方法如下:(1)参照mmessagemmachinetmt7ultratranscriptionkit试剂盒说明进行cas9质粒体外转录,获得cas9质粒的mrna。(2)参照megashortscripttmt7transcriptionkit试剂盒说明将上述grna表达载体进行体外转录,获得sgrna1的mrna和sgrna2的mrna。(3)经licl沉淀回收后,nanodrop3000测定核酸浓度,rna凝胶电泳检测体外转录质量。2、mc1r敲除a375细胞株构建、鉴定用含10%胎牛血清的dmem高糖培养基,于5%co2,37℃恒温培养a375细胞;取对数期细胞用电转法将转录所得的cas9和sgrna1、sgrna2的mrna电转入a375细胞中,电转条件如下:bio-radgenepulserxcell电转仪,250v,950uf,200ω,2mm电转杯,细胞量1.0×106个,cas9的mrna1500ng和sgrna1、sgrna2的mrna各500ng,转染后继续培养48小时,消化收集细胞。将电转后的细胞稀释成单克隆接种至96孔板中培养,待单个细胞扩增到一定数量后,进行96孔板传代,继续单克隆的培养,将其中一块96孔板做pcr鉴定。扩增区域示意图如图1所示,扩增区域引物如下:扩增区域1(退火温度62.0℃):上游引物:tgacgaggggaggggtgaa(seqidno:7)下游引物:gcttagttcatggtgctgcca(seqidno:8)扩增区域2(退火温度62.0℃):上游引物:tcaaggaggtgctgacatgc(seqidno:9)下游引物:gaggcagggatttcacctcc(seqidno:10)扩增区域3(退火温度62.0℃):上游引物:tgacgaggggaggggtgaa(seqidno:11)下游引物:gaggcagggatttcacctcc(seqidno:12)结果显示,3个克隆均为纯合子,完全敲除了mc1r基因编码区,结果见图2、3和4所示。图2结果显示3个单克隆区域1均未扩增出条带,野生型有目的条带,说明3个单克隆在此区域检测为纯合子。图3结果显示3个单克隆区域2均未扩增出条带,野生型有目的条带,说明3个单克隆在此区域检测为纯合子。图4结果显示3个克隆均有570bp左右的单一条带,说明3个克隆均为纯合子且已敲除mc1r基因。选取编号为1g8的克隆进行基因组测序,测序结果如图5所示:该克隆成功敲除mc1r基因编码区。实施例4mc1r敲除细胞株无菌、形态及功能鉴定用含10%胎牛血清的dmem高糖培养基,于5%co2,37℃恒温培养实施例3中获得的1g8细胞株,观察细胞形态及生长情况,并用按标准进行无菌检测,结果如图6所示,图6显示敲除mc1r基因的1g8细胞株生长状态良好,形态正常,无菌检测合格,证明mc1r基因敲除的a375细胞株生长及形态未受基因敲除影响。取培养好的1g8细胞和野生型的a375细胞,按照2e5/cm2接种6孔板,如上条件培养24h后,对照组为正常培养的a375和1g8细胞,实验组分别添加10-7m终浓度的a-msh,继续培养48h后消化收集细胞,采用碱裂解法提取细胞黑色素,酶标仪于450nm波长处检测吸光度,按照公式计算黑素生成相对抑制率。黑素生成抑制率=实验组od450/a375对照组od450%×100%。结果如图7所示:野生型a375在a-msh刺激下黑素生成显著增加,而敲除mc1r基因的1g8细胞株,无论是否有a-msh刺激,黑素生成均显著降低;即:敲除mc1r的1g8细胞株黑素生成较野生型a375细胞黑素生成显著降低,表明所构建的mc1r基因敲除细胞株功能符合预期要求,mc1r基因敲除细胞株构建成功,可作为后续黑素生成、抑制及相关功能筛选、验证所需细胞模型。以上说明所提供的sgrna1、sgrna2序列所构建出的表达载体可以有效的敲除mc1r基因,可以应用于细胞,具体可用于a375细胞中。上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。序列表<110>陕西慧康生物科技有限责任公司<120>靶向敲除人mc1r基因的sgrna及其构建的细胞株<130>gai19cn4843<160>12<170>siposequencelisting1.0<210>1<211>23<212>dna<213>人工序列()<400>1agaggtgtcgaaatgtcctgggg23<210>2<211>23<212>dna<213>人工序列()<400>2agacggagtgtcccaggagtggg23<210>3<211>28<212>dna<213>人工序列()<400>3caccgagaggtgtcgaaatgtcctgggg28<210>4<211>28<212>dna<213>人工序列()<400>4cccccaggacatttcgacacctctcaaa28<210>5<211>28<212>dna<213>人工序列()<400>5caccgagacggagtgtcccaggagtggg28<210>6<211>28<212>dna<213>人工序列()<400>6ccccactcctgggacactccgtctcaaa28<210>7<211>19<212>dna<213>人工序列()<400>7tgacgaggggaggggtgaa19<210>8<211>21<212>dna<213>人工序列()<400>8gcttagttcatggtgctgcca21<210>9<211>20<212>dna<213>人工序列()<400>9tcaaggaggtgctgacatgc20<210>10<211>20<212>dna<213>人工序列()<400>10gaggcagggatttcacctcc20<210>11<211>19<212>dna<213>人工序列()<400>11tgacgaggggaggggtgaa19<210>12<211>20<212>dna<213>人工序列()<400>12gaggcagggatttcacctcc20当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1