三维树形共轭化合物-碳纳米管复合物、制备方法及应用与流程
本发明具体涉及一种碳纳米管的分散或选择性分离方法,碳纳米管复合物及相应的制备方法与应用,例如在制备复合墨水、薄膜及半导体器件(如场效应晶体管)中的应用,属于光电半导体材料与器件领域。
背景技术:
碳纳米管是一类具有独特结构的一维纳米材料,具有机械性能好,电荷迁移率高,电学性能优异,与柔性基底兼容性好,化学稳定性和热稳定性好等优势,在半导体电子器件中具有非常广泛的应用。
碳纳米管通常采用化学气相沉积(CVD)的方法制备,在目前的制备条件下所得到的单壁碳纳米管均是金属型单壁碳纳米管(m-SWCNTs)和半导体型单壁碳纳米管(s-SWCNTs)的混合物,极大的限制了SWCNTs在半导体电子器件中的广泛应用。将金属型单壁碳纳米管和半导体型单壁碳纳米管进行有效地分离与纯化,获得具有单一导电性能的单壁碳纳米管是制备性能优越的碳纳米管半导体器件的关键。
根据金属型单壁碳纳米管和半导体型单壁碳纳米管在管径大小、手性、物理化学性质上存在的微小差异,相关科研工作者已经开发了多种用于分离纯化碳纳米管的有效方法,如密度梯度高速离心法、化学分离法、凝胶色谱法、电泳法、DNA包覆法和聚合物包覆法。
聚合物包覆方法由于其分离方法简便,分离快速,可溶液法批量加工,分离选择性高,分离性能可精细调节,对碳纳米管自身固有性能影响较小等优势,使其已发展成为碳纳米管研究领域中的一个重要研究方向。
但是,现有技术所采用的用于分离半导体型单壁碳纳米管的聚合物主要为线型结构。迄今为止,采用具有新型三维全共轭空间立体结构的化合物分子分离半导体型单壁碳纳米管的报道尚未有任何报道。筛选和开发具有新的空间结构的共轭化合物对高效地分离半导体型单壁碳纳米管、有效地形成共轭化合物碳纳米管复合物、提升器件性能有重要的意义。
技术实现要素:
本发明的主要目的在于提供一种基于三维树形共轭化合物的碳纳米管分散或选择性分离方法与应用,以克服现有技术中的不足。
为实现前述发明目的,本发明采用的技术方案包括:
在一些实施例中提供了三维树形共轭化合物于选择性分离半导体型碳纳米管中的应用,或者,三维树形共轭化合物作为碳纳米管分散剂的用途。
其中,所述三维树形共轭化合物具有式(I)或(II)所示的结构:
其中,B为共轭链接单元,其选自由五元或六元芳香单元形成的具有支化结构的单元,
FG为端基功能修饰单元,其结构中含有如式(III)所示的吡咯并吡咯二酮单元:
m和n为自然整数,m为支化共轭单元B的支化度,其值为2或者3,n为三维树形共轭化合物的代数,代表着分子中重复单元迭代次数,其值为1,2,3或者4。
优选的,所述碳纳米管选自半导体型碳纳米管,特别是半导体型单壁碳纳米管,尤其是大管径半导体型单壁碳纳米管。
在一些实施例中提供了一种碳纳米管分散剂,其包括所述的三维树形共轭化合物。
在一些实施例中提供了一种碳纳米管分散方法,其包括:取所述的三维树形共轭化合物及碳纳米管于溶剂中均匀混合,形成稳定均匀的碳纳米管分散液。
在一些实施例中提供了一种选择性分离半导体型碳纳米管的方法,其包括:
将碳纳米管粉体加入三维树形共轭化合物的溶液中并充分混合,使其中的半导体型碳纳米管均匀分散于溶液中,
以及,移除未能分散于溶液中的碳纳米管。
在一些实施例中提供了一种三维树形共轭化合物-碳纳米管复合物,其包含碳纳米管以及至少附着在碳纳米管部分表面上的所述三维树形共轭化合物。
在一些实施例中提供了包含有所述的三维树形共轭化合物-碳纳米管复合物的碳纳米管分散体。
在一些实施例中提供了一种复合墨水,其包括:
所述的三维树形共轭化合物-碳纳米管复合物,
以及,至少一种溶剂,用以与所述复合墨水的其余组分配合形成稳定的均匀液相分散体系。
优选的,所述复合墨水还包含有机半导体。
在一些实施例中提供了一种复合墨水的制备方法,其包括:
将所述的三维树形共轭化合物和碳纳米管于溶剂中均匀混合,形成均匀碳纳米管分散液,
以及,对所述均匀碳纳米管分散液进行高速离心处理,所获清液即为所述复合墨水。
在一些实施例中提供了一种薄膜,其包含所述的三维树形共轭化合物-碳纳米管复合物。
在一些实施例中提供了一种薄膜的制备方法,其包括:采用印刷和/或涂布方式将所述的分散体或所述的复合墨水施加到基底上,形成所述薄膜。
其中,所述涂布和/或印刷方式包括旋转涂膜、刮刀涂布、狭缝涂布、喷墨印刷、丝网印刷、凹版印刷、柔版印刷、柔版转印方式中的任意一种或两种以上的组合,但不限于此。
进一步的,所述的制备方法还包括薄膜后处理步骤,所述薄膜后处理步骤包括清洗和退火操作。
在一些实施例中提供了一种装置,其包含所述的三维树形共轭化合物,所述的三维树形共轭化合物-碳纳米管复合物或者所述的薄膜。
优选的,所述装置选自半导体装置。
优选的,所述装置包括场效应晶体管,所述场效应晶体管的有源层包含所述的三维树形共轭化合物-碳纳米管复合物或者所述的薄膜。
进一步的,所述场效应晶体管主要由源电极、漏电极、栅电极、介电层和有源层构成。
与现有技术相比,本发明至少具有如下优点:
1)提供了具有三维共轭空间立体结构的新型树形化合物分子,其分子结构单一确定,可克服聚合物批次之间的差异性,可重复性好,且可应用为分散剂而良好的选择性分散半导体型碳纳米管,特别是半导体型单壁碳纳米管;
2)通过调整溶剂种类、碳纳米管浓度和三维树形共轭化合物浓度等,还可有效实现三维共轭空间立体结构的新型树形化合物选择性包覆分离大管径半导体碳纳米管的目的;
3)提供的三维树形共轭化合物-碳纳米管复合物加工工艺简单,所需化合物量相对较少,分离过程所用时间短、离心速度低,成本低廉;
4)利用本发明方法所获大管径半导体碳纳米管可构建出性能优越的印刷薄膜晶体管半导体器件;
5)本发明的三维树形共轭化合物-碳纳米管复合物具有良好的半导体性能,可被应用于大规模商业化生产高性能可印刷半导体碳纳米管墨水和高性能的印刷电子器件。
附图说明
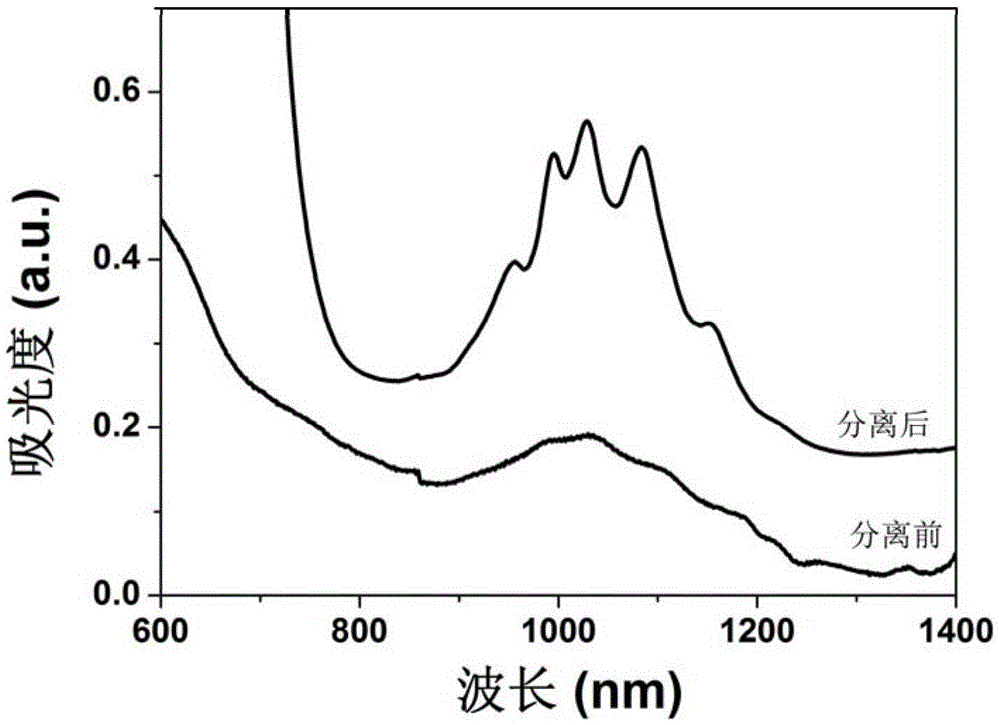
图1和图2分别是本发明实施例4、实施例5(P2-CNT,三维树形共轭化合物(6T-DPP、9T-DPP),甲苯)所获半导体碳纳米管(以下简称“S-CNT”)的吸收光谱图;
图3和图4分别是本发明实施例6、实施例7(P2-CNT,三维树形共轭化合物(6T-DPP、9T-DPP),甲苯)所获S-CNT在不同测试条件下的拉曼光谱图;
图5和图6分别是本发明实施例1(P2-CNT,三维树形共轭化合物(9T-DPP),甲苯)构建的薄膜晶体管的性能曲线图。
具体实施方式
鉴于现有技术中的不足,本案发明人经长期研究和大量实践,得以提出本发明的技术方案。如下将对该技术方案、其实施过程及原理等作进一步的解释说明。
本发明的一个方面提供了一种三维树形共轭化合物,其具有式(I)或(II)所示的结构:
其中,B为共轭链接单元,其由五元或六元芳香单元形成的具有支化结构的单元,FG为端基功能修饰单元,其结构中含有如式(III)所示的吡咯并吡咯二酮单元:
m和n为自然整数,m为支化共轭单元B的支化度,其值为2或者3,n为三维树形共轭化合物的代数,代表着分子中重复单元迭代次数,其值为1,2,3或者4。
其中,R1选自氢原子或取代或未取代的C1~C20的烷基或C1~C20的杂烷基。
其中,吡咯并吡咯二酮(DPP)结构单元FG具有原料廉价易得,强受电子能力,高摩尔消光系数,高电荷迁移率以及易于通过定向化学修饰调节物理化学性能等优点。
优选地,上述的含有吡咯并吡咯二酮单元的端基功能修饰单元FG选自如下结构中的一种,但不局限于下述结构:
其中,R1选自氢原子或取代或未取代的C1~C20的烷基或C1~C20的杂烷基。
在一些实施例中,上述的含有吡咯并吡咯二酮单元的端基功能修饰单元FG中还引入端位的封端单元,钝化一端的反应活性。具体的封端单元可以如下,但不限于下述结构:
其中,R2选自取代或未取代的C1~C20的烷基或C1~C20的杂烷基。
以上述(III-1)单元为例,在一些实施例中,结合了封端单元的不对称FG功能修饰单元的结构可以选自但不限于下述结构:
其中R1,R2为C1~C20的直链或支链烷基或C1~C20的杂烷基。
在一些实施例中,为了构建具有分子结构单一确定性的三维树形共轭化合物,本发明所提供的支化共轭链接单元B具有(IV-1),(IV-2),(IV-3)或(IV-4)所述的结构中的一种:
其中,Y选自苯环或噻吩单元,D选自苯环、噻吩环、由2~5个五元或六元芳香单元形成稠环单元或的由2~4个五元或六元芳香单元形成共轭短链单元。
其中,Y可以选自苯环,可以通过不同位置的取代形成不同的支化结构,包括:
通过重复上述支化结构单元IV-1-1可以构建出如式(IV-1-1-G1)-(IV-1-1-G3)所示的以下支化共轭构建单元,如:
其中外围苯环单元的活性位点与具有吡咯并吡咯二酮单元的功能修饰单元(FG)相连接,而处于内核的苯环上的活性位点为氢原子(如式(I)所示),或者他们二者之间相连形成具有式(II)所示结构的化合物分子。
例如,基于上述IV-1-1-G3单元的具有式(I)和式(II)的分子结构如下:
也可以通过重复上述支化结构单元(式(IV-2-1))构建出以下支化共轭构建单元,如:
其中外围苯环单元的活性位点与具有吡咯并吡咯二酮单元的功能修饰单元相连接,而处于内核的苯环上的活性位点为氢原子(如式(I)所示),或者他们二者之间相连形成具有式(II)所示结构的化合物分子。
例如,基于上述IV-2-1-G3单元的具有式(I)和式(II)的分子结构如下:
Y也可以选自噻吩,通过α,β-双取代的方式形成具有式(IV-1-2)所示的支化结构:
利用重复上述支化结构单元IV-1-2可以构建出如式(IV-1-2-G1)~式(IV-1-2-G3)所示的支化共轭构建单元,如:
其中外围噻吩单元的α-位与具有吡咯并吡咯二酮单元的功能修饰单元相连接,而内核的噻吩α-位为氢原子(如式(I)所示),或者他们二者之间相连形成具有式(II)所示结构的化合物分子。
例如,基于上述IV-1-2-G4单元的具有式(I)和式(II)的分子结构如下:
为了降低分子内不同基团单元之间的空间位阻作用,在重复单元Y之间还可以引入共轭单元D来提高重复单元Y之间的距离。
所述的共轭单元D单元可以选自苯环、噻吩环,也可以选自由2~5个五元或六元芳香单元形成稠环单元,包括但不限于以下结构:
其中,X1=S或O,X2=O,S,C,N,Si或Se及其附属的C1~C20的烷基或C1~C20的杂烷基,R3选自取代或未取代的C1~C20的烷基或C1~C20的杂烷基。
或者D选自于由2~4个五或六元芳香单元形成共轭短链单元,包括但不限于以下结构:
其中,R4,R5独立地选自氢原子,取代或未取代的C1~C20的烷基或C1~C20的杂烷基。
优选的Y与D单元为噻吩单元。相应的支化共轭链接单元B具有如下结构:
式中,外端噻吩α-位(p-a及p-b)与端基功能修饰单元FG相连,或者通过与内端噻吩α-位(c-a)相连形成具有式(V-G2),(V-G3),或(V-G4)的高代数支化共轭链接单元:
其中外围噻吩单元的α-位与具有吡咯并吡咯二酮单元的端基修饰单元相连接,而内核的噻吩α-位为氢原子(结构式I),或者他们二者之间相连形成具有结构式II的分子。
以端位FG为具有III-1-EG1的结构单元,支化结构B为V为,迭代数n=2和n=3例,本发明所构建的具有支化结构的共轭化合物(即所述的三维树形共轭化合物)的分子结构包括:
本发明的一个方面还提供了合成所述三维树形共轭化合物的方法,其包括:采用金属催化缩合反应制备所述三维树形共轭化合物。其中,所述的金属催化缩合反应包括Suzuki缩合、stille缩合等,但不限于此。
在一些实施例中,具有前述式(I)所示的三维树形共轭化合物可通过反应体系(1)表示的反应步骤的方法制备合成。
反应体系(1):
上式中,m为支化共轭单元B的支化度,其值为2或者3;n,n1,n2为三维树形共轭化合物的代数,代表分子中重复单元迭代次数,其值为大于或等于0的整数。n1≧1,n2≧0,n=n1+n2。当n2=0时,式(b)的结构如式(b,n2=0)所示。
V2-FG
(b,n2=0)
在一些实施例中,具有式(II)所示的三维树形共轭化合物可通过包括以反应体系(2)表示的反应步骤的方法制备合成。
反应体系(2):
上式中,m为支化共轭单元B的支化度,其值为2或者3;n,n1,n2为三维树形共轭化合物的代数,代表分子中重复单元迭代次数,其值为大于或等于0的整数,n1≧1,n2≧0,n=n1+n2。当n2=0时,式(b)的结构如式(b,n2=0)所示。
V2-FG
(b,n2=0)
在一些实施例中,具有式(II)所示的三维树形共轭化合物也可通过包括以反应体系(3)表示的反应步骤的方法制备合成。
反应体系(3):
上式中,m为支化共轭单元B的支化度,其值为2或者3;n为三维树形共轭化合物的代数,代表分子中重复单元迭代次数,其值为1,2,3或者4。
如下将对上述的各反应体系及相应反应步骤进行详细的说明。
在上述的各反应体系及反应步骤中,带有反应活性基团V1的树枝状共轭化合物分子与带有反应活性基团V2的树枝状共轭化合物分子的反应是通过Suzuki交叉偶联反应或stille 交叉偶联反应实现的。
例如,前述反应活性基团V1及V2可以选自于以下三组活性反应基团,但不局限于此。
活性反应基团组1:
-F,-Br,-I。
活性反应基团组2:
R4选自含碳数为1-4的直链或支链烷基。
活性反应基团组3:
R5选自含碳数为1-4的直链或支链烷基。
在一些实施例中,当利用Suzuki缩合方法制备所述三维树形共轭化合物时,V1及V2的组合(V1,V2)包括(选自活性反应基团组1,选自活性反应基团组3)或(选自活性反应基团组3,选自活性反应基团组1)。
在一些实施例中,当利用stille缩合方法制备化合物时,V1及V2的组合(V1,V2)包括(选自活性反应基团组1,选自活性反应基团组2)或(选自活性反应基团组2,选自活性反应基团组1)。
更为优选的,V1选自-Br,-I;V2选自式(g)所示的活性反应基团。
所述的金属催化剂指的是可溶性的金属钯配合物,例如可选自但不限于:Pd(PPh3)4,Pd(OAc)2,Pd2(dba)3,Pd2(dba)3·CHCl3,Pd(dppf)Cl2,更优选的催化剂可以为Pd(PPh3)4,Pd2(dba)3,Pd2(dba)3·CHCl3。
在一些实施例中,当选用不含配体的Pd催化剂Pd2(dba)3或Pd2(dba)3·CHCl3时,催化体系中还需要添加磷配体,以提高反应效率,所述的磷配体包括但不限于:PPh3或HP(tBu)3BF4,较佳的,例如磷配体与催化剂Pd原子的摩尔比例可以为2:1。
在一些实施例中,当选用Suzuki催化缩合制备方法时,反应体系中需要加入无机金属碱溶液。所述的金属碱包括碳酸盐,如:Na2CO3,NaHCO3,K2CO3,KHCO3;磷酸盐,如:K3PO4,K2HPO4;羧酸盐,如:KOAc,NaOAc,但均不限于此。
其中,适用的溶剂可包括但不限于甲苯、乙二醇二甲醚、四氢呋喃、1,4-二氧六环、DMF,DMSO、二氯甲烷、三氯甲烷。优选的溶剂可以为四氢呋喃或三氯甲烷,但不局限于此。
本发明的另一个方面还提供了一种三维树形共轭化合物-碳纳米管复合物,其主要由碳纳米管和附着于碳纳米管表面的所述三维树形共轭化合物组成。
本发明的三维树形共轭化合物为包含有吡咯并吡咯二酮单元的具有三维树形立体结构的非线性共轭化合物,其中,具有一定空间扭曲构象结构的B基团的引入是获得非线性树枝状共轭化合物的关键,这些三维树形共轭化合物在空间具有扭曲的树枝状结构,该树枝状扭曲结构的共轭化合物能够有效地包覆在碳纳米管上,使得聚合物与碳纳米管之间的相互作用力得到增强,复合物的稳定性也得到大幅提高。
本发明的另一个方面还提供了所述三维树形共轭化合物作为碳纳米管分散剂的用途,特别是作为半导体型碳纳米管选择性分散剂的用途,其中所述碳纳米管选自半导体型碳纳米管,特别是半导体型单壁碳纳米管,尤其是大管径半导体型单壁碳纳米管。
本发明的另一个方面还提供了一种碳纳米管分散方法,其包括:取所述的三维树形共轭化合物及碳纳米管于溶剂中均匀混合,形成稳定均匀的碳纳米管分散液。
本发明的另一个方面还提供了一种选择性分离半导体型碳纳米管的方法,其包括:
将碳纳米管粉体加入三维树形共轭化合物的溶液中并充分混合,使其中的半导体型碳纳米管均匀分散于溶液中,
以及,移除未能分散于溶液中的碳纳米管。
其中,所述碳纳米管粉体可以是商业途径获取的碳纳米管粉体或利用本领域已知的各种方式制备的碳纳米管粉体,其通常可以包括半导体型碳纳米管和金属型碳纳米管等。
其中,所述溶液中采用的溶剂可以选自任何能够溶解但不破坏所述三维树形共轭化合物分子结构的溶剂,特别是有机溶剂,例如三氯甲烷、四氢呋喃、甲苯、邻二甲苯、对二甲苯、间二甲苯、丙酮、三甲苯,氯苯和二氯苯等。
在一些实施例中,所述选择性分离半导体型碳纳米管的方法具体包括:
将碳纳米管粉体加入三维树形共轭化合物的溶液中并充分混合,使其中的半导体型碳 纳米管与三维树形共轭化合物结合形成复合物而均匀分散于溶液中,而使其中基本不能与三维树形共轭化合物结合的金属型碳纳米管沉降,之后,以离心或过滤等方式移除未能分散于溶液中的金属型碳纳米管等。
本发明的另一个方面还提供了包含有所述的三维树形共轭化合物-碳纳米管复合物的碳纳米管分散体。
较为具体的,例如,本发明提供了一种复合墨水,其包含以下组分:
(a)至少一种所述的三维树形共轭化合物,
(b)至少一种碳纳米管,
(c)至少一种溶剂,特别有机溶剂。
本发明所提供的复合墨水中具有结构式I或结构式II的三维树形共轭化合物能够有效地包覆在半导体型碳纳米管的管壁上形成超分子复合结构,提高半导体型碳纳米管在有机溶剂中的分散性,从而选择性的分散半导体型单壁碳纳米管,获得富集半导体型单壁碳纳米管的分散均匀的墨水。
前述复合墨水中具有结构式I或结构式II的三维树形共轭化合物所含有的直链或支链烷基单元还能够有效地改善三维树形共轭化合物在有机溶剂中的溶解性、调节三维树形共轭化合物与碳纳米管之间的相互作用、降低薄膜中的碳纳米管层的表面缺陷,提高载流子浓度以及载流子迁移率等,从而调节碳纳米管-三维树形共轭化合物薄膜对电极界面的修饰性能,进而改善优化半导体器件的电学性能。
本发明所提供的复合墨水中碳纳米管可采用商业化大管径P2单壁碳纳米管,即采用商业化电弧放电方法得到的大管径单壁碳纳米管。
本发明所提供的复合墨水中采用的有机溶剂,优选为可以溶解三维树形共轭化合物而自身不能分散碳纳米管的有机溶剂,例如可选自三氯甲烷、四氢呋喃、甲苯、邻二甲苯、对二甲苯和间二甲苯中的任意一种或两种及两种以上的组合,但不限于此。
在一些实施案例中,本发明的溶剂中还可以包含丙酮、三甲苯、氯苯、二氯苯中的一种或两种及两种以上,用于分散及稳定所配置的复合墨水,提高所配置复合墨水的加工性能。
在本发明所提供的复合墨水中,由于三维树形共轭化合物的选择性包覆作用,能够很大程度地提高半导体型单壁碳纳米管在三维树形共轭化合物以及有机溶剂中的分散性,同时不会发生明显的团聚,从而能够在保持半导体型单壁碳纳米管原有的力学和电学性能的前提下有效地稳定所配制的墨水。
在本发明所提供的复合墨水中,三维树形共轭化合物-碳纳米管复合物的重量/体积浓度 优选为0.01~20mg/mL。更低的固含量使得复合物加工制备过程中复合物沉积量不足,半导体型单壁碳纳米管不能很好地附着和固定在基材表面,更高的固含量浓度容易导致固体析出,不利于墨水的稳定分散。
在本发明所提供的复合墨水中,三维树形共轭化合物与碳纳米管的重量混合比优选为1:0.1~1:10,过高含量的聚合物将导致聚合物在器件中的残留,弱化半导体型单壁碳纳米管在所加工器件中电荷传输方面的作用,过低的聚合物比例难以有效地在基材上附着固定半导体型单壁碳纳米管,导致无法在沟道中形成致密的碳纳米管网络结构,从而影响所加工器件中电荷的高效传输,降低晶体管的电学性能。更为优选的三维树形共轭化合物与碳纳米管的重量混合比例优选为1:0.2~1:5。
本发明还提供了所述复合墨水的制备方法。其中,所述复合墨水可以采用如下两种制备方法。
例如,将所述的三维树形共轭化合物先溶解于至少一种溶剂中,然后在温度≤0℃的条件下,将所述的商业化大管径P2单壁碳纳米管均匀分散于第一步制备的三维树形共轭化合物溶液中,最后再进行短时高速离心分离,分离出上层清液即获得富集半导体型的大管径的商业化碳纳米管的墨水溶液。
例如,先将所述的三维树形共轭化合物与其他有机半导体材料进行混合,然后一同溶剂于有机溶剂中,然后在温度≤0℃的条件下超声均匀分散,最后再进行短时高速离心分离,分离出上层清液制备形成墨水。
所述的有机溶剂至少可选自于三氯甲烷、四氢呋喃、甲苯、邻二甲苯、对二甲苯或间二甲苯中的任意一种或两种及两种以上的组合,但不限于此。
显然,该复合墨水的制备工艺简单、成本低廉、操作简便,易于批量制备。
其中,溶剂的选择需要结合碳纳米管在不同溶剂中的分散性能、三维树形共轭化合物在不同溶剂中的溶解性能以及后续三维树形共轭化合物-碳纳米管复合物沉积过程对溶剂的要求进行综合选择。
其中,溶剂的极性以及溶解性对三维树形共轭化合物分离半导体型单壁碳纳米管有较大的影响。例如,三氯甲烷虽然对单壁碳纳米管有非常好的溶解性,但大部分分散的碳纳米管在溶液中是以聚集成束的状态存在,不能实现单根半导体型单壁碳纳米管的选择性分散,从而影响了其在器件加工制备方面的应用。以制备碳纳米管薄膜晶体管为例,较为优选的有机溶剂包括甲苯、邻二甲苯、对二甲苯和间二甲苯等自身不能分散碳纳米管的有机溶剂,但这些溶剂对三维树形共轭化合物有良好的溶解性,在这些溶剂中三维树形共轭化合物可以选择性的包覆半导体型单壁碳纳米管形成三维树形共轭化合物-碳纳米管复合物, 实现选择性分离半导体型单壁碳纳米管的目的,获得可用于制备加工碳纳米管薄膜晶体管的富集半导体型的大管径的商业化碳纳米管的墨水溶液。
此外,所选用溶剂的不同,在一定程度上会影响墨水的表面张力,进而影响墨水与基底的接触性能,从而影响所加工制备的碳纳米管薄膜晶体管的性能。优选的,所述有机溶剂可选自于三氯甲烷、四氢呋喃、甲苯、邻二甲苯、对二甲苯、间二甲苯、三氯甲烷和四氢呋喃中的任意一种或两种及两种以上的组合,但不限于此。
前述复合墨水的制备工艺中采用的高速离心操作,其优选的实施方式为:离心速度大于8000rpm,离心速度优选控制在10000~30000rpm;离心时间在20min以上,离心时间优选控制为30~100min。
本发明的一个方面还提供了一种薄膜,其中包含所述三维树形共轭化合物-碳纳米管复合物。
进一步地,本发明还提供了一种制备所述薄膜的简单方法,例如,利用所述的复合墨水,通过涂布或者印刷等方法沉积制备而成。所述涂布方式包括浸渍涂布、滴落涂布、旋转涂布、刮刀涂布、狭缝涂布等;所述的印刷方式包括喷墨打印、丝网印刷、凹版印刷、气流喷印、柔版转印等,但不限于此。
在实际沉积制备复合薄膜的过程中,所述的薄膜通常沉积在基底材料之上。所述的基底材料包括:硅片、玻璃、塑料、纸张以及金属薄片,如:不锈钢、铝箔等,但不限于此。
前述喷墨打印中的打印机可选用挤压式打印墨水的打印机,如Dimatrix的2831、3000、5005、MicroFab和气溶胶喷墨打印机等,但不限于此。
前述印刷制备复合薄膜的过程中,所述墨水中碳纳米管的浓度优选为0.0001~1mg/mL。
本发明还提供了一种薄膜的后续处理方法,所述的后续处理工艺包括溶剂清洗和退火等。
前述的溶剂清洗工序中,溶剂可选自于三氯甲烷、四氢呋喃、甲苯、邻二甲苯、对二甲苯、间二甲苯、三氯甲烷和四氢呋喃中的任意一种或两种及两种以上的组合,但不限于此。
前述的退火工序中,采用的退火温度在200℃以下,优选为25~120℃,退火时间为30~120min,优选为30~60min。
本发明的一个方面还提供了一种装置,其包含所述的三维树形共轭化合物-碳纳米管复合物或者所述的薄膜。
在一些实施方案之中,所述装置可以是半导体装置,其半导体材料层(例如有源层) 包含所述的薄膜。
进一步的,所述半导体装置可以为晶体管,反相器等,但不限于此。
例如,本发明还提供了一种场效应晶体管,其主要由源电极、漏电极、栅电极、介电层和有源层构成,所述有源层含有所述的三维树形共轭化合物-碳纳米管复合物或者所述的薄膜。
进一步的,所述场效应晶体管为薄膜晶体管,其可以利用所述碳纳米管-三维树形共轭化合物复合墨水,通过滴涂、旋涂、浸涂、凹版印刷和喷墨打印等方法构建而成,沉积方式多样,加工工艺简单。
例如,本发明提供了所述三维树形共轭化合物-碳纳米管复合物作为薄膜晶体管的有源层。
本发明可被应用于大规模商业化生产高性能可印刷半导体碳纳米管墨水和高性能的印刷电子器件。
为使本发明更易于理解,下面将结合若干实施例对本发明的技术方案做进一步的阐述说明,应当指出的是,这些实施例仅是对本发明的示范性说明,其中所采用的各种产品结构参数、各种反应参与物及工艺条件均是较为典型的范例,但经过本案发明人大量试验验证,于前文所列出的其它类型的反应参与物及其它工艺条件也均是适用的,并也均可达成本发明所声称的技术效果。以下结合附图及若干优选实施例对本发明的技术方案作进一步的说明。
如下实施例中涉及了一些较为典型的三维树形共轭化合物,其结构式分别如下:
这些典型的三维树形共轭化合物中,具有分枝状结构的树形寡聚噻吩核心构建单元可参考文献(Chem.Eur.J.2012,18,12880–12901)合成。
前述的这些典型的三维树形共轭化合物的具体合成工艺可以参阅下文,(7)、(8)、(9)、(10)、(11)和(16)中的-C8H17为乙基己基,(12)、(13)、(14)、(15)中的-C20H41为辛基十二烷基。
(1)化合物3的合成
称取(化合物1)(2.0g,2.13mmol),化合物2(1.5g,5.1mmol),Pd2(dba)3.CHCl3(44mg,43μmol),HP(tBu)3BF4(25mg,86μmol)在氮气氛下加入250mL两口圆底烧瓶,向上述两口瓶中加入脱气处理的脱气处理的THF(150mL),然后再向反应体系中逐滴加入K2CO3水溶液(1M,12.0mL,12.0mmol),常温过夜搅拌。反应体系逐渐变为蓝紫色。24小时后,先将溶液中的大部分溶剂蒸发浓缩,再用二氯甲烷/水萃取溶液,收集有机相,用无水硫酸钠干燥有机相,过滤,旋转蒸发除去溶剂,用硅胶色谱柱分离产物,淋洗剂为二氯甲烷:正己烷(1:4),得到2.1g蓝色固体(化合物3),产率94%。其表征数据如下:
1H NMR(CDCl3,400MHz):δ=8.92(d,J=4.04Hz,1H),δ=8.84(d,J=3.04Hz,1H),7.60(d,J=4.40Hz,1H),7.24-7.27(m,1H),7.22(d,J=4.12Hz,1H),7.14(d,J=3.44Hz,1H),6.74(d,J=3.60Hz,1H),4.03(d,J=2.08Hz,2H),4.01(d,J=2.00Hz,2H),2.80-2.84(m,2H),1.91-1.96(m,2H),1.66-1.74(m,2H),1.21-1.34(m;70H),0.83-0.92ppm(m,15H)。
MALDI-TOF MS:m/z calcd for C64H102N2O2S3:1026.7,found:1026.0(matrix:DCTB)。
(2)化合物4的合成
在手套箱中称取[Ir(OMe)(COD)]2(62mg,94μmol),HBPin(600mg,4.68mmol),dtbpy(50mg,186μmol),充分搅拌,使三种物质充分混合,形成催化剂体系。称取化合物3(2.4g,2.34mmol)加入100mL两口烧瓶中,向该两口烧瓶中加入50mL干燥的THF。将催化剂体系加入到盛有化合物3的两口烧瓶中,在50℃下反应4小时。当反应冷却至室温,旋蒸掉 溶剂得到粗产物。粗产物通过硅胶色谱柱分离,得到化合物4(2.3g),产率85%。其表征数据如下:
1H NMR(CDCl3,400MHz):δ=8.95(d,J=4.16Hz,1H),δ=8.85(d,J=3.84Hz,1H),7.70(d,J=3.88Hz,1H),7.23(d,J=4.16Hz,1H),7.14(d,J=3.56Hz,1H),6.74(d,J=3.64Hz,1H),4.01-4.07(m,4H),2.80-2.84(m,2H),1.89-1.96(m,2H),1.66-1.74(m,2H),1.37(s,12H),1.21-1.34(m;70H),0.83-0.92ppm(m,15H)。
MALDI-TOF MS:m/z calcd for C64H102N2O2S3:1151.8,found:1151.6(matrix:DCTB)。
(3)化合物6(G1-Dendron-FG1-EG2)的合成
称取(化合物5)(100mg,200μmol),化合物4(530mg,460μmol),Pd2(dba)3·CHCl3(10mg,10μmol),HP(t-Bu)3·BF4(8mg,27μmol)在氮气氛下加入25mL两口圆底烧瓶,向上述 两口瓶中加入脱气处理的脱气处理的THF(10.0mL),然后再向反应体系中逐滴加入K2CO3水溶液(1M,1.2mL,1.2mmol),常温过夜搅拌。反应体系逐渐变为深蓝色。24小时后,先将溶液中的大部分溶剂蒸发浓缩,再用二氯甲烷/水萃取溶液,收集有机相,用无水硫酸钠干燥有机相,过滤,旋转蒸发除去溶剂,用硅胶色谱柱分离产物,淋洗剂为二氯甲烷:正己烷(1:9),得到372mg蓝色固体(化合物6),产率81%。其表征数据如下:
1H NMR(CDCl3,400MHz):δ=8.94(d,J=2.92Hz,1H),δ=8.93(d,J=2.96Hz,1H),8.89(t,J=4.48Hz,2H),7.34(d,J=5.28Hz,1H),7.27(d,J=1.04Hz,1H),7.26(d,J=1.04Hz,1H),7.23(d,J=3.80Hz,1H),7.21(d,J=4.24Hz,3H),7.19(d,J=5.28Hz,1H),7.13(d,J=3.56Hz,2H),7.11(d,J=3.84Hz,1H),7.05(d,J=3.84Hz,1H),6.73(d,J=3.64Hz,2H),4.02(d,J=7.00Hz,8H),2.81(t,J=7.56Hz,4H),1.94-1.99(m,4H),1.66-1.73(m,4H),1.21-1.34(m;140H),0.81-0.93ppm(m,30H)。
MALDI-TOF MS:m/z calcd for C140H208N4O4S9:2297.3,found:2297.6(matrix:DCTB)。
(4)化合物8(G1-Dendrimer-FG1-EG2)的合成
称取化合物4(630mg,546μmol),化合物7(100mg,124μmol),[Pd2(dba)3]·CHCl3(10mg,10μmol),and HP(tBu)3·BF4(8mg,27μmol)在氮气氛下加入50mL两口圆底烧瓶,向上述两口瓶中加入脱气处理的脱气处理的THF(25.0mL),然后再向反应体系中逐滴加入K2CO3水溶液(1M,2.0mL,2.0mmol),常温过夜搅拌。反应体系逐渐变为深蓝色。24小时后,先将溶液中的大部分溶剂蒸发浓缩,再用二氯甲烷/水萃取溶液,收集有机相,用无水硫酸钠干燥有机相,过滤,旋转蒸发除去溶剂,用硅胶色谱柱分离小分子杂质,再用凝胶色谱柱纯化分离得到460mg蓝色固体(化合物8),产率81%。其表征数据如下:
1H NMR(CDCl3,400MHz):δ=8.81(d,J=3.56Hz,4H),δ=8.76(dd,J=4.24Hz,4H),7.15-7.19(m,10H),7.07-7.09(m,8H),7.04(d,J=3.12Hz,4H),6.66(d,J=3.40Hz,4H),3.90(br,16H),2.73(br,8H),1.85(br,8H),1.57-1.65(m,8H),1.21-1.34(m;280H),0.72-0.84ppm(m,60H)。
MALDI-TOF MS:m/z calcd for C280H414N8O8S18:4592.7,found:4592.3(matrix:DCTB)。
(5)化合物10(G2-Dendron-FG1-EG2)的合成
称取化合物9(50mg,24μmol),化合物4(204mg,177μmol),[Pd2(dba)3]·CHCl3(5mg, 4.8μmol),and HP(tBu)3·BF4(4mg,13.8μmol)在氮气氛下加入25mL两口圆底烧瓶,向上述两口瓶中加入脱气处理的脱气处理的THF(10.0mL),然后再向反应体系中逐滴加入K2CO3水溶液(1M,1.0mL,1.0mmol),常温过夜搅拌。反应体系逐渐变为深蓝色。24小时后,先将溶液中的大部分溶剂蒸发浓缩,再用二氯甲烷/水萃取溶液,收集有机相,用无水硫酸钠干燥有机相,过滤,旋转蒸发除去溶剂,用硅胶色谱柱分离小分子杂质,再用凝胶色谱柱纯化分离得到146mg蓝色固体(化合物10),产率75%。其表征数据如下:
1H NMR(CDCl3,400MHz):δ=8.92(d,J=4.08Hz,4H),8.89(d,J=4.12Hz,2H),8.87(d,J=4.16Hz,2H),δ=7.33(d,J=5.24Hz,1H),δ=7.23(s,1H),δ=7.22(s,1H),7.13-7.20(m,12H),7.03-7.12(m,13H),6.70-6.72(m,4H),3.99(br,16H),2.78-2.82(m,8H),1.94(br,8H),1.65-1.71(m,8H),1.20-1.32(m;280H),0.79-0.92ppm(m,60H)。
MALDI-TOF MS:m/z calcd for C292H420N8O8S21:4838.7,found:4838.9(matrix:DCTB)。
(6)化合物12(G2-Dendrimer-FG1-EG2)的合成
称取化合物7(100mg,124μmol),化合物11(1.35g,559μmol),[Pd2(dba)3]·CHCl3(15mg,14μmol),and HP(tBu)3·BF4(9mg,31μmol)在氮气氛下加入250mL两口圆底烧瓶,向上述两口瓶中加入脱气处理的脱气处理的THF(100.0mL),然后再向反应体系中逐滴加入K2CO3水溶液(1M,10.0mL,10.0mmol),常温过夜搅拌。反应体系逐渐变为深蓝色。24小时后,先将溶液中的大部分溶剂蒸发浓缩,再用二氯甲烷/水萃取溶液,收集有机相,用无水硫酸钠干燥有机相,过滤,旋转蒸发除去溶剂,用硅胶色谱柱分离小分子杂质,再用凝胶色谱柱纯化分离得到745mg蓝色固体(化合物12),产率62%。其表征数据如下:
MALDI-TOF MS:m/z calcd for C584H838N16O16S42:9685.4;found:9684.7(matrix:DCTB),HR MS:m/z calcd for C584H838N16O16S42:(100%abundance)9685.3664;found:9685.3677。Mn=10463g/mol,Mw=11119,PDI=1.07。
(7)化合物15的合成
称取(化合物13)(2.0g,3.32mmol),化合物14(1.08g,4.15mmol),Pd2(dba)3.CHCl3(86mg,83μmol),HP(tBu)3BF4(48mg,166μmol)在氮气氛下加入500mL两口圆底烧瓶,向上述两口瓶中加入脱气处理的脱气处理的THF(250mL),然后再向反应体系中逐滴加入K2CO3水溶液(1M,15.0mL,15.0mmol),常温过夜搅拌。反应体系逐渐变为蓝紫色。24小时后,先将溶液中的大部分溶剂蒸发浓缩,再用二氯甲烷/水萃取溶液,收集有机相,用无水硫酸钠 干燥有机相,过滤,旋转蒸发除去溶剂,用硅胶色谱柱分离产物,淋洗剂为二氯甲烷:正己烷(1:3),得到2.0g蓝色固体(化合物15),产率92%。其表征数据如下:
1H NMR(CDCl3,400MHz):δ=8.96(d,1H),δ=8.88(d,1H),7.79(m,2H),7.67-7.70(m,2H),7.55(s,1H),7.43(dd,1H),7.36(m,2H),4.03-4.01(m,4H),1.50-1.60(m,2H),1.21-1.34(m;16H),0.83-0.92ppm(m,12H)。
MALDI-TOF MS:m/z calcd for C38H44N2O2S3:656.3,found:656.4(matrix:DCTB)。
(8):化合物16的合成
在手套箱中称取[Ir(OMe)(COD)]2(62mg,94μmol),HBPin(600mg,4.68mmol),dtbpy (50mg,186μmol),充分搅拌,使三种物质充分混合,形成催化剂体系。称取化合物15(2g,3.05mmol)加入100mL两口烧瓶中,向该两口烧瓶中加入50mL干燥的THF。将催化剂体系加入到盛有化合物15的两口烧瓶中,在50℃下反应4小时。当反应冷却至室温,旋蒸掉溶剂得到粗产物。粗产物通过硅胶色谱柱分离,得到化合物16(2.15g),产率90%。其表征数据如下:
1HNMR(CDCl3,400MHz):δ=8.95(d,1H),8.86(d,1H),7.79(d,1H),7.65-7.72(m,2H),7.52(s,1H),7.40(dd,1H),7.36(m,2H),4.06-4.02(m,4H),1.53-1.63(m,2H),1.24-1.36(m;16H),1.28(s,12H)0.82-0.93ppm(m,12H)。
MALDI-TOF MS:m/z calcd for C44H55BN2O4S3:782.3,found:782.7(matrix:DCTB)。
(9)化合物18的合成
称取(化合物17)(200mg,653μmol),化合物16(1.29g,1650μmol),Pd2(dba)3.CHCl3(34mg,33μmol),HP(t-Bu)3.BF4(20mg,66μmol)在氮气氛下加入100mL两口圆底烧瓶, 向上述两口瓶中加入脱气处理的脱气处理的THF(10.0mL),然后再向反应体系中逐滴加入K2CO3水溶液(1M,2.5mL,2.5mmol),常温过夜搅拌。反应体系逐渐变为深蓝色。24小时后,先将溶液中的大部分溶剂蒸发浓缩,再用二氯甲烷/水萃取溶液,收集有机相,用无水硫酸钠干燥有机相,过滤,旋转蒸发除去溶剂,用硅胶色谱柱分离产物,淋洗剂为二氯甲烷:正己烷(1:2),得到810mg蓝色固体(化合物18),产率85%。其表征数据如下:
1H NMR(CDCl3,400MHz):δ=8.98(m,2H),δ=8.90(m,2H),7.85(m,3H),7.80(m,2H),7.68-7.74(m,4H),7.55(m,2H),7.42(m,2H),7.36-7.38(m,4H),4.06-4.02(m,8H),1.53-1.63(m,4H),1.22-1.35(m;32H),0.84-0.95ppm(m,24H),0.29(s,9H)。
MALDI-TOF MS:m/z calcd for C85H98N4O4S6Si:1458.6,found:1458.8(matrix:DCTB)。
(10)化合物19的合成
称取化合物18(1.0g,685μmol)溶于100ml CH2Cl2中,在0℃氮气氛围下逐滴加入BBr3(1.0Ml,1M,1mmol),滴加完毕后继续搅拌2h,用高真空泵移除其中的易挥发溶剂。再称取pinacol(97mg,820μmol)溶解在100mL THF中,将溶解后的pinacol溶液注入上述反应体系中,常温过夜搅拌。24小时后,先将溶液中的大部分溶剂蒸发浓缩,再用二氯甲烷/水萃取溶液,收集有机相,用无水硫酸钠干燥有机相,过滤,旋转蒸发除去溶剂,用硅胶色谱柱分离产物,淋洗剂为二氯甲烷:正己烷(3:7),得到912mg蓝色固体(化合物19),产率88%。
1H NMR(CDCl3,400MHz):δ=8.99(m,2H),δ=8.92(m,2H),7.86(m,3H),7.82(m,2H),7.67-7.75(m,4H),7.54(m,2H),7.45(m,2H),7.33-7.40(m,4H),4.03-4.09(m,8H),1.54-1.65(m,4H),1.31(s,12H),1.20-1.36(m;32H),0.83-0.94ppm(m,24H)。
MALDI-TOF MS:m/z calcd for C88H101BN4O6S6:1512.6,found:1512.9(matrix:DCTB)。
(11)化合物20的合成制备
称取化合物13(100mg,215μmol),化合物19(1.5g,992μmol),[Pd2(dba)3]·CHCl3(25mg,24μmol),and HP(tBu)3·BF4(15mg,51μmol)在氮气氛围下加入250mL两口圆底烧瓶,向上述两口瓶中加入脱气处理的脱气处理的THF(100.0mL),然后再向反应体系中逐滴加入K2CO3水溶液(1M,10.0mL,10.0mmol),常温过夜搅拌。24小时后,先将溶液中的大部分溶剂蒸发浓缩,再用二氯甲烷/水萃取溶液,收集有机相,用无水硫酸钠干燥有机相,过滤,旋转蒸发除去溶剂,用硅胶色谱柱分离小分子杂质,再用凝胶色谱柱纯化分离得到850mg蓝色固体(化合物20),产率70%。其表征数据如下:MALDI-TOF MS:m/z calcd for C340H362N16O16S24:5692.1,found:5692.5(matrix:DCTB)。Mn=6203g/mol,Mw=6513g/mol,PDI=1.05。
(12)化合物22的合成
称取(化合物21)(200mg,199μmol),化合物2(61mg,207μmol),Pd2(dba)3.CHCl3(10mg,10μmol),HP(t-Bu)3.BF4(6mg,20μmol)在氮气氛下加入100mL两口圆底烧瓶,向上述两口瓶中加入脱气处理的脱气处理的THF(20.0mL),然后再向反应体系中逐滴加入K2CO3水溶液(1M,2.5mL,2.5mmol),常温过夜搅拌。反应体系逐渐变为深蓝色。24小时后,先将溶液中的大部分溶剂蒸发浓缩,再用二氯甲烷/水萃取溶液,收集有机相,用无水硫酸钠干燥有机相,过滤,旋转蒸发除去溶剂,用硅胶色谱柱分离产物,淋洗剂为二氯甲烷:正己烷(1:3),得到87mg蓝色固体(化合物22),产率40%。其表征数据如下:
1H NMR(CDCl3,400MHz):δ=7.78(m,2H),δ=7.69(m,2H),7.52(m,2H),7.40(m,2H),7.32(d,1H),6.74(d,1H),3.90-3.95(m,4H),2.81-2.85(m,2H),1.90-1.98(m,2H),1.66-1.74(m,2H),1.21-1.34(m;70H),0.83-0.92ppm(m,15H)。
MALDI-TOF MS:m/z calcd for C68H105BrN2O2S:1092.7,Found:1093.0(matrix:DCTB)。
(13)化合物24的合成
称取化合物23(100mg,200μmol)、化合物22(546mg,500μmol),,Pd2(dba)3.CHCl3(10mg,10μmol),HP(t-Bu)3.BF4(8mg,27μmol)在氮气氛下加入25mL两口圆底烧瓶,向上述两口瓶中加入脱气处理的脱气处理的THF(20.0mL),然后再向反应体系中逐滴加入K2CO3水溶液(1M,2mL,2mmol),常温过夜搅拌。反应体系逐渐变为深蓝色。24小时后,先将溶液中的大部分溶剂蒸发浓缩,再用二氯甲烷/水萃取溶液,收集有机相,用无水硫酸钠干燥有机相,过滤,旋转蒸发除去溶剂,用硅胶色谱柱分离产物,淋洗剂为二氯甲烷:正己烷(3:7),得到372mg蓝色固体(化合物24),产率82%。其表征数据如下:
1H NMR(CDCl3,400MHz):δ=7.80(m,4H),δ=7.76(d,1H),7.70(m,4H),7.55(m,4H),7.42(m,4H),7.30-7.35(m,4H),7.20(d,1H),7.16(d,2H),6.76(d,2H),4.02(d,J=7.00Hz,8H),2.81(t,J=7.56Hz,4H),1.94-1.99(m,4H),1.66-1.73(m,4H),1.21-1.34(m;140H),0.81-0.93ppm(m,30H)。
MALDI-TOF MS:m/z calcd for C148H216N4O4S5:2273.5,found:2273.0(matrix:DCTB)。
(14)化合物25的合成
在手套箱中称取[Ir(OMe)(COD)]2(66mg,100μmol),HBPin(340mg,2.66mmol),dtbpy(54mg,200μmol),充分搅拌,使三种物质充分混合,形成催化剂体系。称取化合物24(3g,1.32mmol)加入250mL两口烧瓶中,向该两口烧瓶中加入100mL干燥的THF。将催化剂体系加入到盛有化合物24的两口烧瓶中,在50℃下反应4小时。当反应冷却至室温,旋 蒸掉溶剂得到粗产物。粗产物通过硅胶色谱柱分离,得到化合物25(2.69g),产率85%。
1H NMR(CDCl3,400MHz):δ=7.84(m,4H),7.60(s,1H),7.73(m,4H),7.57(m,4H),7.45(m,4H),7.32-7.37(m,4H),7.18(d,2H),6.79(d,2H),4.05(d,J=7.00Hz,8H),2.83(t,J=7.56Hz,4H),1.92-1.98(m,4H),1.64-1.71(m,4H),1.33(s,12H),1.21-1.34(m;140H),0.81-0.93ppm(m,30H)。
MALDI-TOF MS:m/z calcd for C148H216N4O4S5:2399.6,found:2400.0(matrix:DCTB)。
(15)化合物26的合成
称取化合物9(80mg,64μmol),化合物25(695mg,290μmol),[Pd2(dba)3]·CHCl3(10mg,10μmol),and HP(tBu)3·BF4(6mg,19μmol)在氮气氛围下加入250mL两口圆底烧瓶,向上 述两口瓶中加入脱气处理的脱气处理的THF(120.0mL),然后再向反应体系中逐滴加入K2CO3水溶液(1M,3mL,3.0mmol),常温过夜搅拌。24小时后,先将溶液中的大部分溶剂蒸发浓缩,再用二氯甲烷/水萃取溶液,收集有机相,用无水硫酸钠干燥有机相,过滤,旋转蒸发除去溶剂,用硅胶色谱柱分离小分子杂质,再用凝胶色谱柱纯化分离得到392mg蓝色固体(化合物26),产率62%。其表征数据如下:MALDI-TOF MS:m/z calcd for C628H876N16O16S29:9826.0,found:9826.8(matrix:DCTB)。Mn=8963g/mol,Mw=9680g/mol,PDI=1.08。
(16)化合物28的合成
称取化合物13(50mg,107μmol),化合物27(785mg,483μmol),[Pd2(dba)3]·CHCl3(10mg,10μmol),and HP(tBu)3·BF4(6mg,19μmol)在氮气氛围下加入250mL两口圆底烧瓶,向上述两口瓶中加入脱气处理的脱气处理的THF(150.0mL),然后再向反应体系中逐滴加入K2CO3水溶液(1M,5mL,5.0mmol),常温过夜搅拌。24小时后,先将溶液中的大部分溶剂蒸发浓缩,再用二氯甲烷/水萃取溶液,收集有机相,用无水硫酸钠干燥有机相,过滤,旋转蒸发除去溶剂,用硅胶色谱柱分离小分子杂质,再用凝胶色谱柱纯化分离得到428mg蓝色固体(化合物28),产率65%。其表征数据如下:MALDI-TOF MS:m/z calcd for C356H362N16O16S32:6139.9,found:6139.5(matrix:DCTB)。Mn=6954g/mol,Mw=7371g/mol,PDI=1.06。
实施例1:三维树形共轭化合物(6T-DPP)-碳纳米管复合墨水的制备
称取2mg商业化大管径碳纳米管P2样品,溶于10毫升的甲苯溶液中,然后加入分子量为4597的三维树形共轭化合物6T-DPP 6mg,在冰浴的条件下用功率为60W的超声清洗机超声分散30分钟,使聚合物可以选择性地和半导体碳纳米管充分作用,得到均匀分散的分散液。再经过15000rpm离心30分钟后,使上述分散液中的碳纳米管束以及未被三维树形共轭化合物包覆的金属性碳纳米管沉积在离心管的底部,分离出上层清液即可获得高纯的富集有大管径半导体碳纳米管的复合墨水。
实施例2:三维树形共轭化合物(9T-DPP)-碳纳米管复合墨水的制备
称取2mg商业化大管径碳纳米管P2样品,溶于10毫升的甲苯溶液中,然后加入分子量为4848的三维树形共轭化合物9T-DPP 6mg,在冰浴的条件下用功率为60W的超声清洗机超声分散30分钟,使聚合物可以选择性地和半导体碳纳米管充分作用,得到均匀分散的分散液。再经过15000rpm离心30分钟后,使上述分散液中的碳纳米管束以及未被三维树形共轭化合物包覆的金属性碳纳米管沉积在离心管的底部,分离上层清液即可获得高纯的富集有大管径半导体碳纳米管的复合墨水。
实施例3:三维树形共轭化合物(18T-DPP)-碳纳米管复合墨水的制备
称取2mg商业化大管径碳纳米管P2样品,溶于10毫升的甲苯溶液中,然后加入分子量为9846的三维树形共轭化合物18T-DPP 6mg,在冰浴的条件下用功率为60W的超声清洗机超声分散30分钟,使聚合物可以选择性地和半导体碳纳米管充分作用,得到均匀分散的分散液。再经过15000rpm离心30分钟后,使上述分散液中的碳纳米管束以及未被三维树形共轭化合物包覆的金属性碳纳米管沉积在离心管的底部,分离上层清液即可获得高纯的富集有大管径半导体碳纳米管的复合墨水。
实施例4:三维树形共轭化合物(6T-DPP)-碳纳米管复合墨水的UV-Vis-IR光谱性能 表征
对三维树形共轭化合物(6T-DPP)-碳纳米管分散液离心后的上清液,利用紫外-可见-近红外(Perkin Elmer Lambda 750)测试其吸收光谱,其结果如图1所示。从吸收光谱图中可以看出,对应半导体碳纳米管的S22吸收峰(900-1200nm)变得非常尖锐,而且吸收背景非常低,说明三维树形共轭化合物(6T-DPP)可以选择性的分散富集半导体碳纳米管。
实施例5:三维树形共轭化合物(9T-DPP)-碳纳米管复合墨水的UV-Vis-IR光谱性能表征
对三维树形共轭化合物(9T-DPP)-碳纳米管分散液离心后的上清液,利用紫外-可见-近红外(Perkin Elmer Lambda 750)测试其吸收光谱,其结果如图2所示。从吸收光谱图中可以看出,对应半导体碳纳米管的S22吸收峰(900-1200nm)变得非常尖锐,而且吸收背景非常低,说明三维树形共轭化合物(9T-DPP)可以选择性的分散富集半导体碳纳米管。
实施例6:三维树形共轭化合物(6T-DPP)-碳纳米管复合墨水的Raman光谱性能表征
利用Raman光谱仪测试上述离心后的上清液的拉曼光谱。图3中表示了其拉曼光谱测试结果。在785nm的激光下,对于未处理的碳纳米管P2中可以观察到与159cm-1金属性碳纳米管相对应的峰,但是经过三维树形共轭化合物(6T-DPP)的选择性包覆以及离心分离得到样品中,159cm-1金属性碳纳米管峰消失。因此,可以认定,依照本发明实施例1的方法能高效地分离出大量大管径的半导体碳纳米管。
实施例7:三维树形共轭化合物(9T-DPP)-碳纳米管复合墨水的Raman光谱性能表征
利用Raman光谱仪测试上述离心后的上清液的拉曼光谱。图4中表示了其拉曼光谱测试结果。在633nm的激光下,对于未处理的碳纳米管P2中可以观察到与金属性碳纳米管相对应的峰(1550-1580cm-1)和与半导体碳纳米管相对应的峰(1590cm-1),但是经过三维树形共轭化合物(9T-DPP)的选择性包覆以及离心分离得到样品中,1590cm-1处的半导体碳纳米管峰变得更加尖锐,同时与未分离的P2碳纳米管相比,金属性碳纳米管相对应的峰半导体碳纳米管峰的峰面积之比变得更小。因此,可以表明,依照本发明实施例2的方法能高效地分离出大量大管径的半导体碳纳米管。
实施例8:三维树形共轭化合物(9T-DPP)-碳纳米管复合墨水的薄膜晶体管制备以及性能表征
取上述实施例2中利用三维树形共轭化合物(9T-DPP)选择性包覆分离出的大管径P2半导体碳纳米管的墨水溶液用滴涂加工的方法制备薄膜晶体管器件。图5为晶体管的电性 能转移特征曲线,从图中可以看出,晶体管的开关比和迁移率分别可以达到4×106和37.63cm2/Vs以上。图6为晶体管的电性能的输出特征曲线,从图中可以看出,该晶体管输出电流随栅电压增加而变小,该晶体管为p型晶体管。该晶体管具有优良的电性能进一步证实通过三维树形共轭化合物的选择性包覆,可以很好的进行分离商业化大管径碳纳米管。
此外,参照实施例1-8的实施方案,本案发明人还以前文列出的化合物20、26及其它化合物进行试验,并得到了相似的测试结果。
应当理解,以上说明及所示的实施例,不可解析为限定本发明的设计思想。在本发明的技术领域里的技术人员可以将本发明的技术性思想根据现有技术和本领域的常识以多样的形式改良变更,这样的改良及变更应理解为属于本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!