一种对基因组的一部分进行富集的方法与相关应用
1.本发明是关于一种对基因组的一部分进行富集的方法与相关应用,属于基因工程技术领域。
背景技术:
2.基因工程检测技术领域,如涉及动植物遗传或育种中所需的全基因组检测,经常会用到对基因组的一部分进行富集分析的方法。举例而言,标记辅助选择育种技术,需要先借助dna分子标记或者生化标记对前景基因(foreground gene)进行跟踪筛选,随后利用全基因组均匀分布的分子标记对遗传背景(genetic background)进行检测,以期获得既含有尽可能小的前景基因片段、又使遗传背景尽可能接近受体品种的新材料,用于后续的育种过程。检测遗传背景的常用技术主要以下几种:简单序列重复(simple sequence repeats,ssr)分子标记检测、dna芯片(dna chip)检测、多重pcr(multiplex pcr)检测、简化基因组测序(reduced-representation genome sequencing)和全基因组重测序(whole genome resequencing)等。其中,
①
ssr分子标记检测技术原理是基于全基因组上广泛分布的短串联重复序列,因重复序列单元数目的差异而在不同样本间产生多态性;进行全基因组ssr标记检测时需要提前在每一个ssr位点的侧翼序列设计一对特异性引物,然后逐个检测所有ssr位点的基因型。
②
dna芯片检测技术主要是通过对此物种核心资源基因组序列的比对分析,选取基因组上普遍存在的差异性位点进行核酸杂交检测,进而判断测试材料的基因组多态性。
③
多重pcr检测技术是对基因组上分布的多个靶位点(几个到几千个)进行统一的单管pcr扩增反应,因每个靶位点需要一对特异性的引物,因此pcr反应体系中包含所有位点的扩增引物。
④
简化基因组测序技术主要是对基因组上特定限制性内切酶酶切位点附近的区域(约占10%)进行高通量测序,这类技术的文库构建过程主要涉及限制性内切酶的选取、酶切、接头连接、pcr扩增以及多步纯化等步骤,一般可以获得几万或上百万个酶切位点附近区域的序列变异信息(取决于测序数据量及内切酶的种类),用于全基因组多态性分析。
⑤
全基因组重测序技术是对整个基因组序列进行测序分析,一般需要先利用超声波、转座酶或片段化酶随机断裂基因组dna,再进行接头连接及后续pcr扩增的文库构建过程,通常可以获得覆盖全基因组的几十万至几百万个位点的变异信息。
3.然而,上述检测遗传背景的方法存在以下缺点:
①
ssr分子标记检测,要求每个位点独立扩增、扩增产物独立通过琼脂糖胶或聚丙烯酰胺凝胶电泳检测,因此检测技术落后,且当样本量大时检测效率低下。
②
dna芯片检测技术,前期需要昂贵的成本用于芯片设计、制作;检测的位点固定,当需要检测其它位点或用于其它物种时,现有芯片需要重新设计制作,因此通用性非常差。
③
多重pcr检测技术,需要先设计每一个靶位点扩增所需的引物;引物序列确定后需要昂贵的成本批量合成这些引物;pcr扩增体系包含全部的引物序列,因此存在引物间相互干扰的问题。
④
简化基因组测序技术,建库流程涉及多步酶切及连接反应,因此过程繁琐,且多步的酶学反应效率在不同样本间难以保持一致、造成测序数据中检测到的变异位点在样本间缺失率高。
⑤
全基因组重测序技术,基于超声波或转座酶方法进行
文库构建的费用较高;一般要求每个测序数据量大(覆盖度5
×
以上),最终所得变异位点数量过多(一般育种中需要几百至几千个均分分布全基因组的标记足以进行背景检测),存在严重的信息冗余。
4.此外,上述检测遗传背景的方法多数不能实现分子标记辅助育种过程中所需要的前景基因和遗传背景的同时检测,前景基因的筛选和遗传背景的检测是两个相互独立的环节,因此整个筛选鉴定过程耗时较长,且操作流程繁琐。
技术实现要素:
5.本发明的一个目的在于提供一种对基因组的一部分进行富集的方法。
6.本发明的另一目的在于提供所述对基因组的一部分进行富集的方法的相关应用,具体包括在物种遗传和育种检测中的应用,例如育种中检测前景基因和遗传背景。
7.一方面,本发明提供了一种对基因组的一部分进行富集的方法,该方法包括:
8.采用序列特异引物(sequence specific primer)和随机简并引物(arbitrary degenerate primer)配对,对待测样本的基因组dna进行热不对称交错式pcr(thermal asymmetric interlaced pcr,tail-pcr)扩增;
9.对pcr扩增产物进行纯化并构建测序文库。
10.根据本发明的具体实施方案,本发明中,所述“序列特异引物”是指该引物中不含有简并碱基,即,其简并度为1。
11.本发明的对基因组的一部分进行富集的方法,可用于全基因组背景基因型的检测(即不关注前景基因),具有类似于简化基因组测序的功效;也可用于前景基因和遗传背景的同时检测。
12.本发明的对基因组的一部分进行富集的方法,对应的测序文库构建过程中,只需要两条引物:一条序列特异的引物和另外一条含随机简并碱基的引物(即随机简并引物),利用序列特异引物在基因组上靶位点的特异性退火和数万个位点的非特异性退火,与随机简并引物的配对,通过一次热不对称交错式pcr扩增可实现对基因组的一部分(1%~10%)进行富集。具体而言,全基因组遗传背景的检测主要依靠引物与dna模板间的非特异性扩增,即只要两条引物在基因组上通过特异或非特异结合位置间的距离合适,中间的片段即可被扩增测序,且这些片段可在全基因组均匀分布。具体而言,本发明的方法中,可对所有的扩增产物进行一定范围内片段的筛选,比如后续选择pe150测序策略时,可考虑筛选目标长度(如250-600bp)范围内的扩增产物进行建库测序;或者,也可以直接在所有扩增产物(包含250-600bp)的两端连接测序接头,待文库构建完毕后,再借助sageelf或者pippin ht等片段分选仪器对可被用于pe150测序的区域(350-700bp,含测序接头)进行分选,而后进行高通量测序。当本发明的方法需要对前景基因进行检测时,前景基因的检测可主要依靠序列特异引物与目标基因的特异性扩增。本发明可利用序列特异引物在基因组上靶位点的特异性退火与数万个位点的非特异性退火,与随机简并引物的配对扩增,能实现通过一次pcr扩增而检测样本中的前景标记和全基因组的背景标记,对于遗传和育种检测有重要应用价值。
13.根据本发明的具体实施方案,本发明的对基因组的一部分进行富集的方法中,所述序列特异引物可根据基因组上任意区域的序列而设计。或者,当本发明的方法需要对前
景基因进行检测时,所述序列特异引物可根据前景基因序列而设计(例如,根据前景基因序列内部的多态性位点附近而设计)。更具体地,序列特异引物结合位置与前景基因内的多态性位点间隔在150bp以内,例如10-150bp。这样可以确保测序read覆盖到多态性位点,也可以保证序列特异引物与前景基因的完全连锁。
14.根据本发明的具体实施方案,本发明的对基因组的一部分进行富集的方法中,所述序列特异引物的长度为18-30nt,优选可以为18-25nt。
15.根据本发明的具体实施方案,本发明的对基因组的一部分进行富集的方法中,所述序列特异引物的5’端可额外添加6-10nt的barcode序列。所述barcode序列可用以区分样品种类。
16.根据本发明的具体实施方案,本发明的方法中,barcode序列中atcg四个碱基分布均匀即可,避免类似连续aaaa的多聚碱基类型。本发明对barcode序列无其它特殊要求,能实现区分不同样品的功能即可。
17.根据本发明的具体实施方案,本发明的对基因组的一部分进行富集的方法中,所述随机简并引物的长度为8-20nt,优选为8-15nt,更优选为10-15nt。优选地,所述随机简并引物的简并度为64-4096,优选为120-3072。
18.根据本发明的具体实施方案,本发明的对基因组的一部分进行富集的方法中,所述随机简并引物的5’端可额外添加6-10nt的barcode序列。所述barcode序列可用以区分样品种类。
19.根据本发明的具体实施方案,本发明的对基因组的一部分进行富集的方法中,pcr扩增时,随机简并引物的量超过序列特异引物的量。
20.根据本发明的具体实施方案,本发明的对基因组的一部分进行富集的方法中,pcr扩增时,反应体系使用高保真dna聚合酶(高通量测序文库用dna聚合酶)。具体可参照所属领域的现有技术操作进行。
21.根据本发明的具体实施方案,本发明的对基因组的一部分进行富集的方法中,pcr扩增时,可根据需要调整引物的退火温度和循环条件。引物的退火温度可根据引物tm值的需要而调整。通常情况下,扩增循环数总共可为20-37(以一次“变性、退火、延伸”计为一个循环单元,计数为1循环数)。pcr循环数也可根据pcr产物的量进行增减,一般扩增500ng左右,足以用于后续文库构建。
22.根据本发明的具体实施方案,本发明的对基因组的一部分进行富集的方法中,所述热不对称交错式pcr扩增包括在较高退火温度和较低退火温度下交错进行循环反应的过程。
23.在本发明的一些具体实施方案中,本发明的对基因组的一部分进行富集的方法中所述热不对称交错式pcr扩增包括在较高退火温度下与较低退火温度下交错进行6-10轮循环,每轮循环包括2-3个循环单元且其中至少1个在较高退火温度下退火的循环单元以及至少1个在较低退火温度下退火的循环单元,更具体地,每轮循环采用以下(1)至(8)所述方式之一:
24.(1)变性、较高退火温度下退火、延伸、变性、较高退火温度下退火、延伸、变性、较低退火温度下退火、延伸(即每轮循环包括3个循环单元,循环数计为3,进行6-10轮循环的循环数计为18-30);
2min,较低退火温度下退火30s-1min,72℃延伸1min-3min;6-10轮循环;
40.72℃终延伸5min-10min;
41.其中,各较高退火温度各自独立地为50-60℃,各较低退火温度各自独立地为40-50℃。
42.根据本发明的具体实施方案,本发明的对基因组的一部分进行富集的方法中,对pcr扩增产物进行纯化并构建测序文库的过程可按照所属领域的常规操作进行。本发明中优选地,pcr扩增产物纯化后,进行3’末端加a、连接y型illumina测序接头,完成测序文库的制备。
43.根据本发明的具体实施方案,本发明的对基因组的一部分进行富集的方法还包括:对测序文库进行测序,或者,测序文库经片段分选后用于高通量测序。如前述所提及的,本发明的方法中,可对所有的扩增产物进行一定范围内片段的筛选,比如后续选择pe150测序策略时,可考虑筛选目标长度(如250-600bp)范围内的扩增产物进行建库测序;或者,也可以直接在所有扩增产物(包含250-600bp)的两端连接测序接头,待文库构建完毕后,再借助sageelf或者pippin ht等片段分选仪器对可被用于pe150测序的区域(350-700bp)进行分选,而用进行高通量测序。
44.根据本发明的具体实施方案,本发明的对基因组的一部分进行富集的方法还包括:
45.对测序文库的高通量测序数据进行拆分,比对参考基因组,检测前景基因内、序列特异引物处的reads覆盖情况,以及全基因组上由reads富集、覆盖深度≥3
×
的高深度tags位点的分布情况,这些高深度的tags位点即可用于后续的全基因组基因型检测。
46.另一方面,本发明还提供了所述的对基因组的一部分进行富集的方法在标记辅助选择育种或基因组育种中的应用。具体地,该方法可以是用于对物种进行全基因组遗传背景检测,或是用于对物种进行前景基因和遗传背景检测等。本发明的方法可用于对动植物等物种进行测序。在一些具体实施方案中,所述物种包括但不限于水稻、犬和猪等。
47.在一些具体实施方案中,本发明的对基因组的一部分进行富集的方法,可以用于全基因组背景基因型的检测(即不关注前景基因),具有类似于简化基因组测序的功效。
48.在一些具体实施方案中,本发明的对基因组的一部分进行富集的方法,还可实现对前景基因和遗传背景的同时检测,因此检测过程更加快捷高效。并且,所用序列特异引物位于前景基因内部的多态性位点附近,从而保证序列特异引物的特异性扩增产物与前景基因完全连锁。检测遗传背景时,序列特异引物在基因组上靶位点的特异性退火和数万个位点的非特异性退火,与随机简并引物的配对,扩增获得的tags位点稳定出现,且均匀覆盖整个基因组。与现有的基于简化基因组测序和dna芯片等遗传背景检测技术相比,本发明的方法,无需提前打断基因组、仅使用序列特异引物和随机简并引物对基因组进行一次pcr扩增,即可稳定获得数以万计、均匀覆盖全基因组的高深度tags位点,用于育种材料遗传背景的检测。
附图说明
49.图1显示实施例1中引物pi2_f01与ad1_r03的pcr扩增结果检测。
50.图2显示实施例1中文库构建结果检测。
51.图3显示实施例1中三个文库混合后的片段分选结果检测。
52.图4显示实施例1中三次技术重复对应的测序数据在水稻基因组上的分布。其中,图片a:reads在前景基因内富集(图中蓝色横线表示pi2基因区间位置);图片b:reads在pi2_f01引物结合处富集(图中红色箭头表示序列特异引物结合位置);图片c:三次技术重复中的测序reads在基因组上显著富集,稳定地形成高深度tags;图片d:三次技术重复中共有tags在基因组上的密度分布。
53.图5显示实施例2中引物rf4_f01与ad2_r03的pcr扩增结果检测。
54.图6显示实施例2中三个文库混合后的片段分布范围检测。
55.图7显示实施例2中三个文库混合后的片段分选结果检测。
56.图8显示实施例2中三次技术重复对应的测序数据在水稻基因组上的分布。其中,图片a:reads在前景基因内富集(图中蓝色横线表示rf4基因区间位置);图片b:reads在rf4_f01引物结合处富集(图中红色箭头表示序列特异引物结合位置);图片c:三次技术重复中的测序reads在基因组上显著富集,稳定地形成高深度tags;图片d:三次技术重复中共有tags在基因组上的密度分布。
57.图9显示实施例3中pcr产物纯化及片段分布检测过程中,以dog_139-rep1和pig_421-rep1样品为代表的检测结果。
58.图10显示实施例3中高通量测序文库构建过程中,以dog_139-rep1和pig_421-rep1样品为代表的文库检测结果。
59.图11显示实施例3中片段分选结果。
60.图12显示实施例3中测序数据拆分及tags特征分析过程中,由引物hd1_f01与ad1_r3b随机扩增产生的reads在猪和犬的基因组上显著富集、稳定地形成高深度tags位点。
61.图13显示实施例3中犬样品、猪样品三次重复中共有tags在基因组上的分布位置。
具体实施方式
62.为了对本发明的技术特征、目的和有益效果有更加清楚的理解,现结合具体实施例及对本发明的技术方案进行以下详细说明,应理解这些实例仅用于说明本发明而不用于限制本发明的范围。实施例中,各原始试剂材料均可商购获得,未注明具体条件的实验方法为所属领域熟知的常规方法和常规条件,或按照仪器制造商所建议的条件。
63.除非另外专门定义,本文使用的所有技术和科学术语都与相关领域普通技术人员的通常理解具有相同的含义。
64.本发明通过设计一对引物,对待测材料的基因组dna进行一次pcr扩增,随后经高通量测序及数据分析,可以完成对前景基因和遗传背景的同时检测。其中一对引物包括:结合在前景基因上的正向序列特异引物f,以及反向随机简并引物r。基于正向序列特异引物f与前景基因序列间特异性结合后的pcr扩增,完成前景基因的筛选;基于正向序列特异引物f和反向随机简并引物r在pcr反应中引发的非特异性扩增,完成遗传背景的检测。本发明的方法主要包括:
65.(1)引物设计
66.根据前景基因序列,在其基因内部的多态性位点附近、设计长18-25nt的常规正向序列特异引物f(为区分不同的样品,可在f引物5’端额外添加6-10nt的barcode序列);根据
待测物种的基因组序列特点,设计长8-20nt的反向随机简并引物r(为区分不同的样品,也可在r引物5’端额外添加6-10nt的barcode序列)。在随机简并引物r中,简并碱基位于引物的中间位置,简并度在64-4096之间。
67.(2)pcr扩增
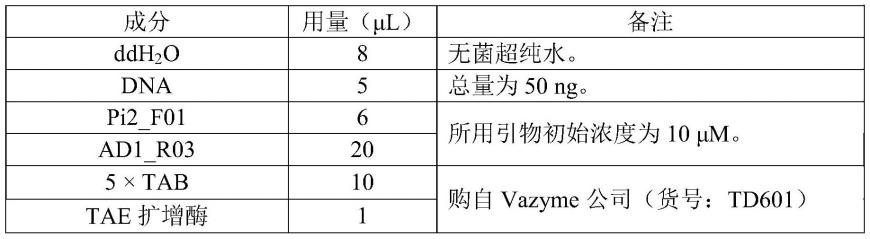
68.以所述引物对待测材料的基因组dna进行一次pcr扩增。其中,基因组dna的提取可参照现有技术。本发明中,针对不同物种或组织的特点,可使用catb法或者商业试剂盒等提取待测样本的基因组dna,要求dna经琼脂糖凝胶检测条带清晰完整、无明显降解和rna污染。
69.在pcr反应体系中,使用高保真的dna聚合酶进行pcr反应,通过调整引物的退火温度和循环条件,同时实现前景基因和遗传背景的检测目的。例如,可根据正向序列特异引物的tm值和最终pcr扩增产物的总量(尤其是其中200-600bp区域的总量)来调整,期望扩增产物总量在500ng左右,方便后续建库,继而进行pe150测序。
70.所述pcr扩增为热不对称交错式pcr扩增,包括两阶段循环反应:第一阶段循环反应主要使更多的(相比于随机简并引物而言)特异引物结合在模板上,进而获得更多的扩增产物;第二阶段循环反应主要使简并引物更容易地(相比于序列特异引物而言)与模板结合,引发扩增反应。具体而言,第一阶段循环反应包括在较高退火温度下进行3-7轮循环,每轮循环包括变性、较高退火温度下退火、延伸。第二阶段循环反应包括在较高退火温度下与较低退火温度下交错进行6-10轮循环,每轮循环包括3个循环单元且其中至少1个在较高退火温度下退火的循环单元以及至少1个在较低退火温度下退火的循环单元。优选地,每轮循环包括:变性、较高退火温度下退火、延伸、变性、较高退火温度下退火、延伸、变性、较低退火温度下退火、延伸。
71.在一些更具体实施方案中,本发明的所述热不对称交错式pcr扩增按照以下条件进行:
72.90-98℃预变性1min-5min;
73.第一阶段循环反应:90-98℃变性20s-2min,较高退火温度下退火30s-1min,72℃延伸1min-3min;3-7轮循环;
74.第二阶段循环反应90-98℃变性20s-2min,较高退火温度下退火30s-1min,72℃延伸1min-3min;90-98℃变性20s-2min,较高退火温度下退火30s-1min,72℃延伸1min-3min;90-98℃变性20s-2min,较低退火温度下退火30s-1min,72℃延伸1min-3min;6-10轮循环;
75.72℃终延伸5min-10min;
76.其中,各较高退火温度各自独立地为50-60℃,各较低退火温度各自独立地为40-50℃。
77.(3)扩增产物纯化及文库构建
78.利用磁珠对扩增产物进行纯化,随后进行3’末端加a、连接y型illumina测序接头等步骤,完成测序文库的制备,文库经片段分选后可用于pe150高通量测序。
79.(4)测序数据分析及基因型鉴定
80.根据样品特异的barcode序列对下机数据进行拆分,可获得每个样品对应的测序数据,随后将其比对参考基因组,检测前景基因内、正向序列特异引物f处的reads覆盖情况(前景基因筛选),以及全基因组上由reads富集、覆盖深度≥3
×
的高深度tags位点的分布
情况,这些多态性的tags位点即可用于后续的全基因组基因型检测(遗传背景检测)。
81.实施例1:利用正向序列特异引物pi2_f01与反向随机简并引物ad1_r03对水稻进行前景基因pi2(抗稻瘟病基因)和遗传背景的同时检测
82.本实施例中,提供了利用正向序列特异引物pi2_f01与反向随机简并引物ad1_r03对水稻进行前景基因pi2(抗稻瘟病基因)和遗传背景进行同时检测的方法。该方法主要过程包括:
83.(1)基因组dna提取及稀释
84.利用ctab法提取水稻品种nipponbare幼嫩叶片的基因组dna,经琼脂糖凝胶检测合格后,用无菌超纯水稀释。50μl反应体系稀释浓度最低应在3.85ng/μl,本实施例中稀释至10ng/μl(qubit浓度)。
85.(2)pcr反应体系配置
86.按照表1中pcr反应体系的成分及用量,使用规格为200μl的pcr薄壁管,在冰上配置总体积为50μl的反应体系,利用移液枪反复吹打10次混合均匀。同时设置3次技术重复(pi2-rep1、pi2-rep2和pi2-rep3)。所用正向序列特异引物pi2_f01的序列为:5
‘‑
taacagccaacctccgaacaacgccaactg-3’(seq id no:1,下划线处10nt为正向序列特异引物所连的barcode序列);反向随机简并引物ad1_r03的序列为:5
‘‑
tcagtgagtcgccvnvnnncgg-3’(seq id no:2,下划线处10nt为反向引物所连的barcode序列;中间的v和n为简并碱基;此引物简并度为2304)。
87.表1、pcr反应体系(50μl)
[0088][0089]
(3)pcr扩增反应程序设置
[0090]
经小型掌上离心机短暂离心后,收集所有50μl液体至pcr管底,随后按照表2,设置pcr扩增反应的程序(需提前105℃热盖),循环数共计26;所用pcr仪为德国耶拿biometra tone 96扩增仪(其它类似仪器均可)。
[0091]
表2、pcr反应程序
[0092][0093]
(4)pcr产物纯化及片段分布检测
[0094]
使用1.8
×
磁珠(90μl;购自vazyme公司,货号:n411-03)对pcr产物进行纯化,随后用30μl无菌超纯水进行洗脱,获得纯化产物。利用qubit 3.0仪器测定纯化后产物的浓度及总量,结果如表3所示。
[0095]
表3、纯化后pcr产物浓度及总量
[0096]
样品浓度(ng/μl)总量(ng)pi2-rep150.81,524pi2-rep235.61,068pi2-rep343.41,302
[0097]
利用qsep100核酸片段分析仪检测纯化后的片段大小分布,三个样品的片段大小分布基本一致,其中pi2-rep1检测结果如图1所示。
[0098]
(5)高通量测序文库构建
[0099]
利用sage pippin ht核酸片段分选仪对上述三次技术重复的纯化产物进行片段分选,回收200-600bp范围内的片段,并用无菌超纯水稀释至20ng/μl。随后取200ng,按照表4配制总体积为60μl的末端修复反应体系,反应程序为:105℃热盖;30℃静置20min;72℃静置20min;4℃结束反应。随后按照表5配制3’末端加“a”及短接头连接反应(共100μl),反应程序为:20℃静置30min。反应完成之后,利用0.8
×
磁珠(80μl;购自vazyme公司,货号:n411-03)对pcr产物进行纯化,用36μl无菌超纯水进行洗脱,获得纯化产物。然后按照表6配制illumina长接头的连接及文库扩增反应体系(总体积为50μl;三个技术重复添加不同的p5xx和p7xx组合,其中,xx代表不同的数字编号,每个重复有一个特定的p5xx+p7xx组合,主要用于区分此样品/重复,便于下机测序数据依此组合进行拆分,对应到每个样品/重复),反应程序按照表7设置,使用德国耶拿biometra tone 96扩增仪完成此步反应(其它类似仪器均可)。pcr扩增结束之后,利用0.9
×
磁珠(45μl;购自vazyme公司,货号:n411-03)进行纯化,用30μl无菌超纯水进行洗脱,获得纯化产物。利用qubit 3.0仪器测定纯化后产物的浓
universal pro dna library prep kit for mgi,货号:ndm608),将本发明中pcr扩增后的产物转化为华大测序所需的文库类型。
[0112]
(6)片段分选及高通量测序
[0113]
由于每个样品在文库扩增过程中已添加了特异的index(即不同的p5xx和p7xx组合),因此可将所有三个文库等量混合在一起(三个文库也可分别进行分选、测序),再次利用sage pippin ht核酸片段分选仪将适用于后续pe150测序的片段(如350-700bp)分选出来(主要用于去除未耗尽的引物序列和小于350bp的短片段),如图3所示(12.8ng/μl;共384ng),利用illumina测序平台进行高通量测序。
[0114]
(7)测序数据拆分及tags特征分析
[0115]
测序数据经拆分后,获得三次技术重复对应的数据量分别为1.28gb、1.31gb和1.16gb(表9)。随后,去除每个样品测序数据中低质量(碱基质量值小于15)的reads,比对水稻nipponbare msu v7.0参考基因组序列(~0.4gb,http://rice.uga.edu/),利用可视化软件igv(https://igv.org/)可以看见测序reads不仅在前景基因pi2内的引物结合处显著富集(图4中的图片a、图片b),而且三次技术重复中由引物pi2_f01与ad1_r03随机扩增产生的reads在基因组上显著富集、稳定地形成高深度tags位点(图4中的图片c)。在获得参考基因组上碱基覆盖深度数据之后,可以在三次技术重复中检测到全基因组范围内depth≥3
×
的高深度tags位点数目分别为19,814、20,103和19,266个;其中,三次技术重复间检测到的共有tags数目为12,209个,这些tags在三次技术重复检测到的总tags数目中的占比介于60.73-63.37%之间(表9),说明本发明中即使是引物引发的随机扩增,但扩增产物依然具有较高的稳定性和可重复性。当查看这些共有tags在基因组上的分布位置时,相邻tag间平均间距为30.55kb,标准差为42.97kb,可见共有tags均匀分布于全基因组的所有染色体上(图4中的图片d)。
[0116]
表9、样品测序数据量及tags数目检测结果
[0117][0118]
综上,本发明中利用正向序列特异引物pi2_f01与反向随机简并引物ad1_r03进行一次pcr扩增,不仅可以完成对前景抗稻瘟病基因pi2的筛选,而且可以稳定获得数目众多、均匀分布于全基因组的高深度tags,用于遗传背景检测。
[0119]
实施例2:利用正向序列特异引物rf4_f01与反向随机简并引物ad2_r03对水稻进行前景基因rf4(水稻育性恢复基因)和遗传背景的同时检测
[0120]
本实施例中,提供了利用正向序列特异引物rf4_f01与反向随机简并引物ad2_r03对水稻(与实施例1相同的水稻品种nipponbare)进行前景基因rf4(水稻育性恢复基因)和遗传背景进行同时检测的方法。该方法主要过程包括:
[0121]
(1)基因组dna提取及稀释
[0122]
同实施例1中(1)。
[0123]
(2)pcr反应体系配置
[0124]
按照表1中的成分(其中的引物替换为本实施例的引物)及用量,使用规格为200μl的pcr薄壁管,在冰上配置总体积为50μl的反应体系,利用移液枪反复吹打10次混合均匀。同时设置3次技术重复(rf4-rep1、rf4-rep2和rf4-rep3)。所用正向序列特异引物rf4_f01的序列为:5
‘‑
taacagccaactgcttaca aagtgaggtggtgt-3’(seq id no:3,下划线处10nt为正向序列特异引物所连的barcode序列);反向随机简并引物ad2_r03的序列为:5
‘‑
tcagtgagtcgccbnbnnncgg-3’(seq id no:4,下划线处10nt为反向引物所连的barcode序列;中间的b和n为简并碱基;此引物简并度为2304)。
[0125]
(3)pcr扩增反应程序设置
[0126]
参见实施例1中(3)。
[0127]
(4)pcr产物纯化及片段分布检测
[0128]
使用1.8
×
磁珠(90μl;购自vazyme公司,货号:n411-03)对pcr产物进行纯化,随后用30μl无菌超纯水进行洗脱,获得纯化产物。利用qubit 3.0仪器测定纯化后产物的浓度及总量,结果如表10所示。
[0129]
表10、纯化后pcr产物浓度及总量
[0130]
样品浓度(ng/μl)总量(ng)rf4-rep147.11,413rf4-rep249.41,482rf4-rep350.01,500
[0131]
利用qsep100核酸片段分析仪检测纯化后的片段大小分布,三个样品的片段大小分布基本一致,其中以rf4-rep1样品为代表的检测结果如图5所示。
[0132]
(5)高通量测序文库构建
[0133]
分别取上述纯化产物5μl,按照表11配制总体积为60μl的末端修复反应体系,反应程序为:105℃热盖;30℃静置20min;72℃静置20min;4℃结束反应。随后按照表5配制3’末端加“a”及短接头连接反应(共100μl),反应程序为:20℃静置30min。反应完成之后,利用0.8
×
磁珠(80μl;购自vazyme公司,货号:n411-03)对pcr产物进行纯化,用36μl无菌超纯水进行洗脱,获得纯化产物。然后按照表6配制illumina长接头的连接及文库扩增反应体系(总体积为50μl;三个技术重复添加不同的p5xx和p7xx组合),反应程序按照表12设置,使用德国耶拿biometra tone 96扩增仪完成此步反应(其它类似仪器均可)。pcr扩增结束之后,利用0.9
×
磁珠(45μl;购自vazyme公司,货号:n411-03)进行纯化,用26μl无菌超纯水进行洗脱,获得纯化产物。利用qubit 3.0仪器测定纯化后产物的浓度,结果如表13所示。
[0134]
表11、末端修复反应体系(60μl)
[0135][0136]
表12、文库扩增pcr反应程序
[0137][0138]
表13、纯化后文库浓度及总量
[0139]
样品浓度(ng/μl)总量(ng)rf4-rep111.0286.0rf4-rep211.1288.6rf4-rep311.3293.8
[0140]
其它类似的文库构建试剂(如vazyme公司的vahts universal pro dna library prep kit for illumina,货号:nd608)也可用于本发明中pcr扩增后的文库构建过程;除利用illumina测序平台外,也可使用适用于华大测序平台的试剂(如vazyme公司的vahts universal pro dna library prep kit for mgi,货号:ndm608),将本发明中pcr扩增后的产物转化为华大测序所需的文库类型。
[0141]
(6)片段分选及高通量测序
[0142]
由于每个样品在文库扩增过程中已添加了特异的index(即不同的p5xx和p7xx组合),且纯化后总量近似相等(表13,均值
±
标准差为:289.47
±
3.24),因此可直接将所有三个文库全部混合在一起,经qsep100核酸片段分析仪检测混合后的片段范围分布,如图6所示。利用sage pippin ht核酸片段分选仪,将适用于后续pe150测序的片段(如350-700bp)从混合后的总文库中分选出来,如图7所示(1.75ng/μl;共52.5ng),利用illumina测序平台进行高通量测序。
[0143]
(7)测序数据拆分及tags特征分析
[0144]
测序数据经拆分后,获得三次技术重复对应的数据量分别为0.28gb、0.29gb和0.33gb(表14)。随后,去除每个样品测序数据中低质量的reads,比对水稻nipponbare msu v7.0参考基因组序列(~0.4gb,http://rice.uga.edu/),利用可视化软件igv(https://igv.org/)可以看见测序reads不仅在前景基因rf4内的引物结合处显著富集(图8中的图片a、图片b),而且三次技术重复中由引物rf4_f01与ad2_r03随机扩增产生的reads在基因组上显著富集、稳定地形成高深度tags位点(图8中的图片c)。在获得参考基因组上碱基覆盖深度数据之后,可以在三次技术重复中检测到全基因组范围内depth≥3
×
的高深度tags位点数目分别为18,578、18,504和20,939个;其中,三次技术重复间检测到的共有tags数目为12,350个,这些tags在三次技术重复检测到的总tags数目中的占比介于58.98-66.74%之间(表14)。当查看这些共有tags在基因组上的分布位置时,相邻tag间平均间距为30.02kb,标准差为38.51kb,可见共有tags均匀分布于全基因组的所有染色体上(图8中的图片d)。
[0145]
表14、样品测序数据量及tags数目检测结果
[0146][0147]
综上,本发明中利用正向序列特异引物rf4_f01与反向随机简并引物ad2_r03进行一次pcr扩增,不仅可以完成对前景水稻育性恢复基因rf4的筛选,而且可以稳定获得数目众多、均匀分布于全基因组的高深度tags,用于遗传背景检测。
[0148]
实施例3:利用正向序列特异引物hd1_f01与反向随机简并引物ad1_r3b对动物样本进行全基因组遗传背景检测
[0149]
本实施例中,提供了利用正向序列特异引物hd1_f01与反向随机简并引物ad1_r3b,分别对两份动物dna样本(一份金毛寻回犬dog_139和一份杜洛克猪样品pig_421)进行全基因组遗传背景检测。该方法主要过程包括:
[0150]
(1)基因组dna提取及稀释
[0151]
利用fastpure blood dna isolation mini kit v2试剂盒(购自vazyme公司,货号:dc111-01),按照官方说明书,提取dog_139和pig_421样品的血液dna,经琼脂糖凝胶检测合格后,用无菌超纯水稀释。50μl反应体系稀释浓度最低应在3.85ng/μl,本实施例中稀释至10ng/μl(qubit浓度)。
[0152]
(2)pcr反应体系配置
[0153]
按照表1中pcr反应体系的成分及用量,使用规格为200μl的pcr薄壁管,在冰上配置总体积为50μl的反应体系,利用移液枪反复吹打10次混合均匀。每份样品同时设置3次技术重复(dog_139-rep1、dog_139-rep2和dog_139-rep3;pig_421-rep1、pig_421-rep2和pig_421-rep3)。所用正向序列特异引物hd1_f01的序列为:5
‘‑
taacagccaa aggacggaggtggccgggatggt-3’(seq id no:5,下划线处10nt为正向序列特异引物所连的barcode序列);反向随机简并引物ad1_r3b的序列为:5
‘‑
tcagtgagtc gccvavngncgg-3’(seq id no:6,下划线处10nt为反向引物所连的barcode序列;中间的v和n为简并碱基;此引物简并度为144)。
[0154]
(3)pcr扩增反应程序设置
[0155]
参见实施例1中步骤(3)。
[0156]
(4)pcr产物纯化及片段分布检测
[0157]
使用1.8
×
磁珠(90μl;购自vazyme公司,货号:n411-03)对pcr产物进行纯化,随后用50μl无菌超纯水进行洗脱,获得纯化产物。利用qubit 3.0仪器测定纯化后产物的浓度及总量,结果如表15所示。
[0158]
表15、纯化后pcr产物浓度及总量
[0159]
样品浓度(ng/μl)总量(ng)dog_139-rep130.01,500.0dog_139-rep234.11,705.0dog_139-rep328.51,425.0pig_421-rep127.81,390.0
pig_421-rep231.81,590.0pig_421-rep325.01,250.0
[0160]
利用qsep100核酸片段分析仪检测纯化后的片段大小分布,三个样品的片段大小分布基本一致,其中以dog_139-rep1和pig_421-rep1样品为代表的检测结果如图9所示。
[0161]
(5)高通量测序文库构建
[0162]
按照实施例2中步骤(5)进行文库构建和纯化,最终用21μl无菌超纯水进行洗脱,获得纯化产物。利用qubit 3.0仪器测定纯化后产物的浓度,结果如表16所示。利用qsep100核酸片段分析仪检测纯化后的文库片段大小分布,犬和猪对应的三个重复样品的文库峰形基本一致,其中以dog_139-rep1和pig_421-rep1样品为代表的文库检测结果如图10所示。
[0163]
表16、纯化后文库浓度及总量
[0164]
样品浓度(ng/μl)总量(ng)dog_139-rep133.4701.4dog_139-rep226.2550.2dog_139-rep327.8583.8pig_421-rep128.8604.8pig_421-rep227.4575.4pig_421-rep325.6537.6
[0165]
(6)片段分选及高通量测序
[0166]
按照实施例2中步骤(5)进行文库混合和片段分选,将适用于后续pe150测序的片段(如400-800bp)从混合后的总文库中分选出来,如图11所示(2.05ng/μl;共61.5ng),利用illumina测序平台进行高通量测序。
[0167]
(7)测序数据拆分及tags特征分析
[0168]
测序数据经拆分后,dog_139三次技术重复对应的数据量分别为1.02gb、1.13gb和1.29gb;pig_421三次技术重复对应的数据量分别为1.14gb、1.08gb和1.48gb(表17)。随后,去除每个样品测序数据中低质量的reads,分别比对到常用的犬参考基因组(~2.4gb,https://www.ncbi.nlm.nih.gov/assembly/gca_008641055.3),以及猪参考基因组(~2.5gb,https://www.ncbi.nlm.nih.gov/assembly/gcf_000003025.6/)上,利用可视化软件igv(https://igv.org/)可以看见三次技术重复中由引物hd1_f01与ad1_r3b随机扩增产生的reads在猪和犬的基因组上显著富集、稳定地形成高深度tags位点(图12)。
[0169]
表17、样品测序数据量及tags数目检测结果
[0170][0171]
在获得参考基因组上碱基覆盖深度数据之后,可以在犬dog_139样品的三次技术重复中检测到全基因组范围内depth≥3
×
的高深度tags位点数目分别为110,930、115,395和122,240个,其中三次技术重复间检测到的共有tags数目为71,885个,这些共有tags在三次技术重复检测到的总tags数目中的占比介于58.81-64.80%之间(表17)。对于猪样品pig_421,三次技术重复中检测到全基因组范围内depth≥3
×
的高深度tags位点数目分别为118,255、111,705和133,166个,其中三次技术重复间检测到的共有tags数目为74,937个,这些共有tags在三次技术重复检测到的总tags数目中的占比介于56.27-67.08%之间(表17)。当查看犬样品三次重复中共有tags在基因组上的分布位置时(图13),相邻tag间平均间距为33.43kb,标准差为210.64kb;同样,当查看猪样品三次重复中共有tags在基因组上的分布位置时(图13),相邻tag间平均间距为32.39kb,标准差为227.52kb。可见,这些共有tags可以较均匀地分布于犬和猪的所有染色体上。
[0172]
综上,本发明对基因组的一部分进行富集的方法,不仅可以用于前景基因和遗传背景的同时检测,而且可作为一种新的基因型鉴定技术,应用于动植物全基因组的基因型检测。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1