特异性靶向CLCN7的sgRNA及其应用的制作方法
本发明涉及生物技术领域,尤其是涉及一种特异性靶向clcn7的sgrna及其应用。
背景技术:
石骨症(osteopetrosis,op)也称大理石骨病、骨硬化症和粉笔样骨,是一种与破骨细胞分化或功能异常为主要病变的骨代谢异常疾病,也是一种典型的人类遗传病,临床表现为全身骨硬化、骨塑形异常、贫血、感染、肝脾肿大、骨折以及视神经萎缩及耳聋等。该疾病的性别差异不明显,各年龄阶段均可发病。根据石骨症的遗传方式将其分为常染色体隐性遗传和常染色体显性遗传两大类,进一步根据临床表现优分别可以细分为7型和2型。
clc型氯离子通道是一类广泛分布的阴离子通道,参与细胞内囊泡的酸化、肌肉细胞的电压稳定性等多种生理过程,其突变或功能异常会导致多种疾病的发生。clc-7是clc家族的重要成员,主要位于后期内涵体、溶酶体和破骨细胞的竖起膜。已有实验证明,在小鼠和人体内,损失clc-7会导致石骨症。clc-7的编码基因为clcn7。人体内的clcn7突变可以引起三种类型的石骨症,包括婴儿型恶性常染色体隐性遗传石骨症、中间型常染色体石骨症和ⅱ型常染色体显性遗传石骨症。目前,石骨症的有效治疗手段较为有限,其中同种异体造血干细胞移植是治疗严重石骨症的有效方法之一,但该方法受到多种因素限制,未能在良性石骨症治疗中得到应用。因此,有必要利用现有的基因编辑工具对clcn7基因进行敲除或修饰等研究,进一步解析该基因的相关功能,有效指导当前石骨症的科研和治疗工作的开展。
技术实现要素:
本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明利用crispr技术针对clcn7基因设计并筛选获得了特异性靶向clcn7的sgrna,以及该sgrna的相关应用。
第一方面,本发明的一个实施例提供了一种特异性靶向clcn7的sgrna,该sgrna的核苷酸序列包括以下任一种:
sg1rna:gccggagugucagcggcguu(seqidno.1);
sg2rna:gagcgggacuucgucuccgca(seqidno.2)。
本发明实施例的胰岛分离装置至少具有如下有益效果:
为了实现对clcn7基因的编辑,首先要根据该基因的识别序列设计特异性sgrna,但是值得注意的是sgrna在识别靶位点时容易出现碱基错配或者形成dna/rna凸起,导致crispr/cas9系统产生脱靶效应。因此,在设计sgrna时通过对其潜在脱靶位点的预测,减少sgrna与脱靶位点的碱基配对数:靠近pam区至少有2个碱基不配对,避免有连续或间隔的4个碱基配对,以此来提高sgrna特异性。遵循上述原则,在clcn7基因的外显子区设计了两条长20bp的sgrna,其靶点序列的间隔为3bp,其后端紧邻pam(前间区序列邻近基序,ngg)。
通过该sgrna构建表达crispr/cas9能够有效对clcn7实现基因的编辑,从而为下一步clcn7致病位点突变的修复提供参考。
第二方面,本发明的一个实施例提供了一种编码该sgrna的dna分子。
根据本发明的一些实施例的dna分子,该dna分子为双链dna,其正向序列和反向序列的核苷酸序列如下:
sg1rna-f:gccggagtgtcagcggcgtt(seqidno.3);
sg1rna-r:aacgccgctgacactccggc(seqidno.4);
sg2rna-f:gagcgggacttcgtctccgca(seqidno.5);
sg2rna-r:tgcggagacgaagtcccgctc(seqidno.6)。
第三方面,本发明的一个实施例提供了一种重组载体,该重组载体包括上述的sgrna或dna分子。而该dna分子可以转录为sgrna,该重组载体上的sgrna能够对靶基因clcn7进行编辑。该该重组载体上还可以包括针对用于表达的宿主细胞所选择的调节元件,如启动子、增强子、转录终止信号等。
根据本发明的一些实施例的重组载体,该重组载体为质粒载体或病毒载体。质粒载体可以是任选的质粒,病毒载体可以是任选的病毒,如慢病毒、腺病毒等。
第四方面,本发明的一个实施例提供了一种重组细胞,该重组细胞包括上述的重组载体。该重组细胞通过本领域熟知的方式将上述重组载体导入到细胞内部,使sgrna能够在该重组细胞中表达。
第五方面,本发明的一个实施例提供了一种组合物,该组合物包括上述的重组载体。包含该重组载体的组合物可以产生sgrna,从而在特定的条件下对目标的clcn7基因进行基因编辑。该组合物可以以制剂或试剂盒形式存在。该组合物除包含上述的重组载体外,还可以包含一定量的辅料,如水等溶剂/稀释剂,其它辅药等。
第六方面,本发明的一个实施例提供了一种clcn7基因的编辑方法,该编辑方法包括以下步骤:
利用上述sgrna特异性识别并结合目的基因片段,
将核酸酶引导到目的基因片段后对目的基因片段进行剪切;
该编辑方法不适用于疾病的诊断和治疗。
上述sgrna特异性识别结合clcn7基因的目的片段后,核酸酶在pam序列引导下与clcn7基因的目的片段结合,对该目的片段其进行剪切。随后,该目的片段可以进一步通过非同源末端连接修复基质修复后产生移码突变敲除目的基因,或者将外源dna片段插入切口处实现定点敲入。
第七方面,本发明的一个实施例提供了上述sgrna在clcn7基因的特异性切割、特异性敲除、特异性敲除中的应用。通过该sgrna可以在基因编辑过程中实现对clcn7基因的特异性切割、特异性敲除、特异性敲除,从而修复clcn7基因的致病突变位点的修复。
附图说明
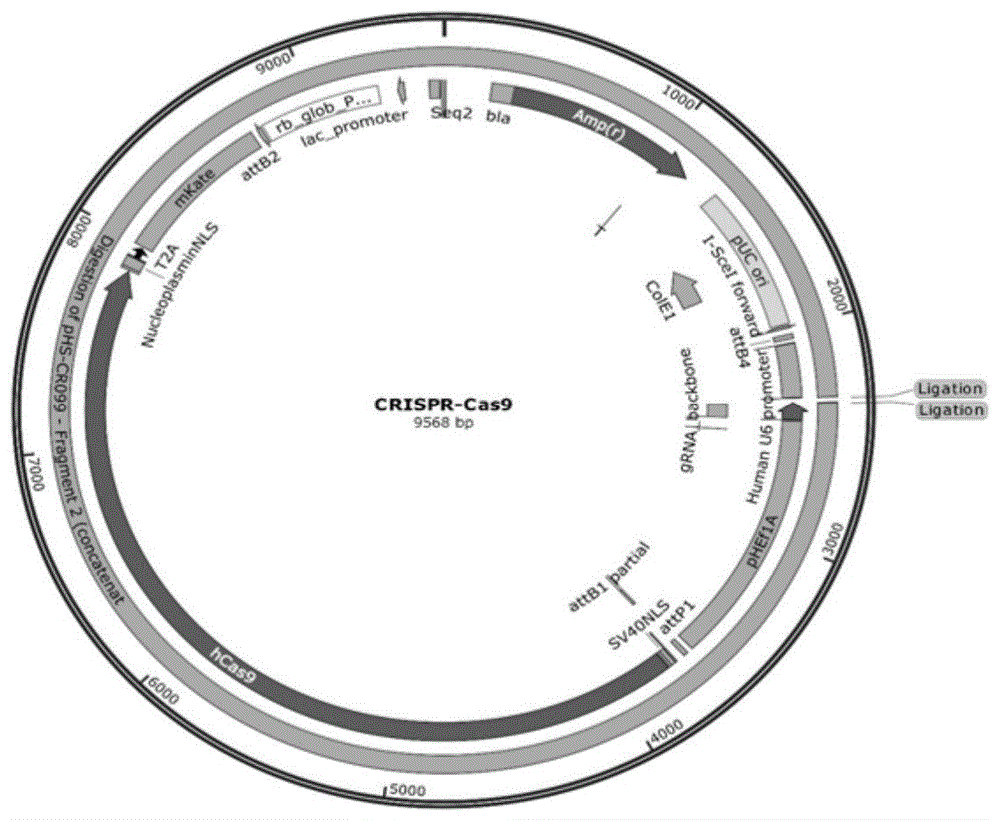
图1是本发明的一个实施例的crispr/cas9质粒图谱;
图2是本发明的一个实施例的琼脂糖凝胶电泳检测sg1rna和sg2rna条带的结果;
图3是本发明的一个实施例的酶切结果的鉴定;
图4是本发明的一个实施例的重组质粒的鉴定;
图5是本发明的一个实施例的重组质粒clcn7-sg1rna-crispr/cas9的测序结果;
图6是本发明的一个实施例的重组质粒clcn7-sg2rna-crispr/cas9的测序结果。
具体实施方式
以下将结合实施例对本发明的构思及产生的技术效果进行清楚、完整地描述,以充分地理解本发明的目的、特征和效果。显然,所描述的实施例只是本发明的一部分实施例,而不是全部实施例,基于本发明的实施例,本领域的技术人员在不付出创造性劳动的前提下所获得的其他实施例,均属于本发明保护的范围。
实施例1
本实施例提供一种特异性靶向clcn7基因的sgrna以及包括该sgrna的重组质粒。
本实施例中各材料来源如下:
crispr/cas9质粒购自北京赛贝生物技术有限公司,质粒图谱见图1。
感受态细胞dh5α购自天根生化科技(北京)有限公司。
bspqi高保真限制性内切酶和t4连接酶购自neb公司。
dna凝胶回收试剂盒和质粒小提试剂盒及top10感受态细胞购自天根生化科技(北京)有限公司。
高保真pcr酶购自宝日医生物科技(北京)有限公司。
dna引物由苏州金唯智生物科技有限公司提供,dna序列测序由北京六合华大基因科技有限公司完成。
本实施例中特异性靶向clcn7基因的sgrna以及包括该sgrna的重组质粒的制备方法如下:
1.sgrna的设计
根据https://www.nlm.nih.gov/网站确认clcn7基因序列(s1);根据http://crispr.mit.edu/网站设计在clcn7基因序列设计sgrna序列。其中sgrna的设计原则为:起始碱基为g,靶位点后序列的pam序列为ngg;forward链5′端添加cacc,reverse链的5′端添加aaac,并经过bspqi酶切后形成的粘性末端进行互补形成闭环质粒。
表1.clcn7-sgrna序列和基因组检测引物序列
2.重组质粒的构建
利用bspqi对crispr/cas9质粒进行酶切,通过agarose凝胶电泳分离目的条带后,由胶回收方法获得线性化载体;对sg1rna和sg2rna进行退火实验后,分别使用t4dna连接酶将退火后的产物连接至线性crispr/cas9载体,实验反应条件:16℃,过夜。
将连接产物转化至top10感受态细胞中,涂布在含有氨苄青霉素抗性的lb平板上,进行克隆筛选,并使用pcr的方法(采用表1中的引物对cc-cx-f、cc-cx-r扩增)鉴定目片段是否已插入,最后挑取阳性克隆进行sanger测序,确认插入片段的序列。
本实施例的结果如下:
1sg1rna和sg2rna的退火
将合成的sg1rna-f/sg1rna-r和sg2rna-f/sg2rna-r序列,稀释至10μm后,两对引物以1:1比例进行混合,在95℃下孵育10min,移至室温下自然冷却2h以上,琼脂糖凝胶电泳结果如图2所示,图2中m为dnamarker(dl2000),1泳道是目的片段sg1rna,2泳道是目的片段sg2rna。
从图2中分析发现双链寡核苷酸片段sg1rna和sg2rna电泳条带与目的片段大小一致。
2.crispr/cas9质粒酶切
质粒crispr/cas9序列上具有bspqi酶切位点(参考图1),采用bspqi酶切断质粒并且去磷酸化后,琼脂糖电泳检测结果如图3所示。图3中m为dnamarker(dl10000),1泳道是限制性内切酶bspqi酶酶切质粒crispr/cas9获得的dna片段。
结合图3,酶切获得的dna片段为9.5kbp,与预期大小一致。
3.重组质粒clcn7-sg1rna-crispr/cas9和clcn7-sg2rna-crispr/cas9构建
在重组质粒clcn7-sg1rna-crispr/cas9和clcn7-sg2rna-crispr/cas9的构建过程中,分别将线性化载体与寡核苷酸使用t4连接酶连接,并转化至top10细胞中,并分别挑取8个克隆,扩培后提取质粒,使用表1中的dna引物cc-cx-f和cc-cx-r进行pcr分析,电泳结果如图4所示。图4中,m为dnamarker(dl2000),泳道1~8为重组质粒clcn7-sg1rna-crispr/cas9的电泳结果,泳道9~16为重组质粒clcn7-sg2rna-crispr/cas9的电泳结果。从图4中可以看出,除了重组质粒clcn7-sg1rna-crispr/cas9的6号克隆和clcn7-sg2rna-crispr/cas9的16号克隆外均为阳性克隆。
选取阳性克隆进行sanger测序,结果如图5和图6所示。图5和图6分别表示重组质粒clcn7-sg1rna-crispr/cas9的测序结果和重组质粒clcn7-sg2rna-crispr/cas9的测序结果,图5和图6中加注背景的序列为插入序列。结合图5和图6可以看出,阳性克隆成功插入了目的序列sg1rna和sg2rna,插入的碱基序列与预期一致,该结果表明证实重组质粒clcn7-sg1rna-crispr/cas9和clcn7-sg2rna-crispr/cas9已构建成功。
实施例2
将实施例1中的重组质粒clcn7-sg1rna-crispr/cas9和clcn7-sg2rna-crispr/cas9电转入小鼠胚胎干细胞,g418筛选后得到阳性克隆。
取未转入重组质粒的小鼠胚胎干细胞作为对照,将阳性克隆细胞与对照细胞分别放入细胞裂解液裂解,取裂解液上清扩增clcn7基因。
结果显示,阳性克隆细胞的扩增片段长度明显小于对照细胞的扩增片段长度,该结果表明,实施例1所提供的重组质粒能够对clcn7基因序列进行剪切,从而实现基因编辑。
实施例3
一种组合物,包括实施例1中的重组载体。利用该组合物可以实现对clcn7基因的编辑,用于clcn7基因的敲除或修饰工作。
上面结合附图对本发明实施例作了详细说明,但是本发明不限于上述实施例,在所述技术领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下作出各种变化。此外,在不冲突的情况下,本发明的实施例及实施例中的特征可以相互组合。
sequencelisting
<110>深圳市人民医院
<120>特异性靶向clcn7的sgrna及其应用
<130>1
<160>8
<170>patentinversion3.5
<210>1
<211>20
<212>rna
<213>人工序列
<400>1
gccggagugucagcggcguu20
<210>2
<211>21
<212>rna
<213>人工序列
<400>2
gagcgggacuucgucuccgca21
<210>3
<211>20
<212>dna
<213>人工序列
<400>3
gccggagtgtcagcggcgtt20
<210>4
<211>20
<212>dna
<213>人工序列
<400>4
aacgccgctgacactccggc20
<210>5
<211>21
<212>dna
<213>人工序列
<400>5
gagcgggacttcgtctccgca21
<210>6
<211>21
<212>dna
<213>人工序列
<400>6
tgcggagacgaagtcccgctc21
<210>7
<211>20
<212>dna
<213>人工序列
<400>7
catcttgtggaaaggacgaa20
<210>8
<211>20
<212>dna
<213>人工序列
<400>8
cccgcatttacaagactatc20
- 还没有人留言评论。精彩留言会获得点赞!
- 肿瘤靶向核磁共振/荧光双模态成像造影剂及其制备和应用的制造方法与工艺
- pH氧化还原双敏感的PAMAM靶向纳米给药载体及其制备方法与制造工艺
- 具有卵巢肿瘤靶向D构型多肽及其基因递释系统的制造方法与工艺
- 靶向survivin纳米抗体及其制备方法和应用与制造工艺
- 用于PSMA靶向的成像和放射疗法的金属/放射性金属标记的PSMA抑制物的制造方法与工艺
- 一种线粒体靶向的粘度荧光探针及其制备方法和应用与制造工艺
- 一种兼具荧光可视、磁靶向和pH敏多功能的复合空心微球药物载体及其制备方法与制造工艺
- 一种用于修饰微泡的多肽以及靶向GBM的药物制剂的制造方法与工艺
- 白果内酯作为脑靶向增效剂在制备防治脑肿瘤药物的应用的制造方法与工艺
- 机器人辅助精确靶向穿刺软组织目标柔性针夹紧固定装置的制造方法