一种扩链剂及其制备方法和应用与流程
本发明属于高分子材料领域,具体涉及一种扩链剂及其制备方法和应用。
背景技术:
聚氨酯(pu)是最重要的聚合物之一,由于其优异的性能,已在涂料,密封剂,粘合剂,泡沫和复合材料等领域得到广泛的应用。目前制备聚氨酯用的亲水扩链剂大多是二羟甲基丁酸(dmba)、2,2-二羟甲基丙酸(dmpa)等几种。这些亲水扩链剂在有机溶剂中的溶解度较低,导致制备pu时使用大量的溶剂,产生的挥发性有机化合物(voc),包括甲苯、丁酮等,对环境和施工人员造成极大的伤害。另一方面,现在石化资源面临枯竭,导致传统亲水扩链剂的价格波动比较大。因此制备新型的、环境友好的、价格低廉的亲水扩链剂势在必行。
近年来许多科研工作者致力于合成新的扩链剂。其中光点击反应由于其高效快速、反应条件简单、易于纯化等优点在制备多元醇、扩链剂等方面得到广泛的应用。desroches等通过油酸与巯基乙醇进行光点击反应,研究了硫醇-烯反应的参数和副作用,并成功制备了菜籽油基多元醇,并且所得聚氨酯具有与市售聚氨酯相同的耐热性。付长清等(江西师范大学)用植物油和巯基丙酸进行巯基-烯点击反应,成功合成了植物油基多元羧酸新型亲水扩链剂。
目前报道的制备新型亲水扩链剂的专利较少,例如公开号为cn106046288a的中国发明专利公开了一种亲水扩链剂的制备及其应用,公开号为cn101240057a的中国发明专利公开了一种磺酸型亲水扩链剂的制备方法,以上专利都是成功制备亲水扩链剂并在聚氨酯方面得以应用的实例,其性能优异,有望取代传统扩链剂。但是以上实例都是以石油基为基材,不满足可持续发展的要求。
因此,开发一种减少或者代替石油类不可再生资源的新型亲水扩链剂具有重要的研究意义和经济价值。
技术实现要素:
本发明的目的在于克服现有技术中制备聚氨酯时选用的亲水扩链剂均是石化资源为基材,不环保的缺陷或不足,提供一种亲水扩链剂。本发明提供的扩链剂同时具有交联作用的羟基(至少3个)和亲水作用的羧基或叔胺基,且在常温下为液态,具有明显的相容性的优势;将其作为亲水扩链剂用于制备聚氨酯时,可赋予水性聚氨酯乳液良好的储存稳定性,高生物含量(高达90%),且水性聚氨酯的力学性能得到提升,具有相当或优于常规商业的扩链剂(例如dmpa和dmba)制备得到的水性聚氨酯材料的水平。
本发明的另一目的在于提供上述扩链剂的制备方法。
本发明的另一目的在于提供上述扩链剂在制备聚氨酯中的应用。
为实现上述发明目的,本发明采用如下技术方案:
一种扩链剂,具有如式(ⅰ)或式(ⅱ)所示结构:
其中,r、r1、r2和r3独立地选自烷基、取代烷基、苯基、取代苯基、杂环基、烷基杂环基或羰基。
本发明提供的扩链剂同时具有交联作用的羟基和亲水作用的羧基或叔胺基,且在常温下为液态,具有明显的相容性的优势;将其作为亲水扩链剂用于制备聚氨酯时,可赋予水性聚氨酯乳液良好的储存稳定性,具有高达90%的生物含量,且水性聚氨酯的力学性能得到提升,具有相当或优于常规商业的扩链剂(例如dmpa和dmba)制备得到的水性聚氨酯材料的水平。
本发明所指的羰基为-cor4,r4为烷基(例如c1~4的烷基)、苯基或烷基取代苯基。
优选地,所述烷基为c1~10的烷基;所述取代烷基为c1~10的取代烷基;
优选地,所述杂环基或烷基杂环基中的杂原子为氧。
优选地,所述取代烷基、取代苯基中的取代基独立地选自羟基、氰基、酯基或羧基中的一种或几种。
优选地,所述r、r1、r2和r3独立地选羰基、烷基杂环基、甲基或乙基。
更为优选地,所述羰基为-coch3、-coch2ch3、
更为优选地,r1为-coch3;r2为或-coch3;r3为乙基。
上述扩链剂的制备方法,包括如下步骤:将蓖麻油
一般情况下,巯基使植物油的双键完全转化的时间通常至少需要3个小时。本发明以蓖麻油和n-乙酰-l-半胱氨酸及其衍生物为原料进行巯基-烯光点击反应,可在常温下较短时间内就实现双键完全转化,并且得到的扩链剂可制备性能优异的水性聚氨酯。
例如,以n-乙酰-l-半胱氨酸(nac)
另外,本发明的制备方法以可再生植物油为原料,来源广泛,符合绿色环保的理念;其中未用到任何石化能源材料,缓解了石化能源过度消耗的压力;以天然n-乙酰-l-半胱氨酸及其衍生物为原料,安全,无毒;并且天然n-乙酰-l-半胱氨酸及其衍生物的气味小,克服了传统巯基酸气味较大的缺点。
优选地,所述蓖麻油中的双键和n-乙酰-l-半胱氨酸及其衍生物中的巯基的摩尔比为1:1~6。
更为优选地,所述蓖麻油中的双键和n-乙酰-l-半胱氨酸及其衍生物中的巯基的摩尔比为1:4。
优选地,所述光引发剂为异丙基噻吨酮itx、2-羟基-2,2-二甲基苯乙酮1173、苯乙酮、二苯甲酮、4-苯甲酰苯甲酸、2-苯甲酰苯甲酸、2-苯甲酰基苯甲酸甲酯、4,4'-双(二甲基氨基)-二苯甲酮,4,4'-双(二乙基氨基)-二苯甲酮、1,4-二苯甲酰基苯、二苯基已二酮、羟基环己基苯基酮、2-乙基蒽醌、光产碱剂酮肟酯、钴-胺络合物、甲酰胺、季铵盐、9-芴氨基甲酸酯或3-硝基戊烷基氨基甲酸酯中的一种或几种。
更为优选地,所述光引发剂为2-羟基-2,2-二甲基苯乙酮1173。
优选地,所述光引发剂的用量为蓖麻油和n-乙酰-l-半胱氨酸及其衍生物的质量总和的3~5%。
优选地,所述反应的时间为10~60min。
更为优选地,所述反应的时间为15~30min。
最为优选地,所述反应的时间为15min。
优选地,所述蓖麻油、n-乙酰-l-半胱氨酸及其衍生物和引发剂于有机溶剂中混合溶解。
更为优选地,所述有机溶剂为乙酸乙酯、二氯甲烷、石油醚、乙醚或四氯甲烷中的一种或几种。
优选地,所述反应后还包括萃取,干燥,过滤,蒸发,干燥的步骤。
具体地,所述反应后的处理过程如下:将反应后的产物用乙酸乙酯萃取,然后用无水硫酸镁干燥,过滤并旋转蒸发以除去乙酸乙酯,然后在45℃真空环境下干燥过夜。
上述化合物作为扩链剂在制备水性聚氨酯中的应用也在本发明的保护范围内。
本发明还请求保护一种水性聚氨酯的制备方法,包括如下步骤:将多元醇、二异氰酸酯和催化剂混合均匀,加入具有如式(ⅰ)或式(ⅱ)所示结构的扩链剂,反应即得所述水性聚氨酯。
本发明提供的制备方法中选用的扩链剂同时具有交联作用的羟基和亲水作用的羧基或叔胺基,且在常温下为液态,具有明显的相容性的优势,可用于制备水性聚氨酯乳液;制备得到的水性聚氨酯乳液具有良好的储存稳定性,高生物含量(高达90%)和优异的力学性能,具有相当或优于常规商业的扩链剂,例如dmpa和dmba,制备得到的水性聚氨酯材料的水平。
本领域常规的多元醇、二异氰酸酯和催化剂均可用于本发明中,反应条件也可为常规的控制条件。
优选地,所述多元醇为聚酯多元醇、聚醚多元醇或植物油基多元醇中的一种或几种。
更为优选地,所述多元醇为聚丙二醇ppg、聚碳酸酯二醇pcdl、蓖麻油co及其衍生物中的一种或几种。
优选地,所述二异氰酸酯为异氟尔酮二异氰酸酯ipdi、甲苯二异氰酸酯tdi、二苯基甲烷二异氰酸酯mdi、1,6-己二异氰酸酯hdi或l-赖氨酸二异氰酸酯ldi中的一种或几种。
更为优选地,所述二异氰酸酯为异氟尔酮二异氰酸酯ipdi。
优选地,所述催化剂为月桂酸二丁基锡dbtdl、辛酸亚锡、辛酸锌或三亚乙基二胺中的一种或几种。
更为优选地,所述催化剂为月桂酸二丁基锡dbtdl。
优选地,所述反应的温度为50~90℃。时间为60~180min。
优选地,以所述多元醇、二异氰酸酯和扩链剂结构中oh或nco摩尔值计,摩尔比为1.0:1.5~2.5:0.5~1.5;所述乳化后得到的水性聚氨酯的固含量为5~50%。
优选地,所述催化剂用量为反应物总质量的0.1~1%。
优选地,所述中和剂为三乙胺、醋酸、盐酸、乙醇酸、赖氨酸中的一种,中和度为60~150%。
所述反应的具体过程为:将多元醇和二异氰酸酯混合,在50~90℃下搅拌,然后加入催化剂进行预反应;再将扩链剂溶解在丁酮mek,搅拌,反应10~60min。反应后加入丁酮以降低体系的粘度。然后冷却至室温,利用中和剂(例如三乙胺tea、酒石酸、果酸等)中和,搅拌,用蒸馏水以400~800rpm的速度乳化60~120min时,旋转蒸发除去mek,可得15~30wt.%的水性聚氨酯乳液。
一种水性聚氨酯,通过上述制备方法制备得到。
上述水性聚氨酯在制备涂料、密封剂、粘合剂、泡沫或复合材料中的应用也在本发明的保护范围内。
上述水性聚氨酯在制备水性聚氨酯涂膜中的应用也在本发明的保护范围内。
一种水性聚氨酯涂膜,通过如下过程制备得到:将上述水性聚氨酯倒入模具中,干燥,即得所述水性聚氨酯涂膜。
与现有技术相比,本发明具有如下有益效果:
本发明提供的扩链剂同时具有交联作用的羟基(至少3个)和亲水作用的羧基或叔胺基,且在常温下为液态,具有明显的相容性的优势;将其作为亲水扩链剂用于制备聚氨酯时,可赋予水性聚氨酯乳液良好的储存稳定性,高生物含量(高达90%),且水性聚氨酯的力学性能得到提升,具有相当或优于常规商业的扩链剂,例如dmpa和dmba,制备得到的水性聚氨酯材料的水平。
附图说明
图1为实施例1制备扩链剂的反应路线图和设备示意图;
图2为实施例1提供的扩链剂nacco-p的ftir光谱图(图2a)、凝胶渗透色谱(gpc)图(图2b)和1h-nmr图(图2c);
图3为不同光引发剂的引发原理;
图4为实施例1提供的扩链剂nacco-t的1h-nmr图(图4a)、ftir光谱图(图4b)、凝胶渗透色谱(gpc)图(图4c)和转化率随时间变化图(图4e);
图5为实施例1提供的扩链剂nacco-m的1h-nmr图(图5a)、ftir光谱图(图5b)、凝胶渗透色谱(gpc)图(图5c)和转化率随巯基/碳碳双键的摩尔比的变化图(图5e);
图6为实施例3中水性聚氨酯乳液的制备原理图;
图7为实施例3提供的水性聚氨酯乳液的粒径分布图(图7a)和透射电镜图(图7b);
图8为实施例3提供的水性聚氨酯涂膜材料的外观(图8a)、热重分析(图8b)、示差扫描量热分析(图8c)和动态热机械分析(图8d);
图9为实施例3提供的水性聚氨酯涂膜材料的机械性能测试结果;
图10为实施例3提供的水性聚氨酯涂膜材料的水接触角(图10a)和二碘甲烷接触角(图10b)。
具体实施方式
下面结合实施例进一步阐述本发明。这些实施例仅用于说明本发明而不用于限制本发明的范围。下例实施例中未注明具体条件的实验方法,通常按照本领域常规条件或按照制造厂商建议的条件;所使用的原料、试剂等,如无特殊说明,均为可从常规市场等商业途径得到的原料和试剂。本领域的技术人员在本发明的基础上所做的任何非实质性的变化及替换均属于本发明所要求保护的范围。
对各实施例提供的扩链剂、水性聚氨酯乳液或水性聚氨酯涂膜材料进行如下表征。
(1)傅里叶变换红外光谱法(ft-ir)
采用美国赛默飞公司的nicoletis10傅立叶变换红外光谱对扩链剂的官能团进行表征。该光谱扫描范围为400-4000cm-1。
(2)核磁共振波谱法(nmr)
采用bruker公司的av600核磁共振波谱仪对样品进行氢谱分析。利用四甲基硅烷(tms)作为内标物,二甲基亚砜-d6为溶剂,鉴定扩链剂的分子结构。碳碳双键的转化率通过下式进行计算:
其中d和d’分别是氢谱中原植物油和所得产物的图谱中的碳碳双键的积分面积。
(3)凝胶渗透色谱(gpc)
采用岛津公司的prominencegpc系统测定样品分子量。系统配备rid-10a示差折光检测器以及shodexkf804l和kf802.5色谱柱。以四氢呋喃为流动相,流速和柱温分别为0.3ml/min和30℃。以聚苯乙烯为标准样。
(4)质谱(lc-ms)
采用absciexapi3200获得产物的相对分子量。
(5)酸值、羟值的测定
根据aocsofficialmethodte1a-64法对扩链剂中的羧基进行测定。具体操作如下:向锥形瓶中加入1g的扩链剂和15g的无水乙醇并溶解,并加入3-5滴酚酞指示剂,然后用0.5mol/l氢氧化钾溶液滴定。
根据unilever法对试样中的羟基含量进行测定。具体操作如下:向锥形瓶中加入10g的乙酸酐和吡啶混合物(质量比为1:9)以及1.0g多元醇。混合物在90-100℃反应1h后,加入25ml吡啶和10ml去离子水。继续反应20min后,产物在酚酞为指示剂下用0.5mol/l氢氧化钾溶液滴定。空白测定以相似的步骤进行。
(6)透射电子显微镜(tem)
采用荷兰的tecnai12,netherlandsfei透射电子显微镜对水性聚氨酯乳液的形貌和粒径进行表征。将水性聚氨酯乳液用蒸馏水稀释成0.1%质量浓度的液体在显微镜下观察。
(7)水性聚氨酯乳液稳定性
使用上海托莫斯科学仪器公司的tomos3-18离心机,以8000rpm的速度离心样品30min来表征的水性聚氨酯乳液稳定性。
(8)粒径分布和zeta电位
用英国马尔文仪器有限公司的zeta-sizernanozse测量水性聚氨酯乳液的粒径分布和zeta电位,在测试前将样品稀释至约0.01wt%。
(9)接触角
使用上海中晨数字技术设备有限公司的powereachjc2000c1接触角仪,在室温下测量固定涂膜样品的水和二碘甲烷接触角。每个样品重复三次取平均值。
(10)热重分析(tga)
采用triosdiscoverytga550热分析仪对样品进行测定。样品在氮气保护下以10℃/min的速率从30℃加热至700℃。在测试中使用5-10mg样品,样品在测试前先进行干燥处理。
(11)机械性能
采用日本岛津公司shimadzuags-x万能拉力试验机进行测定。测试速度为100mm/min。样品规格为25mm×10mm(长×宽)。每个样品测试4个平行样取平均值。
(12)动态机械分析(dma)
采用耐驰公司的dma242c动态机械分析仪进行测定。使用拉伸模式,频率设定为1hz。样品的20mm×6mm(长×宽)。样品从-60℃以5℃/min的速率升至140℃。玻璃转化温度(tg)由tanδ峰值对应的温度得到。
(13)差示扫描量热(dsc)
采用德国的dsc214polyma差示扫描量热仪对样品的玻璃化转变温度进行测定。测试过程中先以10℃/min的升温速率将温度从30℃升温至100℃,再以10℃/min的降温速率将温度降低至-60℃消除热力时。测试的动态温度为-60℃以5℃/min的升温速率升温至100℃进行测定。
实施例1
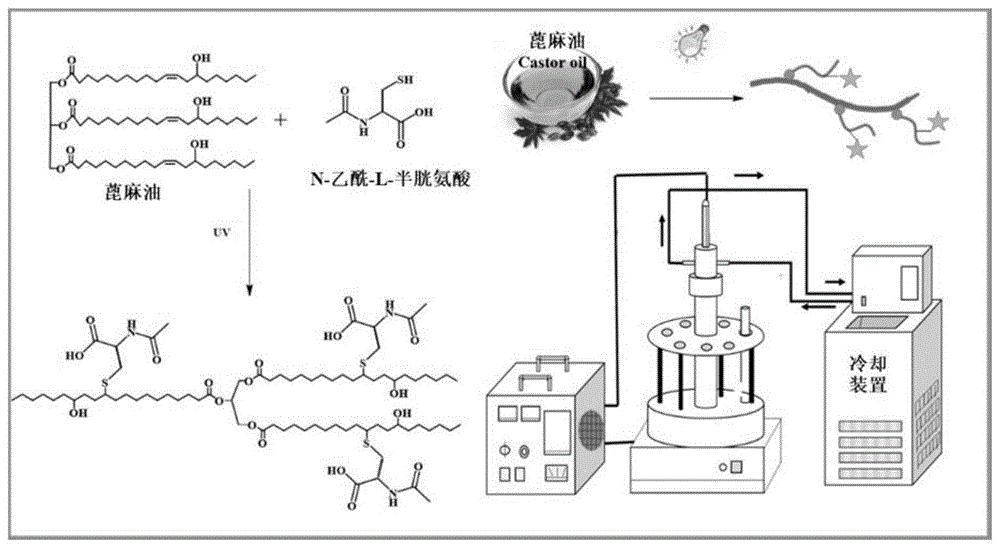
本实施例提供一系列的扩链剂nacco,通过蓖麻油(co)和n-乙酰-l-半胱氨酸(nac)的点击反应制备得到,具体过程如下。
首先,将适当比例的nac,co和光引发剂(4wt.%)溶解在无水乙醇中并转移至石英管中。在350w功率的紫外线照射下,反应一定的时间(反应设备如图1)。之后,产物用乙酸乙酯萃取,并用无水硫酸镁干燥。将产物过滤并旋转蒸发以除去乙酸乙酯,然后在45℃真空环境干燥过夜,得到扩链剂nacco。
具体地,选用两种光引发剂(1173,itx),6个不同反应时间(5min,10min,15min,20min,25min和30min)和6个不同巯基/双键摩尔比(1:1、2:1、3:1、4:1、5:1和6:1)进行,获得的扩链剂分别命名为nacco-p,nacco-t和nacco-m,其中p,t和m分别是光引发剂、时间和巯基/双键摩尔比,具体条件如表1。
表1扩链剂的制备条件
*np代表不选用扩链剂。
利用1hnmr,gpc和ftir光谱对产物扩链nacco的结构进行了全面表征
图2为对nacco-p的表征结果。
如图2a,为ftir光谱图。从图可知,有光引发剂itx、1173参与反应的情况下,和原蓖麻油相比,位于3008cm-1处的碳-碳双键的特征峰消失,位于1655cm-1处出现了c=o键的拉伸振动和nh键的形变振动的特征峰,并且在1535cm-1处出现了酰胺的特征峰.除此之外,产物中位于3400cm-1-3300cm-1处的羟基特征峰向右移动,可能是因为n-h键的引入造成的。以上均初步说明经过光点击反应nac成功引入到蓖麻油的双键中。
如图2b,为gpc图。从图可知,与原蓖麻油相比,得到的产物的保留时间向左移动(保留时间位于17~18.5min之间),说明分子量在增大,并且以1173为引发剂时保留时间最短,说明分子量最大,反应最完全。并且在14.5-17min处还观察到少量的低聚物,这很可能是蓖麻油的二聚体。一般在紫外线照射下,聚合反应是根据自氧化机理通过形成氢过氧化物和环状过氧化物而生成的。其中多元醇中的低聚物可以提供聚氨酯的超支化分子结构。
如图2c,为1h-nmr图。从图可知,与原蓖麻油相比,所得扩链剂主链中位于5.2-5.5ppm处的碳-碳双键特征峰在60min内减少甚至完全消失。并且,图谱中新产生的位于4.0ppm处的–ch基团,位于2.8ppm处的–ch2基团和位于1.8ppm处的–ch3基团特征峰来自于n-乙酰基-l-半胱氨酸,表明通过硫醇-烯光点击反应成功地将n-乙酰-l-半胱氨酸接枝到蓖麻油的脂肪酸链上。此外,将蓖麻油中位于5.1-5.2ppm处的–ch2-ch-ch2特征峰的积分面积定义为1,用作归一化的标准,对产物进行积分计算碳碳双键的转化率。结果表明,以1173为光引发剂得到的产物表现出最高的反应效率,其中c=c双键的转化率为98%,产率为93.78%,而以itx为引发剂时反应效率最低,其转化率为64.36%,产率是72.9%(具体数据如表2所示)。itx和1173是典型的氢捕获引发剂,可以通过光转化形成三重态。三重态和三重态提取的功能分子中c-h键的氢原子进一步转化为自由基,并通过与功能分子自由基偶联完成接枝反应。事实上,光引发剂1173在紫外光的引发下会在每个分子上产生两个自由基,这有助于在传播过程中形成两个噻吩基;而光引发剂itx仅通过氢捕获反应生成一个自由基。在没有光引发剂的情况下,通过sh基团的断裂仅形成一个自由基(如图3b-d)。这个结果表明以1173为光引发剂具有最高的反应效率。
表2不同条件下的扩链剂的双键转化率、产率和分子量
图4为对nacco-t的表征。
如图4a,为1h-nmr图。从图可知,随着反应时间的增加,核磁图中位于5.2-5.5ppm之间的c=c双键的特征峰逐渐变小直至在15min时完全消失。
如图4c,为gpc图。从图可知,15min之前产物的保留时间随着时间的增加变小,随后不变。
如图4b,为ftir光谱图。从图可知,产物出现了新的特征峰,这些均说明在15min时反应完全。
扩链剂的双键转化率、产率和分子量如表2。
图5为对nacco-m的表征结果。从图5可知,巯基/碳碳双键的摩尔比为4:1时为最佳的反应条件。
从上述图表可知,碳碳双键完全转化的条件为:反应时间为15min,在光引发剂1173的存在下巯基/双键摩尔比为4:1,为最佳条件。
对该条件(最佳条件)下得到的扩链剂的理化性能进行测定,结果如表3。
表3最佳条件下的扩链剂的理化性能
在此条件下得到了性能优异的全生物基扩链剂,其酸值和羟值分别为87.82±1.57mgkoh/g和184.86±2.38mgkoh/g,反应效率最高,能耗最少。
实施例2~9
本实施例提供一系列的扩链剂,通过蓖麻油和n-乙酰-l-半胱氨酸衍生物的点击反应制备得到,n-乙酰-l-半胱氨酸衍生物结构参数如表4所示。
表4n-乙酰-l-半胱氨酸衍生物结构
蓖麻油和n-乙酰-l-半胱氨酸衍生物进行点击反应
将n-乙酰-l-半胱氨酸衍生物(衍生物1~8)、co和光引发剂(4wt.%)溶解在无水乙醇中并转移至石英管中。在350w功率的紫外线照射下,反应15min。之后,产物用乙酸乙酯萃取,并用无水硫酸镁干燥。将产物过滤并旋转蒸发以除去乙酸乙酯,然后在45℃真空环境下干燥过夜,得到扩链剂,依次记录为实施例2~9,其性能表征如表5。
表5实施例2~9提供的扩链剂的酸值、羟值和分子量
实施例10
本实施例提供一系列水性聚氨酯乳液,以实施例1提供的nacco(nacco-4:1)作为扩链剂,通过如下过程制备得到。
如图6,多元醇(ppg,pcdl,co,蓖麻油衍生物)和异氟尔酮二异氰酸酯(ipdi)加入配有机械搅拌的双颈烧瓶中,并在78℃的温度下搅拌混合10min。然后,将月桂酸二丁基锡(dbtdl)(多元醇的1%质量分数)加入到混合物中,然后反应20min。随后,将nacco的扩链剂溶解在丁酮(mek)中并滴加到混合物中搅拌,60min内滴加完成。反应物的用量见表6。反应后,将5-10ml的mek倒入混合物中以降低体系的粘度。然后,当温度冷却至室温,将体系用tea中和,搅拌约30min。最后,将混合物用蒸馏水以400~500rpm的速度乳化120min,然后通过旋转蒸发除去过量的mek,以获得固体含量为15%的水性聚氨酯(pu)。所制备的水性聚氨酯分散体称为pu-p,其中p指代不同的多元醇。具体地,pu-co164代表选用的多元醇的羟值为164mgkoh/g的蓖麻油;pu-ppg152代表选用的多元醇的羟值为152mgkoh/g的聚丙二醇;pu-pcdl224代表选用的多元醇的羟值为224mgkoh/g的聚碳酸脂二醇;pu-scp代表选用的多元醇为由蓖麻油和大豆油酸制备得到的多元醇,剩下的以此类推。
表6多元醇的用量
注:a:多元醇的羟基摩尔当量;b:扩链剂的羟基摩尔当量。
将上述水性聚氨酯乳液倒入硅胶模具中并在室温下干燥,得到薄膜用于进一步分析。在测试之前,将所有样品在60℃下干燥超过12h。
测定结果如下。
(1)水性聚氨酯乳液稳定性
按前述提供的方法测定稳定性。图7为水性聚氨酯乳液的粒径分布图和透射电镜图;表7为水性聚氨酯乳液的外观、粒径和电位结果。
表7水性聚氨酯乳液的外观、粒径和电位
从图和表可知,聚氨酯乳液的分散体均显示出优异的储存稳定性,其zeta电位低于-35.15mv(表7所示)。并且在3000rpm速度下离心30min后,未观察到样品沉淀或分层。此外,所有分散液在实验室环境中长期存放超过2年后状态仍保持不变。根据所用的不同多元醇,分散体表现出不同的外观(图7)。多元醇的羟值越大,分散体越透明和稳定。例如,由羟值为208mgkoh/g的蓖麻油衍生物获得的水性聚氨酯分散体显示出蓝色半透明外观,而由羟值为149mgkoh/g的scp获得的水性聚氨酯分散体显示出乳白色外观。透射电镜结果表明,聚氨酯乳液可能在内部自组装为纳米颗粒,这可能是由于扩链剂中的羧酸基团赋予了表面电荷。pu-scp分散体的粒径为150.00±20.00nm,而pu-co164和pu-co208分散体分别显示100.00±10.00nm和50.00±5.00nm的粒径。由石油基多元醇(ppg和pcdl)制备的聚氨酯分散体显示出相似的趋势。这些水性聚氨酯分散体粒径的差异归因于交联密度、亲水性、链的刚性和预聚物的粘度等因素。
(2)聚氨酯乳液涂膜的热力学性能
按前述提供的方法测定聚氨酯乳液涂膜的热力学性能,测定结果如图8和表8。
由图和表可知,所有薄膜材料均显示透明外观(图8a)。这些生物基水性聚氨酯的最高生物含量可高达90%,而固体含量可高达45%。根据图中的tga表征(图8b)显示,所有水性聚氨酯涂膜材料都经历三个降解过程:从100℃到250℃的质量损失对应于不稳定的氨基甲酸酯基团的分解。从250℃至500℃之间的重量损失对应于脂肪酸链的断裂。值得注意的是,由生物基多元醇和聚丙二醇(ppg)制备的水性聚氨酯涂膜的热稳定性随羟值的增加而降低,这是因为高羟值导致的氨基甲酸酯和较高不稳定硬段的增加。然而,随着羟值的增加,由聚碳酸酯二醇(pcdl)水性聚氨酯涂膜的热稳定性增加。这可能与多元醇pcdl224的较高羟值导致的较高的交联密度有关。水性聚氨酯涂膜在高于500℃时的质量损失是因为材料的进一步热氧化。通常,将开始降解的温度,即质量损失5%的温度(t5)和降解到中点的温度(t50)作为热稳定性的参数(具体数据展示在表8中)。随着羟值从149增加到208mgkoh/g,pu-scp和pu-co膜的t5值从236.01℃变208.49℃,从t50从346.38℃上升至334.23℃。由多元醇ppg制备的水性聚氨酯表现出类似的热行为。这可能是因为较高不稳定硬段和增加的交联密度之间竞争的结果。然而,pu-pcdl的t5和t50随着羟值的增加而增加,这是因为交联密度增加的结果。由生物基多元醇,聚酯和聚醚多元醇制成的水性聚氨酯涂膜材料比通过本体反应制备的材料(pu-nacco)表现出更高的热稳定性。而且,由聚酯多元醇制得的水性聚氨酯涂膜材料比由聚醚多元醇制得的材料具有更好的热稳定性。这是因为氨基甲酸酯nh基团和酯基形成的更强的氢键。
表8水性聚氨酯涂膜材料的热力学性能
注:pcdl代表聚碳酸酯二元醇,224、112分别表示其羟值;ppg代表聚丙二醇,152、284分别表示其羟值;co代表蓖麻油,208、164分别代表其羟值;scp为由大豆油酸和蓖麻油反应得到的植物油基多元醇;nacco代表本工作制备的内乳化剂,pu-nacco则代表由nacco为多元醇制备的水性聚氨酯,其他以此类推。
dma(图8c)和dsc(图8d)结果显示,所有涂膜材料在dsc曲线中均显示出一个tg,并且不存在熔融或结晶转变,这与所得水性聚氨酯涂膜材料的无定形性质有关。随着羟值从149mgkoh/g(scp)上升至208mgkoh/g(co208),所得水性聚氨酯涂膜的tg值从20.69℃上升到31.98℃。对于石油基水性聚氨酯薄膜也观察到了相同的趋势。随着羟值从112mgkoh/g(pcdl112)增加到284mgkoh/g(ppg284),tg值从-24.30℃上升到33.01℃,这是因为交联密度随着多元醇的羟值的增加而增加,因此玻璃化转变温度随之增加。下表总结了水性聚氨酯涂膜材料的tg结果,其中dsc测定的tg值比dma测定的tg值低约10-20℃,这与两种测试方法原理不同有关。
(3)聚氨酯乳液涂膜的力学性能
按前述提供的方法测定聚氨酯乳液涂膜的力学性能,测定结果如图9和表9。
表9聚氨酯乳液涂膜的力学性能
注:“/”表示无法进行测量的数据。
图9a展示了水性聚氨酯涂膜材料的应力-应变行为。由多元醇pcdl224,ppg284,co208和scp制备的水性聚氨酯涂膜材料表现出明显的硬塑料应力应变行为,在断裂前具有应变软化和应变硬化行为。由多元醇ppg152和co164制备的水性聚氨酯涂膜材料仅表现出典型弹性体聚合物的弹性区域和屈服行为。显然,随着多元醇羟值的增加,由生物基多元醇制备的水性聚氨酯涂膜材料(pu-scp涂膜除外)的拉伸强度(无论是屈服强度还是应变应力)增加,杨氏模量和韧性以及断裂伸长率降低。例如,pu-co208膜的拉伸强度,杨氏模量和韧性分别为18.56mpa,210.99mpa,37.02mp,而pu-co164膜的相应值为10.67mpa,64.14mpa和18.24mpa。同时,当羟值从164mgkoh/g增加至208mgkoh/g时,断裂伸长率从255.81%降低至241.56%(数据如表9)。通常,高交联密度会导致更好的拉伸强度和降低的断裂伸长率。此外,尽管多元醇co164的羟值比scp高,但由多元醇scp制备的水性聚氨酯涂膜表现出比用多元醇co164得到的水性聚氨酯膜更好的机械性能。这可能与多元醇scp的特定预交联和超支化结构有关,它可以抑制硬段的聚集并促进硬段和软段的兼容性。
对于由石油基多元醇制备的水性聚氨酯膜,观察到类似的拉伸应力-应变行为。(pu-pcdl112太软,无法进行拉伸测试)。随着多元醇羟值的增加,水性聚氨酯的拉伸强度,杨氏模量和韧性都增加,而断裂伸长率减小。所有的水性聚氨酯薄膜均表现出高于145%的断裂伸长率。此外,由多元醇pcdl制备的水性聚氨酯具有比由多元醇ppg制备的水性聚氨酯更好的机械性能。并且pu-pcdl224表现出最高的拉伸强度,杨氏模量和韧性,这是因为它在氨基甲酸酯和酯基之间形成了更多的氢键与pu-ppg相比,形成更多的氢键可使pu-pcdl具有更高的相混合度和更好的晶体结构。
为了进一步评估本实施例提供的水性聚氨酯的优越性,我们在机械性能方面与其他类似产品进行了比较。根据使用的不同类型的扩链剂,对基于植物油基多元醇的水性聚氨酯进行了分类(图9b)。通常,用2,2-双(羟甲基)丙酸(dmpa)作为扩链剂制备的水性聚氨酯与用n-甲基二乙醇胺(mdea)制备的样品相比,具有更好的机械性能。由2,2-双(羟甲基)丁酸(dmba)和实施例1提供的扩链剂制备的水性聚氨酯的机械性能与使用dmpa作为扩链剂制备的样品的机械性能相当。此外,值得注意的是,与使用其他扩链剂制备的样品相比,使用生物基多元醇和实施例1提供的扩链剂制备的水性聚氨酯表现出更优异的拉伸强度和韧性。用pcl以外的石油基多元醇制备的水性聚氨酯的统计数据(图9c)显示,与使用其他扩链剂制备的样品相比,使用实施例1提供的扩链剂制备的样品在拉伸强度和断裂伸长率方面表现出显著优势。此外,值得强调的是,所得到的水性聚氨酯涂膜与其他基于溶剂型的聚氨酯涂膜相比具有相当甚至更高的机械性能。这些现象可以通过与dmpa,dmba和其他扩链剂相比,实施例1提供的扩链剂与多元醇和ipdi具有更好的相容性来解释,从而使聚氨酯的结构更加均匀。此外,长而柔软的脂肪酸扩链剂(实施例1提供的扩链剂)中的碳链可防止聚氨酯中硬段的聚集,并促进软段和硬段的良好相容性。
(4)聚氨酯乳液涂膜的亲、疏水性
按前述提供的方法测定聚氨酯乳液涂膜的亲、疏水性,测定结果如图10和表10。
表10水性聚氨酯涂膜的接触角数据
图10展示了实施例1提供的扩链剂、实施例3的不同多元醇的水性聚氨酯涂膜材料与水和二碘甲烷的接触角。具体的接触角结果示于表10中。来自植物油基多元醇的水性聚氨酯膜显示出随着多元醇的羟值的增加而减小的接触角(pu-scp除外)。pu-co164的水接触角为81.63°,而pu-co208为78.45°。这可能是由于亲水性离子基团和交联密度影响水性聚氨酯薄膜的疏水性。随着羟值的增加,水性聚氨酯膜的交联密度增加,这导致接触角增加。但是,亲水性离子基团随着扩链剂含量的增加而增加,导致亲水性增强。亲水性的提高补偿了由交联密度的增加引起的亲水性的降低,从而导致所得膜的接触角的减小。对于来自聚醚多元醇(ppg)和聚酯多元醇(pcdl)的水性聚氨酯观察到类似的趋势。水和二碘甲烷接触角均随羟基数的增加而减小。例如,随着羟值从152mgkoh/g增加到284mgkoh/g,pu-ppg的水接触角从91.40°减小到75.48°,同样,二碘甲烷接触角从44.08°减小到37.40°。值得注意的是,ppg284的oh数最高的pu具有最低的水和二碘甲烷接触角。该现象证明了上述结论是正确的,亲水性离子基团支配了所得薄膜的接触角。
由上述可知,(1)扩链剂的结构以及其良好的相容性赋予水性聚氨酯乳液良好的储存稳定性。多元醇的官能度对乳液的外观和粒径具有明显的影响,随着多元醇羟值的增加,分散体外观由牛奶白逐渐变澄清透明,乳液粒径逐渐变小。
(2)实施例3得到的水性聚氨酯涂膜材料和商业dmpa和dmba得到的涂膜材料相比具有相当甚至更优的力学性能。随着多元醇羟值的增加,聚氨酯涂膜材料的交联密度增加,热力学稳定性和机械性能更加优异。此外得到的水性聚氨酯涂膜材料的生物基含量高达90%。
(3)随着多元醇羟值的增加,实施例3得到的水性聚氨酯涂膜材料的接触角逐渐变小,说明表面润湿性增加。其中pu-ppg152具有最高的接触角91.40°。
实施例11~18
本实施例提供一系列水性聚氨酯乳液,分别以实施例2~9提供的植物油基产物作为扩链剂,通过如下过程制备得到。
将ppg和二异氰酸酯混合,在50~90℃下搅拌,然后加入催化剂进行预反应;然后,分别将实施例2~9制备植物油基扩链剂溶解在丁酮(mek)中并滴加到混合物中搅拌,扩链反应10~60min。反应物的用量见表11。反应后加入丁酮以降低体系的粘度。然后冷却至室温,利用中和剂中和,搅拌,加入蒸馏水以400~800rpm的速度乳化,旋转蒸发除去过量的mek,以获得固体含量为15~35%的水性聚氨酯(pu)。
表11实施例11~18水性聚氨酯乳液的配方参数
注:a:多元醇的羟基摩尔当量;b:扩链剂的羟基摩尔当量。
表12实施例11~18水性聚氨酯乳液的制备工艺参数
按照前述的方法测定上述水性聚氨酯乳液稳定性,测试结果如表13。由表可知,除了实施例11的粒径较大之外,其他实施例的粒径都较小,反应物比例的增大对粒径的影响较大,并且除了实施例18,所有实施例样品的zeta电位绝对值均大于34,说明乳液稳定性较好。
表13实施例11~18水性聚氨酯乳液的粒径、zeta电位
将上述聚氨酯乳液倒入硅胶模具中并在室温下干燥,得到薄膜用于进一步分析(热力学性能)。在测试之前,将所有样品在60℃下干燥超过12h。
实施例11~18提供的水性聚氨酯涂膜材料均有较好的热力学和力学性能。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!