聚合物发泡体及其制备方法与流程
本发明是关于一种用来制备聚合物发泡体的方法,更具体地,是关于一种透过压力诱导流动(pressure-inducedflow,PIF)步骤来制备聚合物发泡体的方法。
背景技术:
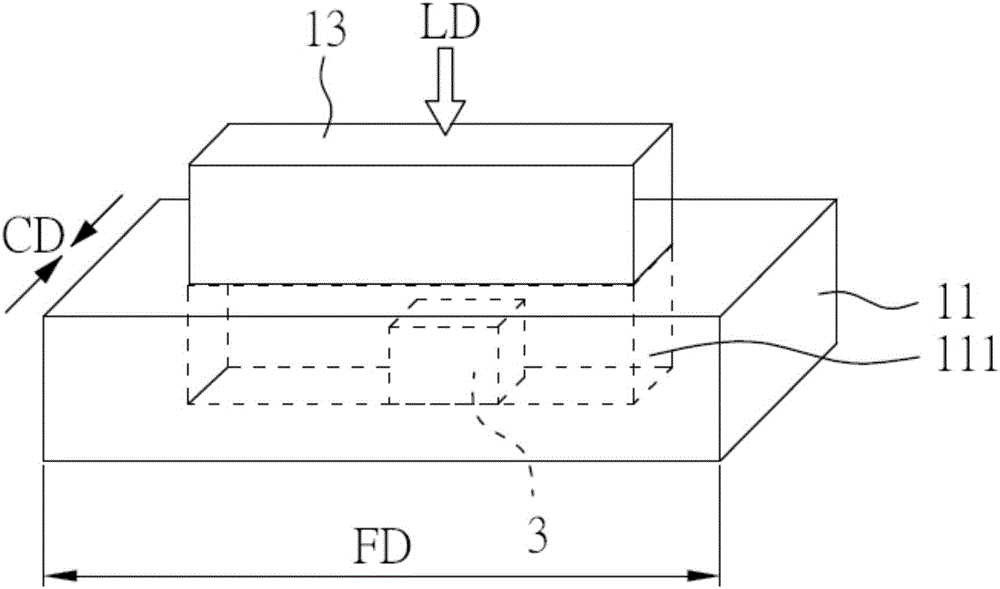
:聚合物发泡体,特别是聚丙烯(polypropylene)发泡体,有着许多任务业上的应用,全球聚合物发泡体市场已见证了过去几年间的快速成长。聚丙烯(PP)作为最广泛使用的商业聚合物之一,有着许多理想的特性,譬如高熔点、良好的可塑性、低密度、优良的耐化学性以及食品安全性,并且容易回收。这些杰出的特性及低材料成本使得聚丙烯(PP)在各种工业应用中相较于其他热塑性塑料,在生产塑料及发泡体产品方面更具竞争性。然而,如同其他商品聚合物,聚丙烯(PP)的低热稳定性及低机械强度阻碍其在许多工程及结构方面的应用。此外,除非有长链接支、聚合物复合、或使用化学交联来改质聚丙烯(PP),否则在熔体发泡步骤中,聚丙烯(PP)的低熔融强度会导致泡体(cell)破裂及低发泡倍率。然而,因为低熔融强度及高结晶度使得聚丙烯(PP)非常难以发泡。许多方法如:聚合物共混物、复合材料、及共聚合已被用于获得良好的可发泡性的聚丙烯(PP)及聚丙烯(PP)发泡体。但因为材料成本显着增加、且发泡体产品的机械强度于比聚丙烯(PP)及其他被广泛使用的发泡体材料如聚苯乙烯(PS)来的差,使得上述方法的应用性受到局限。因此,亟需提供一种新颖的制备聚合物发泡体,特别是聚丙烯(PP)发泡体的方法,其能够以最有希望及最可行的方式制备聚合物发泡体;并且因此,所获得的聚合物发泡体能够成功地被应用于工业产品上。技术实现要素:本发明的一目的在于提供一种透过一压力诱导流动(pressure-inducedflow,PIF)步骤以及一发泡步骤制备聚合物发泡体的方法。本发明的另一目的在于提供一种由本发明的方法制得的聚合物发泡体。此外,本发明的又一目的在于提供一种透过压力诱导流动(PIF)步骤制备具有共连续结构(co-continuousstructure)的聚合物片材的方法。于本发明的一例子中,制备聚合物发泡体的方法包括以下步骤:提供一聚合物本体;在一第一特定温度及一第一特定压力下,于一特定持压时间,在该聚合物本体上进行一压力诱导流动(PIF)步骤以获得一聚合物片材;以及在一第二特定温度及一第二特定压力下,于一饱和时间内,在该聚合物片材上注入一发泡剂进行一发泡步骤以获得聚合物发泡体。于本发明的另一例子中,制备具有共连续结构(co-continuousstructure)的聚合物片材的方法包括以下步骤:提供一聚合物复合物,其包括聚合物颗粒以及涂层材料,且这些聚合物颗粒的表面上涂布有涂层材料;以及在一第一特定温度及一第一特定压力下,于一特定持压时间,在该聚合物复合物上进行一压力诱导流动(PIF)步骤以获得一具有共连续结构的聚合物片材。此外,当上述获得的具有共连续结构的聚合物片材,在一第二特定温度及一第二特定压力下,于一饱和时间内,注入一发泡剂来进行一发泡步骤时,可获得聚合物复合物发泡体。于本发明的另一例子中,所获得的聚合物发泡体包括:一聚合物本体,该聚合物本体中具有多个泡体(cell),其中该聚合物发泡体具有发泡密度介于0.03g/cm3至0.25g/cm3间;以及当该聚合物发泡体的压缩应变量(compressivestrain)是介于10%及70%之间时,该聚合物发泡体具有压缩强度(compressivestrength)介于0.2MPa至0.7MPa间。现有未经改质的聚合物,特别是纯聚丙烯(neatpolypropylene),因为其低熔融强度及高结晶度的缘故而难以发泡。于本发明中提供一种新颖的方法,其中聚合物是透过压力诱导流动(PIF)步骤及发泡步骤而发泡。借由利用本发明的方法,该聚合物可在较不严苛的条件下(较低压力及较高温度)、及饱和时间短的情况下发泡以获得具有高性能的低密度聚合物发泡体,但本发明不限于此。因此,相较于传统的制备聚合物发泡体的步骤,本发明的方法中的压力及饱和时间可降低;因此,本发明的方法更适合在工业应用中制造聚合物发泡体。此外,相较于由相同组成物形成的传统复合物及发泡体,当该聚合复合物(其包括聚合物颗粒及涂布在该聚合物颗粒上的涂层材料)经压力诱导流动(PIF)步骤及选择性地经发泡步骤处理时,所获得的具有共连续结构的聚合物片材或发泡体结构可展现出优异的机械和物理特性。此外,相较于未经压力诱导流动(PIF)步骤制得的传统聚合物发泡体,本发明的聚合物发泡体同时具有低发泡密度及高强度。因此,以本发明的方法制备的聚合物发泡体可进一步地应用在各种工业产品中。于本发明的聚合物发泡体及方法中,该聚合物本体可简单地由聚合物制得,或者该聚合物本体可为一种聚合物复合物,该聚合物复合物包括聚合物颗粒及涂布在该聚合物颗粒表面上的涂布层。当本发明方法中使用的聚合物本体为上述的聚合物复合物时,该聚合物复合物可由下述步骤制备:提供并混合该聚合物颗粒以及该涂层材料以获得一混合物;且加热该混合物以获得该聚合物复合物,其中该聚合物复合物包括该聚合物颗粒及由涂层材料所形成的涂布层。将该混合物加热之后,所获得的聚合复合物具有一蜂巢状结构(honeycomb-likestructure)。在此,该聚合物颗粒及该涂层材料可在介于140℃至160℃之间的温度下被混合。为了帮助混合,可加入研磨剂如氧化锆增韧氧化铝(zirconiatoughenedalumina),如此可使混合更加均匀及彻底。此外,可在100℃至300℃的温度下加热该混合物;且可在11.7MPa至17.3MPa的压力下加热该混合物。在此,所使用的加热方法可为热气体、电加热、红外线或微波。此外,所获得的聚合物复合物可进一步透过筛子过筛以除去研磨剂以及剩余的涂层材料。再者,该聚合物颗粒上的涂布层厚度并无特别限制,可约为1μm至1000μm。此外,根据将涂层材料涂布在聚合物颗粒上的步骤、温度及/或压力,该涂布层可完全地或部分地嵌入该颗粒的表面的内侧,或者可附着于该颗粒的表面上。于本发明的方法中,该聚合物本体可在高于室温的温度(譬如,210℃)以及预定压力(譬如1700psi)下,选择性地预压成扁平片、膜状、或其他形状。然而,此步骤并非本发明的方法的必须步骤,该聚合物本体可在未进行上述预压步骤的情况下直接进行压力诱导流动(PIF)步骤。于本发明的方法中,在压力诱导流动(PIF)步骤中的该第一特定温度取决于聚合物本体的类型,且较佳系低于该聚合物本体的熔点。该第一特定温度较佳为介于100℃至160℃。该第一特定温度更佳为介于110℃至150℃。最佳地,当聚合物本体是由PP制成时,上述的第一特定温度为适用于PIF步骤的温度。于本发明的方法中,在压力诱导流动(PIF)步骤中的该第一特定压力可介于20MPa至420MPa。该第一特定压力较佳可介于20MPa至200MPa。该第一特定压力更佳可介于20MPa至50MPa。当用于本发明的方法的压力诱导流动(PIF)步骤为超声波辅助压力诱导流动(UAPIF)步骤时,该第一特定压力可被进一步降低。在压力诱导流动(PIF)步骤中,持压时间并没有特别的限制,只要该聚合物本体能够变形即可。该持压时间较佳为介于10秒至300秒。该持压时间更佳为介于10秒至30秒。在本发明的方法中,上述的压力诱导流动(PIF)步骤进行完毕后,聚合物本体的结晶区域可转变为一种共连续的“砖和泥”状结构。在本发明的方法中,在发泡步骤中的该第二特定温度可介于120℃至180℃。该第二特定温度较佳可介于130℃至160℃。该第二特定温度更佳可介于140℃至155℃。在本发明的方法中,在发泡步骤中的该第二特定压力可介于11MPa至20MPa。该第二特定压力较佳可介于11.7MPa至17.3MPa。该第二特定压力更佳可介于13.8MPa至17.2MPa。在发泡步骤中,饱和时间并没有特别的限制,只要该聚合物片材可以确实的发泡即可。该饱和时间可随着聚合物片材的厚度而改变。于本发明中,该饱和时间较佳为10分钟至2小时。于本发明的方法之发泡步骤中,所使用的发泡剂并无特别限制,可为任何气体或是能够释放气体的材料。该发泡剂的例子包括但不限于:戊烷(pentane)、异戊烷(isopentane)、环戊烷(cyclopentane)、CO2、N2、氮系材料(nitrogen-basedmaterial)、或其组合。较佳地,适合本发明的方法之发泡剂为CO2。该发泡剂最佳为超临界二氧化碳(supercriticalCO2)。在此,该发泡剂可被导入用来进行发泡步骤的反应器中,或者利用单一的装置,譬如挤出机或捏合机,在进行PIF或发泡步骤之前导入聚合物本体。于本发明的聚合物发泡体中,该聚合物发泡体可具有发泡密度介于0.03g/cm3至0.25g/cm3。较佳为,该发泡密度介于0.04g/cm3至0.10g/cm3之间。因此,本发明中所获得的聚合物发泡体系一低密度聚合物发泡体。于本发明的聚合物发泡体中,当聚合物发泡体放置在120℃的条件时,该聚合物发泡体的一尺寸变化的绝对值介于0%至60%,且较佳为介于0%至40%之间。于本发明的聚合物发泡体中,该聚合物发泡体可具有结晶度介于10%至30%。较佳为,该发泡体的结晶度介于15%至25%之间。于本发明的聚合物发泡体中,这些泡体(cells)可具有200μm至300μm的平均泡体尺寸。较佳为,这些泡体的平均泡体尺寸介于240μm至260μm之间。于本发明的聚合物发泡体及方法中,该聚合物本体或该聚合物颗粒的种类并无特别的限制,且可为半结晶热塑性塑料或热塑性弹性体。半结晶热塑性塑料或热塑性弹性体的具体实例可为聚烯烃(polyolefins),其可包括但不限于,至少一种选自由聚乙烯(polyethylene)、聚丙烯(polypropylene)(包括均聚物、嵌段共聚物、或无规则共聚物)、聚氯乙烯(polyvinylchloride)、聚碳酸酯(polycarbonates)、聚酰胺(polyamides)、聚对苯二甲酸乙二酯(polyethyleneterephthalate)、聚对苯二甲酸丁二酯(polybutyleneterephthalate)以及聚乳酸(polylacticacid)所组成的群组。该热塑性弹性体的实例可包括,但不限于:聚烯烃共混物、弹性体合金(TPE-v或TPV)及热塑性聚酰胺。此外,这些聚合物颗粒之形状及尺寸并无特别限制。于本发明的方法中,该聚合物本体较佳为未经改质的聚合物。该聚合物本体或该聚合物颗粒更佳包括纯聚丙烯(neatPP)。上述“纯聚丙烯”意指未经改质的PP。此外,该聚合物复合物的涂层材料可为一有机物质、一无机物质、或其组合。该有机物质的实例包括,但不限于至少一种选自由碳纳米颗粒、碳微粒、石墨烯、石墨烯氧化物、碳黑、碳纳米纤维、碳纳米管、及石墨所组成的群组。该无机物质的实例包括,但不限于至少一种选自由粘土、云母、玻璃纤维、硅酸盐、金属颗粒、SiO2、MgO、CaO、滑石粉、TiO2,ZnO及MnO所组成的群组。附图说明图1A是本发明一实施例所使用的PIF装置的示意图;图1B是PP样品经PIF步骤处理前及处理后的示意图;图2A是本发明实施例1的PIF步骤后部分PP样品的示意图;图2B是本发明实施例1的发泡步骤后部分PP发泡体的示意图;图3是本发明实施例1在150℃及34.5MPa下经PIF步骤处理的PP样品的XRD;图4是本发明实施例1的发泡压力与发泡密度的关系;图5是本发明实施例1中以不同发泡压力处理PP发泡体的热特性的示意图;图6是本发明实施例1中,scCO2饱和时间对于发泡密度的效果;图7A至7C是本发明实施例1中,在不同压力及不同饱和时间条件下形成之PIFPP发泡体的SEM;图8是本发明实施例1的各种发泡体的压缩行为的示意图;图9是本发明实施例1的各种发泡体的热稳定性的示意图;图10A是本发明实施例2所获得的PP/MWCNT复合物的示意图;图10B是本发明实施例1中,经PIF步骤后部分PP/MWCNT片材的示意图;图10C是本发明实施例2中,经发泡步骤后部分PP/MWCNT发泡体的示意图;图11A及11B分别为本发明实施例2经PIF步骤处理之前及之后的PP样品的偏光显微镜图像;图12A是本发明实施例2所获得的原始PP、PIFPP、UAPIFPP、以及PIFPP发泡体的DSC曲线;图12B是本发明实施例2所获得的原始PP以及PIFPP的XRD;图12C是UAPIF步骤中的温度上升;图13A至13C分别表示本发明实施例2中,经由PIF或未经PIF所获得的PP及PP/MWCNT复合物的拉力、弯曲、及冲击强度的示意图;图14是本发明实施例2所获得的纯PP及UAPIF-PP/MWCNT复合物的弯曲特性示意图;图15是本发明实施例2所获得的纯PP发泡体及PIFPP发泡体的发泡倍率的示意图;图16是纯PP及PIF-PP发泡体的热机械分析(TMA)曲线;图17A及17B分别为经由PIF步骤及未经PIF步骤的衍生自纯PP及PP/MWCNT纳米复合发泡体的高密度及低密度PP发泡体的压缩应力与压缩应变量曲线;图18A及18B是本发明实施例2中纯PP及PIFPP的TMA曲线;以及图19A及19B是本发明实施例2中,以UAPIF步骤处理后纯PP及PP/MWCNT3wt%复合的弯曲应力。【附图标记说明】11槽状模具111模穴13柱塞210聚合物颗粒220涂布层230泡体3试片3’片材310试片330泡体LD负载方向CD限制方向FD流动方向具体实施方式为使本发明的目的、技术方案和优点更加清楚明白,以下结合具体实施例,并参照附图,对本发明作进一步的详细说明。图1A是本发明所使用的PIF装置的示意图。该用于本发明的PIF装置主要包括:一槽状模具11,该槽状模具11具有模穴111;以及一柱塞13,该柱塞13具有与模穴111大致相同的尺寸。在PIF步骤中,将样品3放置在模穴111中,再以柱塞13加热挤压;随后,样品3产生形变,以流动方向(FD)流入,且被限制在限制方向(CD)并在负载方向(LD)压缩。图1B是样品处理前及经PIF步骤后的示意图。在经PIF步骤处理前的样品3具有一立方体形状;但本发明并不限于此。经PIF步骤后,该样品3被挤压成片材3’。在接下来的实施例中,模穴111的尺寸为100×12×12mm,但本发明不限于此。实施例1-仅以聚丙烯(PP)制备聚合物发泡体鉴定发泡密度:所获得的PP样品的质量密度ρf系根据ASTMD792量测,其系利用沉降片在水中秤重聚合物发泡体。ρf计算如下:其中a为试样在空气中的表观质量,b为试样完全浸入水中的表观质量。扫描电子显微镜(SEM):以SEM观察得到的PP发泡体的形态(PhilipsXL30)。该样品浸泡在液态氮中30分钟并断裂。该断裂面被喷镀一层金以透过SEM进一步观察。热机械分析(TMA):利用TMA(TAInstrumentsTMA2940)观察PIF及发泡体样品的热稳定性。在穿透模式下以5℃/分的扫描速度测定样品从30℃至180℃的尺寸变化。示差扫描量热计(DSC):TAQ200DSC用来定性经PIF步骤或未经PIF步骤的PP发泡体熔融行为。扫描范围系从20至200℃,10℃/分的速率。样品被切割为6-10mg的薄片来进行DSC定性。X射线衍射(XRD):利用具有Cu-kα射线源且X射线波长为的X射线衍射仪(BrukerD8AdvanceXRD)研究晶体结构。在40V及50mA下,从5°至45°以4°/分钟速率扫描该样品。压缩试验:发泡体的压缩强度系以Instron5569先进材料测试系统(Instron5569AdvancedMaterialsTestingsystem)在室温下根据标准ASTMD695进行量测。实验PP及PIF条件对于发泡密度的影响在此,表1中列示了所使用的具有不同熔融流动指数(MFI)的不同种类PP。将尺寸为50mm(长)×12mm(宽)×2mm(高)的PP样品放置于图1A所示的PIF装置中,并且在表1中列示的各种不同温度、不同压力、以及不同持压时间下变形。图2A系经PIF步骤后部分PP样品310的示意图。PIF步骤后,将所制得的样品放置在高压容器中,随后在该高压容器中注入纯度为99.99%的scCO2。当样品在一特定温度及压力下饱和一定时间后,瞬间降压以实现PP发泡。图2B系发泡步骤后部分PP样品310的示意图。如图2B所示,发泡步骤后,在PP样品310中形成了发泡体(cell)330。本实施例中,检测所制得的PP样品的发泡密度并将结果总结在下表1中。表1发泡条件:155℃,13.8MPa,2小时;N/A:无法发泡PP1:纯PP,PC366-3(均聚聚丙烯),由李长荣化学工业股份有限公司提供PP2:纯PP,PT100(均聚聚丙烯),由李长荣化学工业股份有限公司提供PP3:纯PP,6331(均聚聚丙烯),由李长荣化学工业股份有限公司提供PP4:纯PP,HP600S(均聚聚丙烯),由李长荣化学工业股份有限公司提供DowPP:H349-02(均聚聚丙烯),由陶氏化学公司提供如表1所示,可以看出具有低MFI的PP表现出较佳的发泡特性。随着MFI增加,代表熔体强度降低,PP仍然可以发泡,但发泡密度增加了。商业化的高熔体强度聚丙烯(HMSPP)(WB140购自北欧化工)也用来作为比较:在未经PIF时亦可良好的发泡,但发泡体强度及热稳定性不佳,如图8及图9所示。比较如PP纤维及双向拉伸聚丙烯(BOPP)薄膜这类定向而具有“串晶结构(shish-kebabstructure)”的PP产品。表1的结果显示PP纤维及BOPP均无法发泡,意即仅有定向的晶体结构不足以进行PP发泡。显然,由PIF步骤所形成的细长“砖和泥(brickandmud)”状晶体结构对于达成PP发泡是必须的。为了实行PIF,温度必须低于PP的熔点,因此在整个PIF步骤中PP仍然保持固体状态。在适当的压力及温度下,PP非晶区沿着施加压力的垂直方向变形以延长PP的球晶区域(spherulitecrystaldomains),在施加的温度及压力之间存有折衷关系。在低温下,需要使用较高的压力;若使用较高的温度,即使在低压下也是能够达成类似的效果。下文中,选择表1所列示的PP1进一步优化PIF及发泡步骤。PIF条件列示在表2中,进行前述的发泡步骤,同样地维持发泡条件于13.8MPa及155℃,2小时。表2PIF条件PIF时间发泡密度(g/cm3)110℃,414MPa300秒0.043110℃,414MPa10秒0.073150℃,34.5MPa300秒0.052150℃,34.5MPa10秒0.094如表2所示,在150℃及34.5MPa时,经PIF步骤处理的发泡样品的密度非常接近110℃及414MPa时的情况。并且,PIF持压时间对于发泡密度影响往往不大。意味着许多任务业有关的PIF条件,34.5MPa取代414MPa压力且10秒取代300秒的持压时间已足以达到低密度PP发泡体。换言之,PIF的周期时间可以缩短许多。图3是在150℃及34.5MPa条件下以PIF步骤处理的PP样品的XRD图,且此结果显示34.5MPa及150℃与414MPa及110℃所观察到的球晶变形相似。发泡压力对于发泡密度的影响发泡步骤中,发泡压力是一个关键因素。不同的发泡压力用来研究对于发泡密度的影响。以下,选择表1中的PP1,PIF条件为150℃、34.5MPa以及10秒,且发泡条件为155℃、2小时及不同的发泡压力(8.3、11.7、13.1、13.8、15.5、及17.3MPa)。如图4所示,发泡密度突然下降发生在13.8MPa。当压力低于13.8MPa时,发泡密度较高。高于13.8MPa时,随着压力增加,发泡密度基本维持不变。在此,发泡PP样品的结晶度亦以DSC检测并将结果汇总在表3。表3如上表3所示,可观察到发泡体结晶度的明显改变。13.8MPa以下时,PP发泡体的结晶度相似,而当压力达到13.8MPa时,可观察结晶度明显地下降。习知熔融温度会随CO2的饱和压力的增加而降低。显然当压力低于13.8MPa时,PP的熔融温度下降,但仍然高于155℃的发泡温度。在此情况下,用于发泡的非晶区仍然很小,因此发泡密度高。然而,当CO2饱和压力达到13.8MPa或以上时,熔融温度变得接近155℃,大量的结晶熔融成为非晶相。净结果是当CO2饱和压力释放时,发泡密度突然地下降。较高的发泡压力同时也意味着较高的压力释放速率,因此在发泡步骤中有更多的泡体成核并成长。图5系以不同的发泡压力处理的发泡PP的热特性示意图。由图5的结果可见,当CO2饱和压力低于13.8MPa时,结晶熔融峰几乎没有变化。随着CO2饱和压力增加至13.8MPa并超过13.8MPa的阈值时,在155℃的等温CO2饱和步骤中出现小的熔融峰肩,代表不完美的晶体熔化以及再结晶。剩下未熔化的那些结晶可为α1至α2结晶的完善形式,其具有较高的熔化温度,能够在155℃的高CO2饱和压力下留存。剩余的细长的“砖”状晶体结构对于PP的发泡是重要的。在吸热发泡步骤中,冷却步骤非常的快速,因此,熔化晶体以及可能的一些非晶相发生再结晶及重新排列,导致在较高的CO2饱和压力的结晶度略有增加,如表3所示。CO2饱和时间对于发泡密度的影响发泡步骤中,CO2饱和时间可影响发泡密度。因为批次发泡通常需较长气体饱和时间,是一种耗费时间的步骤,故缩短饱和时间对于工业规模的批次发泡制程是非常关键的。研究不同的饱和时间对于经PIF步骤的PP的发泡密度的影响,并将结果显示在图6中,其中发泡条件为155℃及13.8MPa。如图6所示,饱和时间长对应低发泡密度,代表着更高的发泡倍率。然而,对于给定的样品尺寸而言,当饱和时间长于10分钟时可观察到发泡密度的高原期。此结果代表当压力释放时,经PIF步骤的PP能够保持CO2并避免CO2从PP基质中逸散。对于在短的批次发泡周期时间内达成低密度发泡体是有利的。PIFPP发泡体的形态透过SEM观察PIFPP发泡体的形态,结果如图所示。图7A至7C是利用SEM观察在11.7MPa压力2小时条件下所形成的PIFPP发泡体(如图7A所示),当CO2饱和压力低于13.8MPa的关键值时,仅有少数的小泡体存在于发泡体中且发泡密度高。当CO2饱和压力达到13.8MPa的关键值2小时,可获得泡体密度为2.75×106的非常低密度发泡体,如图7B所示。而当在13.8MPa压力下2小时可观察到发泡体中具有均匀分布的泡体,且那些泡体的平均尺寸为261.5μm。如图7C所示,从在13.8MPa压力10分钟发泡的PIFPP发泡体的SEM结果看来,较短的饱和时间可达到类似于具有稍高泡体密度(5.2×106)以及稍小泡体尺寸(255μm)的PP发泡体。PP发泡体的压缩行为发泡体的压缩强度是实际应用中要考虑的一个重要因素。在13.8MPa、155℃进行发泡步骤后,测试PP发泡体的压缩行为,结果如图8所示。以饱和时间2小时及10分钟下的PIFPP发泡体作为比较。相较于纯PP发泡体及WB140HMSPP,PIFPP发泡体具有较高的压缩强度。在不同饱和时间形成的PIF发泡体则具有相似的压缩强度。PIFPP发泡体的热稳定性热稳定性也是决定发泡体应用性的最重要因素之一。图9为所获得的TMA结果。为了进行比较,将用于咖啡杯的高熔体强度PP(HMSPP)及用于热绝缘的PS发泡体一并进行测试。显然在相似的发泡密度下,PIFPP发泡体具有较高的热稳定性且在温度达到100℃以上时发泡体不会崩溃。代表经PIF步骤的PP发泡体具有较高的工作温度以及较佳的压缩强度,可满足多种不同的工作环境并具有取代传统EPP及PS发泡体的潜力,特别是主要需考虑化学毒性的那些用来作为食物容器的应用。从实施例的结果看来,经PIF的PP可在34.5MPa的低压下以及10秒的低持压时间下进行。XRD分析中,观察到的球晶的变形,代表即使在如此温和的PIF条件下,PP中仍形成有定向的「砖和泥」结构。此外,从本实施例的结果,为了在155℃发泡,13.8MPa的临界CO2饱和压力对于达到低密度PP发泡体是必须的。此外,对于2-mm厚的样品而言,亦发现用于PIFPP发泡的CO2饱和时间可大幅度地从2小时缩短为10分钟。相较于传统的PP发泡体,PIFPP发泡体表现出更高的压缩强度及较佳的热稳定性。因此,PP发泡体可透过本发明有希望且可行的方法被制造。实施例2-制备包括PP及多壁碳纳米管(MWCNT)复合的聚合物发泡体鉴定所有用于本实施例的定性方法与实施例1所述的相似,差异点列示于下。SEM:实施例1与本实施例之间的区别在于本实施例是将样品浸泡在液态氮中10分钟并断裂。DSC:实施例1与本实施例之间的区别在于本实施例是将样品切割为10-15mg的薄片来进行DSC定性。XRD:实施例1与本实施例之间的区别在于本实施例是从5°至70°以0.4°递增扫描该样品。偏光显微镜:利用Leitz1720CryostatMicrotome制备PP样品薄片。将PP样品降温至-15℃,接着切割为25至50μm的薄片。该25μm的薄片利用偏光显微镜观察PIF之前以及之后PP样品的晶体结构。而50μm的薄片则利用光学显微镜观察PP样品,确定其“砖和泥”结构。机械性质测试:根据ASTM标准将所有的样品以适当尺寸进行机械性质测试。利用INSTRON5569先进材料试验系统,在室温下测试样品的弯曲、拉力、以及压缩特性。样品的艾佐德冲击强度(Izodimpactstrength)系利用TMI悬臂梁式冲击试验仪(TMIIzodimpacttester)在室温下测定。样品的电阻则是在室温下以Keithley6514静电计(Keithley6514electrometer)测得。实验熔体流动指数为2.0g/10min的线性PPH349-02系来自美国的陶氏化学公司。在氮气中、常压下、10℃/min扫描速率的条件下,其结晶度及熔融温度为37.62±0.02%及164±0.85℃。PP颗粒的原始尺寸介于2-2.5mm间,并被研磨成直径不大于0.3mm的更小颗粒。于本发明的其他实施例中,该研磨颗粒的直径为0.01mm至0.3mm。直径为10-15nm且管长介于0.1-10μm的GraphistrengthC100多壁碳纳米管购自GraphiSTRENGTH先进材料公司(GraphiSTRENGTHAdvancedMaterials)。将研磨PP颗粒(60g)、MWCNT(0.6-2.0g)以及研磨介质(150g)放置于一玻璃容器(1000ml)中,于135℃进行机械搅拌(300rpm)。30分钟后,以不同尺寸的筛将混合物分类,获得经MWCNT涂布的PP。图10A是本发明所获得的PP/MWCNT复合物的聚合物本体示意图,其中该聚合物本体包括一由PP形成的聚合物颗粒210以及一由MWCNT形成的涂布层220,且涂布层220形成于聚合物颗粒210之上。接下来,将经涂布的PP颗粒放置在具有两特氟龙脱模纸(Teflonmoldreleasepapers)及间隔(直径8cm、厚度3mm)的两块铝板之间。将上述组合放置在10MPa、预热至200℃的模压设备(press)。10分钟后,该模压设备自然地冷却至室温且获得蜂巢状结构的PP/MWCNT纳米复合物。为了进行比较,利用双螺杆挤出机混练PP/MWCNT(LeistrizModel2570,L/D=40,D=27mm)。为有足够的混合,挤出机以同向旋转双螺杆构造在相对高的旋转速度下(60rpm)运行。加热温度保持在200℃。纯PP及PP/MWCNT复合物试片则是在200℃利用模压成型制得(Carver3853)。蜂巢状PP/MWCNT纳米复合物被加工成所欲的几何形状,譬如3mmx12mmx25mm的试片。随后,将试片插入模穴中,譬如12mmx12mmx100mm的模穴,然后进行PIF步骤。加热模具到特定温度,譬如110℃时,再对该模具及试片施以一个非常高的静压力(staticpressure),譬如400MPa。借由施加压力以将试片导入而流动。除了经PIF步骤处理的试片外,利用Branson921aes(BransonUltrasonicsCorp.)的超声波振动仪器进行超声波辅助PIF(UAPIF)步骤。除了对试片施加超声波振动探针外,其余步骤与PIF相同。在施加压力(譬如20.7MPa)、超声波的振动频率(譬如20khz)、以及超声振动的时间(譬如4秒)的情况下,使得试片变形或流动。所有经PIF及UAPIF步骤的试片均自然冷却。在PIF或UAPIF步骤后,获得PP/MWCNT片材,且其示意图如图10B所示。图10A及10B中,包括有由PP形成的聚合物颗粒210以及形成在聚合物颗粒210上之由MWCNT形成之涂布层220之该聚合物本体,系在经过前述的PIF或UAPIF步骤后,经模塑而形成聚合物片材;其中该聚合物片材包括多个如图10A所绘示的聚合物本体彼此相互连结。接下来,在进行PIF或UAPIF前或后,利用批次发泡步骤由PP及PP/MWCNT复合物制造出发泡体。该PP纳米复合物放置在预热至发泡温度(130-160℃)的钢制槽体中。待温度达平衡后,将腔室密封并在13.8MPa压力下,利用注射器帮浦将二氧化碳气体注入该高压槽体中。将温度和压力保持2小时使CO2扩散,接着在2-3秒内释放压力以诱导泡体成核及发泡。发泡步骤后即可获得PP/MWCNT发泡体,其示意图如图10C,其中多个泡体230系形成在聚合物颗粒210中。为了比较,透过模压成型制备的纯PP及复合PP/MWCNT纳米复合物亦透过相同的方式进行。以下将对本实施例的结果进行详细说明。“砖和泥”结构和传统的球晶结构不同,强迫PP试片在PIF下以固体状态流动使得晶体沿着PIF的方向取向。从PP试片在400MPa及110℃经PIF处理前及处理后的偏振光显微镜(POM)结果,未经PIF的PP试片表现出传统的球晶结构,尺寸约在30-50μm,如图11A所示;而经PIF的PP试片,其晶体沿着PIF的方向取向,如图11B所示。此外,经PIF的晶体结构改变以示差扫描量热计(DSC)及X射线衍射(XRD)定性。在此,定性PP、PIFPP(PIF条件:110℃,400MPa)、UAPIFPP(PIF条件:110℃,20.7MPa)以及PIFPP发泡体(发泡条件:13.8MPa,155℃)。DSC结果如图12A,显示经PIF后,熔融峰及结晶度均微幅增加,分别从165.1增加为168.74℃、30.2%增加至32%。图12B的XRD光谱(其中非PIFPP-0°及非PIFPP-90°的曲线重迭)显示非PIFPP的曲线在0°及90°的扫描角度几乎相同,代表在非PIFPP没有较佳的晶格方向。PP试片经PIF步骤(PIF条件:110℃,400MPa)后差异明显,代表在此步骤时材料中产生了定向晶体结构。此乃整个球晶变形并涉及片状结晶(lamellae)的直接证据。一般来说,对称的球晶形状变成沿着流动的方向伸长的形状。当PP颗粒经MWCNT涂布时,该MWCNT涂布层亦可在高压下沿着PIF方向流动。该MWCNT涂布层沿着PIF方向定向并形成清楚的共连续“砖和泥”结构。在此,该巨观的“砖和泥”结构是由MWCNT涂布层(“砖”)以及在该涂布层中的PP树脂(“泥”)所组成;以及定向的PP晶体为“砖”而不定向的区域为“泥”的巨观的“砖和泥”结构。相对传统具有相同组成成分的复合物而言,这种独特的“砖和泥”结构可提供优异的机械及物理性能,甚至大幅改善低熔融强度PP发泡性不佳的缺点。根据上述结果,高压的应用可在固体状态下于半结晶聚合物及嵌段共聚物系统中诱导类熔体行为以形成“砖和泥”结构。当那些材料被放置在压力下时,硬质塑料开始熔解而变软,创造出如同包括冰和水的浆液一般流动的混合物,使材料得以模塑为特定形状。当压力释放时,塑料再度硬化。半结晶聚合物(如PP)中的晶体结构也在此PIF步骤中变形。球晶可经历非常大的变形,且其片状结晶可破裂成小片并彼此分散于赤道区域中(equatorialregion);然而在极区(polarregion),片状结晶系定向以平行于大规模变形的附载方向。机械特性经CNTs涂布的PP强度非常低(数据未示),但PIF能够大幅地增加其机械特性。图13A比较经PIF及未经PIF的PP及PP/MWCNT复合物的拉力强度。与纯PP、经模压成型制得的复合PP/MWCNT试片相比,经PIF可使拉力强度分别增加超过250%(>250%)及超过50%(>50%)。经PIF制得之PP的拉力强度亦高于双轴延伸PP,其强度高于注射模塑PP。图13B比较相同试片之间的弯曲强度。同样地,具有“砖和泥”结构的PP/MWCNT纳米复合物有最出色的性能表现。相较于纯PP及经由注射模塑制得的复合PP/MWCNT试片,经PIF制得的试片的弯曲强度分别增加超过60%(>60%)及超过25%(>25%)。在艾佐德冲击强度(Izodimpactstrength)可观察到更佳明显的差异,如图13C所示。具有“砖和泥”结构的PP/MWCNT纳米复合物强度是纯PP试片的500%,并且优于复合PP/MWCNT,超过40%(>40%)。UAPIF让机械性质有类似的提升,但其所需的压力比PIF来得低。在不同的PP及PP/MWCNT试片中,达到200%变形所需要的超声波条件列示在下表4中,其中,在400MPa、110℃时经PIF步骤处理的PP试片变形设定为200%。表4上表4结果显示超声波压力越高,所需的时间就越短。所需的超声波时间只要几秒钟,相较于传统的PIF步骤,导入超声波振动能有效地减少所需压力达至少一个数量级(由400MPa至10-30MPa)。对于PP/MWCNT纳米复合材料而言,在超声波压力下需要较长的步骤时间(更多能量)来达到同样程度的变形。此外,相较于PP/MWCNT复合,经PP/MWCNT涂布的纳米复合物需要稍长的时间(更多能量)。图14比较经UAPIF后,纯PP、PP/MWCNT3wt%MWCNT复合以及PP/MWCNT3wt%MWCNT涂布样品的弯曲应力。如同PIF,UAPIF步骤可显着地改善PP试片的弯曲应力。经UAPIF后,具有“砖和泥”结构的PP/MWCNT纳米复合物的强度优于纯PP样品超过70%(>70%),并且优于3wt%复合PP/MWCNT超过25%(>25%)。具有相对低压力的UAPIF步骤对于大型的PP/MWCNT复合物制造并提升其特性是有利的。此外,在UAPIF下PP试片的温度改变也在此一并进行测试,结果如图12C所示。虽然超声波会加热样品,但当模具温度设定为110℃,最高温度低于PP熔点,换言之,超声波不会熔化砖和泥结构。此外,图12A显示的结果进一步证实经UAPIF处理的PP试片的熔化温度和结晶度仍然类似于经PIF的处理的PP。然而,倘若模具温度、模塑压力、或超声波强度过高时,在UAPIF过程可能会发生熔化,将导致机械特性降低,如图19A及19B所示。因此,选择最佳的UAPIF条件是重要的。为什么超声波可以明显降低PIF所需的压力的确切机制目前还不清楚。然而,我们提出以下解释:为了要在固体状态PIF下形成「砖和泥」结构,需要足够的压力以从橡胶状的非晶相中转移到刚性晶体区域,并导致后者变形或定向。因为晶体结构区域间可彼此相对滑动,而不是被变形或定向,故PIF下需要非常高的静压力以产生足够的压力。另一方面,超声波辅助的PIF,经橡胶状非晶相的压力传播可更有效率而较少有晶体结构区域滑动,因此所需的压力低于PIF压力。前述结果显示超声波可减少PIF步骤中所需的压力。然而,即使未在PIF步骤中导入超声波,PP或PP/MWCNT复合物的晶体区域仍然可以变形并且转变为“砖和泥”结构。如PP,具有3wt%纳米颗粒的复合PP/MWCNT试片是不导电的。而经MWCNT涂布的PP试片因为形成“砖和泥”共连续结构而有导电性。具有3wt%MWCNT涂层的PP电阻约为0.55kΩ。UAPIF稍微增加电阻至0.9kΩ。复合PP/MWCNT复合物中,需要足够的MWCNTs以达到导电的渗透阈值(percolationthreshold)。另一方面,在PIF及UAPIF制得的PP/MWCNT复合物中的蜂巢状共连续结构可使具有3wt%MWCNT的试片具有导电性,这是因为在整个试片中,MWCNTs彼此互连而形成导电通道的缘故。PP/MWCNT“砖和泥”结构复合物的发泡性能经PIF及UAPIF形成的试片的主要缺点在于高残余应力(residualstress)。如图18A及18B所示,热处理时,延伸的PP在流动方向上收缩并在垂直方向上展开,导致严重的试片翘曲。然而,此试片翘曲对于聚合物发泡体的发泡能力不会造成很大的影响。利用超临界二氧化碳(scCO2)的固态批次发泡步骤,以从“砖和泥”结构制备PP复合物发泡体。在许多的发泡应用中,CO2作为物理发泡剂,因为其许多有利特性(即,不易燃、无毒、价格低廉、并且在聚合物有相对高的溶解度)而可取代消耗臭氧层氟发泡剂的使用。固态发泡时,残余的压力可完全的被释放。此外,在150℃、13.8MPaCO2压力时观察到特殊的逐层(layerbylayer)的双峰泡体结构,其具有微结构泡体尺寸(数据未示)。相较之下,纯PP及具3wt%复合MWCNT的PP中仅有少数的泡体形成。经PIF处理的试片具有较低的密度,所形成的发泡体密度为0.65-0.86g/cm3,如下表5所示。表5由于PP的熔融强度低,难以实现具有均匀的泡体结构以及低假比重的发泡体;然而经PIF所产生的“砖和泥”结构可克服上述限制。如图15所示,在155℃、13.8MPaCO2压力时,相较于PIF-PP试片(22.5),由纯PP所获得的发泡体具有更低的发泡倍率(6)。此外,从SEM(数据未示),在155℃、13.9MPaCO2压力时自由发泡而制得的低密度(0.039g/mL)PIF-PP/MWCNT3wt%涂布纳米复合物发泡体具有逐层(layerbylayer)的双峰发泡结构。在155℃发泡造成发泡后晶体的改变。结晶度从32%降低至18.3%,并且有β-cylindrites形成。有趣的是,PIF中固态发泡制得的PP能够改善其热稳定性,如图16所示。在固态批次发泡步骤中,结晶结构会同时影响泡体成核及生长。在泡体成核步骤中,介于片状结晶和非晶区域的接口是一高能区域,其中成核一个稳定的泡体所需的吉布斯自由能(Gibbsfreeenergy)小于均相成核的吉布斯自由能,因此倾向在此界面优先成核稳定的泡体。当此泡体生长时,因为在结晶区域分子键流动性少,所形成的泡体被相邻的片状结晶限制。当发泡温度高于155℃时,因为“砖和泥”结构的熔化,导致CO2饱和PP的熔化温度、纯PP及PIF-PP之间的发泡倍率差异如图15。固体纯PP及衍生自不同PP/MWCNT复合物的PP发泡体的压缩应力与应变量如图17A所示。以10%压缩应变量为例,纯PP(密度为0.76g/cm3)的发泡体的压缩强度仅约10MPa,系固体PP(密度为0.9g/cm3)的约25%。然而,具有0.66g/cm3的低密度的PIF-PP的压缩强度可达25MPa,同时经PIF步骤且经PP/MWCNT涂布纳米复合物发泡体(具有相似的密度)则接近固体PP。在155℃的高发泡温度及13.9MPaCO2压力时,经PIF或未经PIF的PP/MWCNT复合物的发泡密度差异如下表6所示。纯PP发泡体的密度>0.15g/cm3,而具MWCNT纳米颗粒或未具MWCNT纳米颗粒之经由PIF制得的试片,其密度约为0.037-0.04g/cm3。未经PIF的3wt%复合及涂布MWCNT的发泡试片与未经PIF的纯PP发泡体有相似的密度(0.18-0.24g/cm3),代表“砖和泥”结构,而非纳米颗粒,是控制发泡倍率的主要因子。“砖和泥”结构可以防止CO2扩散出试片,并协助支持形成的泡体不坍塌。表6图17B比较经PIF及未经PIF的低密度纯PP及PP/MWCNT纳米复合物发泡体的压缩应力及压缩应变量。在155℃时,发泡体的密度为:纯PP:0.15g/cm3;PIF-PP:0.037g/cm3;PIF-PP/MWCNT3wt%涂层:0.04g/cm3;以及PIF-PP/MWCNT3wt%复合:0.04g/cm3。虽然纯PP的密度高于其余具有“砖和泥”结构的发泡体试片2.5到2.7倍,但纯PP发泡体的压缩应力是最低的。显然,导致较小及封闭的泡体结构的“砖和泥”结构,对于上述明显的性能提升贡献良多。在PIF下,相较于复合试片,包括涂布MWCNT的发泡体提供更好的压缩特性,可能归因于其具有逐层的双峰泡体结构的缘故。总言之,具有“砖和泥”结构的PP/MWCNT纳米复合物系借由在小PP颗粒上涂布MWCNT,然后进行PIF或UAPIF步骤而制得。UAPIF步骤可降低所需的PIF压力,且型态与特性可与进行PIF步骤相媲美,但超声波在本发明中并不是必需的。此外,在PIF或UAPIF步骤后获得的“砖和泥”结构亦可改善低熔融强度PP的发泡性,并在没有任何物理或化学添加物的情况下制造出低密度发泡体。具有涂布的MWCNT时,该“砖和泥”结构使得发泡体具有逐层的双峰泡体结构。相较于具有相同组成成分的传统发泡体,此发泡体可提供优异的机械性质以及更好的热稳定性,并且是可导电的。总之,由实施例1和实施例2的结果可以发现,是否有超声波协助PIF过程是制备聚合物发泡体的关键步骤,因为PIF聚合物片材具有保持CO2并避免CO2从PIF聚合物片材逸出的能力;且因此,可在较短的发泡时间内获得具有低密度的聚合物发泡体。以上所述的具体实施例,对本发明的目的、技术方案和有益效果进行了进一步详细说明,应理解的是,以上所述仅为本发明的具体实施例而已,并不用于限制本发明,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1