一种镍基催化合成3-去氨甲酰基头孢呋辛酸的方法与流程
本发明涉及一种镍基催化合成3-去氨甲酰基头孢呋辛酸的方法,属于医药合成技术领域。
背景技术:
3-去氨甲酰基头孢呋辛酸是合成头孢呋辛酸的中间产物。目前,该化合物的合成方法是先将3-氨基头孢烷酸(7-aca)与n-甲氧亚氨基呋喃乙酰氯(smif-cl)在7位上酰胺化缩合,然后水解去掉3位上的乙酰基得到。其中,7位上的酰胺化缩合反应是工艺合成的关键。该合成方法使用smif-cl作为酰胺化试剂,存在以下问题:1.smif-cl的合成工序繁琐且路线较长,需要先将2-呋喃基-2-甲氧亚氨基乙酸成盐合成呋喃铵盐,再与酰化试剂酰化合成smif-cl,最后经酰胺化合成目标产物。2.smif-cl合成过程中使用三氯氧磷或五氯化磷做酰化试剂,造成呋喃铵盐上z型甲氧亚胺基(c=n-or)在酸性环境中极易发生异构化,生成e型异构体杂质且很难除去。3.由于smif-cl的活泼性较高,室温下极易发生分解,不适合长期储存,给生产造成较大困难。4.工艺能耗较高(合成温度为-20~-40℃),且产生大量废水和磷酸盐固体微废。
技术实现要素:
本发明的目的在于提供一种3-去氨甲酰基头孢呋辛酸的合成方法,以2-呋喃基-2-甲氧亚氨基乙酸为起始原料,经氨基酰胺化反应和碱性水解反应得到3-去氨甲酰基头孢呋辛酸。该方法合成步骤简单,原料易得,目标产物中反式异构杂质生成量少。
根据本发明的一个方面,提供一种镍基催化合成3-去氨甲酰基头孢呋辛酸的方法,包括以下步骤:
将2-呋喃基-2-甲氧亚氨基乙酸和镍基催化剂置于反应装置中,再加入溶剂,搅拌均匀得到第一混合物;
将3-氨基头孢烷酸溶解在氢氧化钠溶液中得到第二混合物;
将所述第二混合物加入所述第一混合物中,进行第一反应,得到包含以下式3的化合物的混合物;
向包含式3的化合物的混合物中加入碱液,进行第二反应,得到3-去氨甲酰基头孢呋辛酸,
在所述第一反应中,2-呋喃基-2-甲氧亚氨基乙酸与3-氨基头孢烷酸的摩尔比为1.05~1.25:1。第一反应温度为10~80℃,第一反应时间为2~10h。
在所述第二反应中,第二反应温度为-10~-15℃,第二反应时间为5~15min。
所述镍基催化剂的摩尔用量为2-呋喃基-2-甲氧亚氨基乙酸的摩尔量的3~40%。
所述镍基催化剂为1,3-(双二苯基膦)丙烷氯化镍、1,2-(双二苯基膦)乙烷氯化镍、二-3-苯基膦氯化镍、醋酸镍中的一种或多种。
优选地,所述方法进一步包括将第二反应完成后的混合物进行过滤,得到固体镍基催化剂和滤液。
优选地,所述方法进一步包括向所述滤液添加酸结晶析出3-去氨甲酰基头孢呋辛酸。
优选地,所述方法进一步包括对滤出的固体镍基催化剂进行洗涤、干燥和回收。
所述溶剂选自乙腈、丙酮、四氢呋喃、n,n-二甲基甲酰胺、二甲基亚砜、二氯甲烷和1,4-二氧六环。
具体地,根据本发明的方法具有如下优点:以2-呋喃基-2-甲氧亚氨基乙酸为起始原料,在镍基催化剂的作用下,一步催化呋喃乙酸直接与7-aca上的氨基进行酰胺化反应,再经3位上碱性水解直接合成3-去氨甲酰基头孢呋辛酸。该方法可以大幅缩短目标化合物的合成步骤,且避免了三氯氧磷或五氯化磷等酰胺化试剂的使用,降低目标产物中反式异构杂质的含量,并且具有工艺操作简单,安全性可靠的特点。
附图说明
参考随附的附图,本发明更多的目的、功能和优点将通过本发明实施方式的如下描述得以阐明,其中:
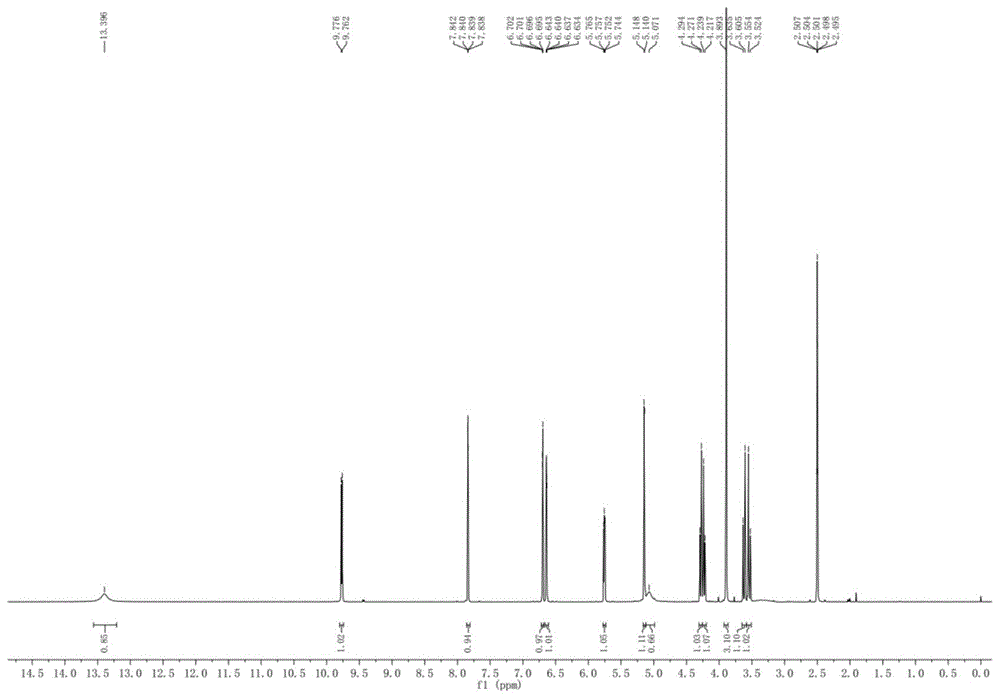
图1是根据本发明实施例4制得的产物化合物的核磁氢谱。
具体实施方式
根据本发明提供的镍基催化合成3-去氨甲酰基头孢呋辛酸的方法,包括以下步骤:
将2-呋喃基-2-甲氧亚氨基乙酸和镍基催化剂和催化剂置于反应装置中,再加入溶剂,搅拌均匀得到第一混合物;
将3-氨基头孢烷酸溶解在氢氧化钠溶液中得到第二混合物;
将所述第二混合物加入所述第一混合物中,进行第一反应,得到以下式3的化合物;
向式3的化合物中加入碱液,进行第二反应,得到3-去氨甲酰基头孢呋辛酸,
以上合成方法的反应过程用以下示意式表示:
根据一个具体实施方式,将2-呋喃基-2-甲氧亚氨基乙酸(化合物1)和镍基催化剂放入到反应装置中,再加入溶剂,在室温下充分搅拌30~40min,得到第一混合物;将3-氨基头孢烷酸(7-aca,化合物2)溶解在氢氧化钠溶液中得到第二混合物;然后将第二混合物加入到第一混合物中。
其中,上述反应装置是密闭的,经过预先干燥。上述溶剂选自乙腈、丙酮、四氢呋喃、n,n-二甲基甲酰胺、二甲基亚砜、二氯甲烷和1,4-二氧六环,优选乙腈、四氢呋喃或n,n-二甲基甲酰胺。溶剂的加入量为2-呋喃基-2-甲氧亚氨基乙酸质量的5~10倍。
上述镍基催化剂为1,3-(双二苯基膦)丙烷氯化镍、1,2-(双二苯基膦)乙烷氯化镍、二-3-苯基膦氯化镍、醋酸镍中的一种或多种。该镍基催化剂的摩尔用量为2-呋喃基-2-甲氧亚氨基乙酸的摩尔量的3~40%,优选5~20%。
上述氢氧化钠溶液优选为百分比为10%的氢氧化钠水溶液。该氢氧化钠溶液与3-氨基头孢烷酸的质量比为2~2.5:1。
在如以上反应式(i)所示的第一反应(氨基酰胺化反应)过程中,2-呋喃基-2-甲氧亚氨基乙酸(化合物1)和3-氨基头孢烷酸(化合物2)在催化剂的作用下,在10~80℃(温度1)下进行2~10h(时间1),得到包含化合物3的混合物。温度1进一步优选为30~45℃。时间1进一步优选为5~7h。
在如以上反应式(ii)所示的第二反应(氨基酰胺化反应)过程中,将第一反应完成后的混合物降温至-10~-15℃(温度2),在碱液的作用下,进行第二反应(水解反应)5~15min(时间2),优选10min,得到包括目标化合物3-去氨甲酰基头孢呋辛酸(化合物4)的混合物。该步骤中所用的碱液优选为质量百分比为10%的氢氧化钠水溶液。在该步骤中,降至-10~-15℃的温度,加入氢氧化钠的目的是为了水解掉化合物3的甲酰基,为酯的水解。降温的目的是为了避免在温度高的情况下主环β-内酰胺环在碱性条件下也发生水解。
进一步地,上述方法还包括将上述包括3-去氨甲酰基头孢呋辛酸(化合物4)的混合物进行过滤,其中,滤出的固体催化剂,再用二氯甲烷洗涤后干燥备用,滤液则继续加入10%盐酸析晶过滤得到3-去氨甲酰基头孢呋辛酸。
通过本发明的上述合成方法,可以镍基催化进行酰胺化反应,以及碱性水解两步直接合成3-去氨甲酰基头孢呋辛酸,并且由于没有使用三氯氧磷或五氯化磷做酰化试剂,体系避免了酸性环境下呋喃铵盐上z型甲氧亚胺基(c=n-or)发生异构化,生成e型异构体的可能性,从而使得目标产物收率高,纯度可达到99%以上,目标产物中反式异构体杂质的含量降低至0.05%以下,并且催化剂回收率高,工艺操作简单,成本低,且安全性高。
以下结合实施例对本发明做进一步描述。
实施例1
向密闭的预先干燥好的反应容器内,加入44g(0.26mol)2-呋喃基-2-甲氧亚氨基乙酸和催化剂7g(0.013mol,5%mol)的nicl2(dppp),再加入280ml的乙腈后,在室温下充分搅拌30分钟后缓慢加入溶解在130g10%氢氧化钠溶液中的65g(0.24mol)7-aca,然后在30℃下继续反应5h。然后,降温至-10℃后加入碱液136g10%氢氧化钠溶液,继续反应10min后过滤。滤出的固体催化剂用二氯甲烷多次洗涤后干燥备用。滤液继续滴加10%盐酸析晶,过滤得到81.25g3-去氨甲酰基头孢呋辛酸。
检测结果:产率125%wt,反式异构杂质0.01%(由液相色谱谱图峰面积计算得到),纯度99.3%。催化剂回收率95%wt。
其中,液相色谱测试条件如下:
仪器:岛津2030系列的hplc系统
检测器:273nmuv
色谱柱:等效柱
进样量:25μl
流速:1ml/min
柱温:30℃
分析方法:非梯度
计算方法:外标法
流动相配制:0.68g乙酸钠,6.01g冰乙酸溶入950ml纯化水中,加入230ml乙腈,调节ph值为3.6,过滤脱气。
ph7.0缓冲液的配制:称取约5.68g磷酸氢二钠,3.63g磷酸二氢钾,用水溶解、稀释至1000ml。
实施例2
向密闭的预先干燥好的反应容器内,加入45g(0.27mol)2-呋喃基-2-甲氧亚氨基乙酸和催化剂28g(0.053mol,20%mol)的nicl2(dppe),再加入300ml的丙酮后,在室温下充分搅拌30分钟后缓慢加入溶解在130g10%氢氧化钠溶液中的65g(0.24mol)7-aca,然后在50℃下继续反应7h。然后,降温至-10℃后加入碱液136g10%氢氧化钠溶液,继续反应10min后过滤。滤出的固体催化剂用二氯甲烷多次洗涤后干燥备用。滤液继续滴加10%盐酸析晶过滤得到85.8g3-去氨甲酰基头孢呋辛酸。
检测结果:产率132%wt,反式异构杂质0.03%(由液相色谱谱图峰面积计算得到,测试条件与实施例1相同),纯度99.2%。催化剂回收率98%wt。
实施例3
向密闭的预先干燥好的反应容器内,加入49g(0.29mol)2-呋喃基-2-甲氧亚氨基乙酸和催化剂5.13g(0.029mol,10%mol)的醋酸镍ni(ch3coo)2,再加入280ml的的n,n-二甲基甲酰胺后,在室温下充分搅拌30分钟后缓慢加入溶解在162.5g10%氢氧化钠溶液中的65g(0.24mol)7-aca,然后在10℃下继续反应10h。然后,降温至-10℃后加入碱液136g10%氢氧化钠溶液,继续反应10min后过滤。滤出的固体催化剂用二氯甲烷多次洗涤后干燥备用。滤液继续滴加10%盐酸析晶过滤得到84.5g3-去氨甲酰基头孢呋辛酸。
检测结果:产率130%wt,反式异构杂质0.01%(由液相色谱谱图峰面积计算得到,测试条件与实施例1相同),纯度99.1%。催化剂回收率97%wt。
实施例4
向密闭的预先干燥好的反应容器内,加入49g(0.29mol)2-呋喃基-2-甲氧亚氨基乙酸和催化剂7.4g(0.014mol,5%mol)的nicl2(dppe),再加入300ml的n,n-二甲基甲酰胺后,在室温下充分搅拌30分钟后缓慢加入溶解在130g10%氢氧化钠溶液中的65g(0.24mol)7-aca,然后在80℃下继续反应2h。然后,降温至-15℃后加入碱液136g10%氢氧化钠溶液,继续反应10min后过滤。滤出的固体催化剂用二氯甲烷多次洗涤后干燥备用。滤液继续滴加10%盐酸析晶过滤得到83.2g3-去氨甲酰基头孢呋辛酸。
检测结果:产率128%wt,未检测到反式异构杂质(由液相色谱谱图峰面积计算得到,测试条件与实施例1相同),纯度99.4%。催化剂回收率99%wt。对制得的目标化合物进行核磁氢谱测定,结果如图1所示。1hnmr(600mhz,cdcl3,δppm):13.40(s,1h),9.77(d,j=7.8hz,1h),7.84(m,1h),6.70-6.69(m,1h),6.64-6.63(m,1h),5.76-5.74(m,1h),5.14(d,j=4.8hz,1h),5.07(s,1h),4.28(d,j=13.8hz,1h),4.23(d,j=13.2hz,1h),3.89(s,3h),3.62(d,j=18.0hz,1h),3.54(d,j=18.0hz,1h)。
结合这里披露的本发明的说明和实践,本发明的其他实施例对于本领域技术人员都是易于想到和理解的。说明和实施例仅被认为是示例性的,本发明的真正范围和主旨均由权利要求所限定。
- 还没有人留言评论。精彩留言会获得点赞!