一种负载型钴基催化剂及其制备方法和使用方法与流程
本发明涉及催化领域,特别地涉及一种负载型钴基催化剂及其制备方法和使用方法。
背景技术:
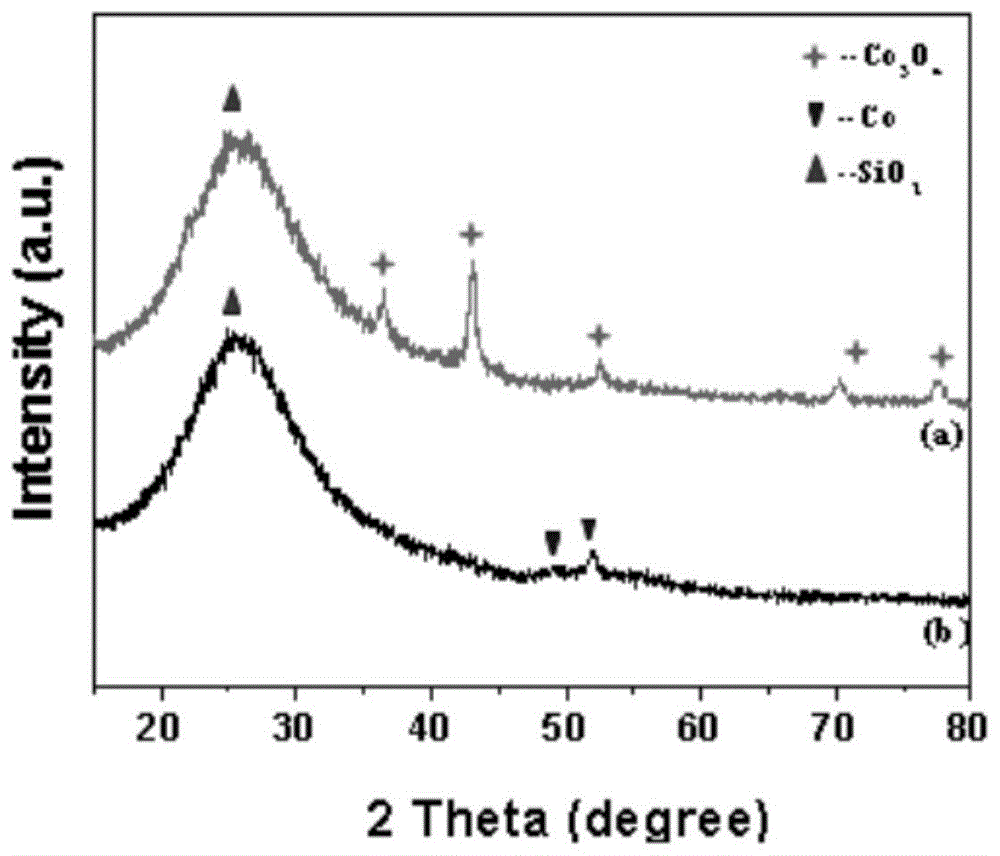
:自1938年德国科学家ottoroelen在奥伯豪森ruhrchemie实验室发现了氢甲酰化反应,并自50年代初建成第一套工业装置以来,烯烃氢甲酰化反应的研究一直久盛不衰。氢甲酰化反应是指以烯烃和合成气(co+h2)为原料,在催化剂作用下生成醛的过程。费托合成反应实现了煤变油的转变,费托产物中含有大量的烯烃,从费托产物中分离出烯烃,随后经氢甲酰化反应得到醛并进一步经还原得到醇,醇可以用来制备增塑剂、表面活性剂等,具有较高的附加值,因此氢甲酰化反应已经是目前工业上最重要的均相反应之一。尽管均相催化剂的反应活性和选择性是多相催化剂无法比拟的,但是很多均相催化剂都仅仅因为催化剂与产物难以分离而无法工业化。近年来,均相催化剂多相化的研究得到了广泛的重视,主要分为两大类:一类是均相固载化,另一类是两相催化。均相固载化包括无机载体固载化、有机聚合物、担载液相催化剂、担载水相催化剂。两相催化包括液/液两相催化、氟两相体系、温控相分离催化、超临界流体两相、离子液体两相、超临界流体-离子液体两相体系等。虽然在这些催化体系中涌现了许多创新性的概念,但是它们或者金属组分流失严重,或者催化剂稳定性较差,或者采用昂贵的有机配体、溶剂,或者催化剂制备过程繁琐、工艺复杂。公开号为cn102617308a的中国专利申请公开了一种烯烃两相氢甲酰化方法,其中,将rh-tppts作为催化剂,采用具有室温可凝固特性的聚醚胍甲磺酸盐离子液体(pgmils)实现该反应的“均相反应,两相分离”。虽然该反应的高碳醛的选择性达85~99%且正构醛与异构醛的摩尔比为2.0~2.4。但是该方法所采用的离子液体价格昂贵且制备复杂,生产成本较高,限制了其工业化应用。公开号为cn104710289a的中国专利申请公开了一种采用固体多相催化剂的用于烯烃氢甲酰化反应的方法,该方法采用的是金属rh与多孔有机聚合物配体络合的方式制备催化剂,所制备的催化剂具有均相催化剂的优点,在反应时催化剂的活性和产物收率都较高,但是聚合物配体本身合成工艺复杂,成本较高。在low-pressurehydroformylationofmiddleolefinsovercoandrhsupportedonactivecarboncatalysts,energy&fuels2003,17,810-816文章中提到了,钴基催化剂用于氢甲酰化反应时很容易形成羰基钴造成催化剂的流失,活性下降,活性金属的流失量可达到20%,并且在提高压力至5.0mpa时,流失量可达50%。这不仅失去了多相催化剂分离与循环利用的优势,也很难在一个反应体系中区分均相催化与多相催化的活性。因此,在多相氢甲酰化反应中,针对催化剂活性金属的流失问题,目前还没有在实际的工业化过程中可利用的较为完善的解决方案。技术实现要素:针对上述技术问题,本发明提供一种在催化反应中能有效抑制活性组分流失,且制备方法简单的负载型钴基催化剂及其制备方法和使用方法。为实现上述目的和额外的其它目的或益处,本发明公开了如下技术方案。在一个方面,本发明涉及一种负载型钴基催化剂,其中,所述催化剂包括以重量份数计的如下成分:活性组分co0.1-10份;载体75-98份;有机酸1-15份;有机溶剂,其重量为所述有机酸的200-1100倍。在另一方面,本发明涉及一种制备上述负载型钴基催化剂的方法,其中,所述方法包括如下步骤:(1)将活性组分co的前体溶解于溶剂中,得到含有活性组分co的溶液;(2)将载体浸渍于所述含有活性组分co的溶液中,室温下随着搅拌进行超声处理,得到混合溶液;(3)将所述混合溶液在真空条件下移除溶剂,得到固体混合物;(4)将所述固体混合物依次进行干燥、焙烧和还原处理,得到半成品催化剂;(5)将有机酸溶解于有机溶剂中,得到有机酸的溶液;(6)将所述半成品催化剂置于所述有机酸的溶液中,搅拌以使所述有机酸包裹所述活性组分co,得到所述负载型钴基催化剂。在又一方面,本发明涉及一种使用上述负载型钴基催化剂催化链状烯烃的氢甲酰化反应的方法,其中,所述方法包括在所述负载型钴基催化剂的作用下,使所述链状烯烃同co和h2反应生成醛。本发明的有益效果:本发明提供的负载型钴基催化剂不仅能显著地抑制活性金属的流失,还具有与均相羰基钴催化剂相媲美的选择性,且反应后与产品分离容易,既避免了均相催化剂所存在的催化剂和产品的分离及循环使用问题,又避免了常规多相催化剂形成羰基钴使钴流失的问题,具有良好的工业应用前景。附图说明图1示出了制备例1中得到的半成品的co/sio2催化剂的xrd图。图2示出了制备例1中得到的半成品的co/sio2催化剂的透射电子显微镜图像。具体实施方式如下的示例性的实施方式仅是用于对本发明的方案进行解释,而并不旨在以任何方式限制本申请的保护范围。在本发明中,除非另有说明,术语“室温”是指20℃-40℃。在一个实施方式中,本发明涉及一种负载型钴基催化剂,其中,所述催化剂包括以重量份数计的如下成分:活性组分co0.1-10份;载体75-98份;有机酸1-15份;有机溶剂,其重量为有机酸的200-1100倍。在本发明的一个示例性的实施方式中,所述催化剂包括重量份数为0.9-9的所述活性组分co。在本发明的一个示例性的实施方式中,所述活性组分co可来源于包括硝酸钴、氯化钴、乙酰丙酮钴和羰基钴的组中的一种或多种,但不限于此。在本发明的一个示例性的实施方式中,所述载体可为选自sio2、α-al2o3、sic、si3n4、c3n4和mcm-48分子筛中的一种或多种,但不限于此。在进一步优选的实施方式中,所述载体的比表面积为10-850m2/g,孔容为0.05-1.0cm3/g,孔径分布在2-30nm的范围内。在本发明的一个示例性的实施方式中,所述有机酸可为选自甲酸、草酸、柠檬酸和酒石酸中的一种或多种,但不限于此。在本发明的一个示例性的实施方式中,所述有机溶剂可选自甲苯、n,n-二甲基甲酰胺(dmf)、二甲基亚砜(dmso)、丙酮、四氢呋喃、乙醇、甲醇或其任意混合物,但不限于此。在另一实施方式中,本发明涉及一种制备上述负载型钴基催化剂的方法,其中,所述方法包括如下步骤:(1)将活性组分co的前体溶解于溶剂中,得到含有活性组分co的溶液;(2)将载体浸渍于所述含有活性组分co的溶液中,室温下随着搅拌进行超声处理,得到混合溶液;(3)将所述混合溶液在真空条件下移除所述溶剂,得到固体混合物;(4)将所述固体混合物依次进行干燥、焙烧和还原处理,得到半成品催化剂;(5)将有机酸溶解于有机溶剂中,得到有机酸的溶液;(6)将所述半成品催化剂置于所述有机酸的溶液中,搅拌以使所述有机酸包裹所述活性组分co,得到所述负载型钴基催化剂。在本发明的一个示例性的实施方式中,所述步骤(1)中的溶剂可选自醇和/或水;所述醇例如选自c1-c4醇,优选乙醇。更优选地,所述活性组分co与所述步骤(1)中的溶剂的质量体积比为1:(400-1000)。在本发明的一个示例性的实施方式中,在所述步骤(1)中,所述活性组分co的前体可为选自硝酸钴、氯化钴、乙酰丙酮钴和羰基钴中的一种或多种,但不仅限于此。在本发明的一个示例性的实施方式中,所述载体与所述活性组分co的质量比为100:(1-10)。在本发明的一个示例性的实施方式中,所述超声处理可采用超声震荡器进行;优选地,所述超声处理的功率为800w,工作频率为40khz,且时间为1-2h。在本发明的一个示例性的实施方式中,可将所述混合溶液在真空条件(优选-0.1mpa)下于313k-343k的温度下以100-300rpm的转速进行旋转蒸发处理以移除所述溶剂,从而得到固体混合物;例如,所述旋转蒸发的温度可为320k-325k,转速可为140-160rpm。在本发明的一个示例性的实施方式中,所述干燥可采用烘箱进行;优选地,所述干燥的温度为333k-393k,干燥的时间为6-12h。在本发明的一个示例性的实施方式中,所述焙烧可采用马弗炉进行;优选地,所述焙烧的温度为573k-773k,焙烧的时间为2-6h。在本发明的一个示例性的实施方式中,所述还原可采用h2气氛的管式炉进行;优选地,所述还原的温度为573k-973k,还原的时间为2-12h。在本发明的一个示例性的实施方式中,所述有机酸和所述有机溶剂的质量比为1:(200-1100)。在本发明的一个示例性的实施方式中,所述有机溶剂可选自甲苯、n,n-二甲基甲酰胺(dmf)、二甲基亚砜(dmso)、丙酮、四氢呋喃、乙醇、甲醇或其任意混合物,但不限于此。另外,在上述的制备方法中采用的载体和有机酸如上文所定义的。在本发明的一个示例性的实施方式中,出于避免有机酸浪费的目的,所述半成品催化剂与所述有机酸的质量比为(50-150):(2-8)。在又一实施方式中,本发明涉及一种使用上述负载型钴基催化剂催化链状烯烃的氢甲酰化反应的方法,其中,所述方法包括在所述负载型钴基催化剂的作用下,使所述链状烯烃同co和h2反应生成醛。在本发明的一个示例性的实施方式中,所述负载型钴基催化剂与链状烯烃的质量摩尔比为(50-150):(2-4)。在本发明的一个示例性的实施方式中,所述链状烯烃可为c3-c10链状烯烃,例如c6-c8链状烯烃,更优选例如可选自1-己烯、2-己烯、1-辛烯和/或2-辛烯,但不仅限于此。在本发明的一个示例性的实施方式中,所述氢甲酰化反应在373-393k下进行3-15h。在本发明中,通过采用以特定的方法制备的负载型钴基催化剂,从而可以在氢甲酰化反应中显著地抑制催化剂中的活性金属的流失,同时还保留了高的选择性,并且该催化剂容易与产品分离,适合工业化应用。实施例以下对本发明的示范性实施例做出说明,其中包括本发明实施例的各种细节以助于理解,应当将它们认为仅仅是示例性的。因此,本领域普通技术人员应当认识到,可以对这里描述的实施例做出各种改变和修改,而不会背离本发明的范围和精神。同样,为了清楚和简明,以下的描述中省略了对公知功能和结构的描述。下述实施例中所用的试剂、材料和装置等,如无特殊说明,均可商业化购买得到。制备例1称取0.75gco(no3)2·6h2o并溶于200g去离子水中,得到活性金属co含量为3wt%的co(no3)2溶液。称取5.0gsio2载体(比表面积150m2/g,孔容0.9m3/g,平均孔径23.4nm)并浸渍于上述溶液中,室温下随着搅拌采用超声震荡器进行超声处理1h(其中,超声输出功率为800w,工作频率为40khz),超声结束后继续搅拌12h,然后将得到的混合溶液在旋转蒸发仪(转速为150rpm)中于真空(负压-0.1mpa)、323k下蒸干溶剂,得到粉末状的固体混合物。将该固体粉末置于烘箱中于333k下干燥12h,然后在马弗炉中于673k下焙烧4h,得到co3o4/sio2。将co3o4/sio2在管式炉中于h2气氛、673k下还原6h,其中氢气流速为50ml/min,还原后的催化剂为半成品催化剂(即co/sio2催化剂)。将6mg柠檬酸溶解于20mmol甲苯中,得到柠檬酸的甲苯溶液,将100mg的上述的半成品催化剂置于如上所述的柠檬酸的甲苯溶液中,于373k下搅拌2h,待柠檬酸充分包裹活性金属co后,即得到负载型钴基催化剂。其中,该制备例得到的半成品钴基催化剂的xrd图和透射电子显微镜图像如图1和图2所示。制备例2称取2.47gco(no3)2·6h2o并溶于200g去离子水中,得到活性金属co含量为10wt%的co(no3)2溶液。取5.0gsio2载体(比表面积150m2/g,孔容0.9m3/g,平均孔径23.4nm),将载体浸入上述溶液中,室温下随着搅拌采用超声震荡器进行超声处理2h(其中,超声输出功率为800w,工作频率为40khz),超声结束后继续搅拌12h,然后将得到的混合溶液在旋转蒸发仪(转速为300rpm)中于真空(负压-0.1mpa)、313k下蒸干溶剂,得到粉末状的固体混合物。将该固体粉末置于烘箱中于363k下干燥12h,然后在马弗炉中于573k下焙烧6h,得到co3o4/sio2,将co3o4/sio2在管式炉中于h2气氛、973k下还原2h,其中氢气流速为50ml/min,还原后的催化剂为半成品催化剂(即co/sio2催化剂)。将8mg草酸溶解于25mmoln,n-二甲基甲酰胺(dmf)中,得到草酸的dmf溶液,将50mg的该半成品催化剂置于如上所述的草酸的dmf溶液中,于373k下搅拌2h,待草酸充分包裹活性金属co后,即得到负载型钴基催化剂。制备例3称取0.75gco(no3)2·6h2o并溶于100g去离子水中,得到活性金属co含量为3wt%的co(no3)2溶液。取5.0gc3n4载体(比表面积11m2/g,孔容0.05m3/g,平均孔径12nm),将此载体浸渍于上述溶液中,室温下随着搅拌采用超声震荡器进行超声处理1h(其中,超声输出功率为800w,工作频率为40khz),超声结束后继续搅拌12h,然后将得到的混合溶液在旋转蒸发仪(转速为100rpm)中于真空(负压-0.1mpa)、343k下蒸干溶剂,得到粉末状的固体混合物。将该固体粉末置于烘箱中于393k下干燥6h,然后在马弗炉中于773k下焙烧2h,得到co3o4/c3n4,将co3o4/c3n4在管式炉中于h2气氛、673k下还原8h,其中氢气流速为50ml/min,还原后的催化剂为半成品催化剂(即co/c3n4催化剂)。将6mg酒石酸溶解于30mmol丙酮中,得到酒石酸的丙酮溶液,将80mg的该半成品催化剂置于如上所述的酒石酸的丙酮溶液中,于373k下搅拌2h,待酒石酸充分包裹活性金属co后,即得到负载型钴基催化剂。制备例4称取0.907g乙酰丙酮钴并溶于100g乙醇中,得到活性金属co含量为3wt%的co(no3)2溶液。取5.0gα-al2o3载体(比表面积78m2/g,孔容0.43m3/g,平均孔径23nm),将此载体浸渍于上述溶液中,室温下随着搅拌采用超声震荡器进行超声处理2h(其中,超声输出功率为800w,工作频率为40khz),超声结束后继续搅拌12h,然后将得到的混合溶液在旋转蒸发仪(转速为150rpm)中于真空(负压-0.1mpa)、323k下蒸干溶剂,得到粉末状的固体混合物。将该固体粉末置于烘箱中于333k下干燥12h,然后在马弗炉中于673k下焙烧6h,得到co3o4/α-al2o3,将co3o4/α-al2o3在管式炉中于h2气氛、573k下还原12h,其中氢气流速为50ml/min,还原后的催化剂为半成品催化剂(即co/α-al2o3催化剂)。将6mg甲酸溶解于30mmol二甲基亚砜(dmso)中,得到甲酸的dmso溶液,将50mg的该半成品催化剂置于如上所述的甲酸的dmso溶液中,于373k下搅拌2h,待甲酸充分包裹活性金属co后,即得到负载型钴基催化剂。制备例5称取1.01g六水合氯化钴并溶于50g乙醇中,得到活性金属co含量为5wt%的cocl2溶液。取5.0gmcm-48分子筛载体(比表面积810m2/g,孔容0.506m3/g,平均孔径2.857nm),将此载体浸渍于上述溶液中,室温下随着搅拌采用超声震荡器进行超声处理1h(其中,超声输出功率为800w,工作频率为40khz),超声结束后继续搅拌12h,然后将得到的混合溶液在旋转蒸发仪(转速为150rpm)中于真空(负压-0.1mpa)、323k下蒸干溶剂,得到粉末状的固体混合物。将该固体粉末置于烘箱中于333k下干燥12h,然后在马弗炉中于673k下焙烧6h,得到co3o4/mcm-48,将co3o4/mcm-48在管式炉中于h2气氛、573k下还原12h,其中氢气流速为50ml/min,还原后的催化剂为半成品催化剂(即co/mcm-48催化剂)。将6mg柠檬酸溶解于25mmol四氢呋喃中,得到柠檬酸的四氢呋喃溶液,将100mg的该半成品催化剂置于如上所述的柠檬酸的四氢呋喃溶液中,于373k下搅拌2h,待柠檬酸充分包裹活性金属co后,即得到负载型钴基催化剂。制备例6称取0.726g八羰基二钴溶于50g乙醇中,得到活性金属co含量为5wt%的co2(co)8溶液。取5.0gsi3n4分子筛载体(比表面积66m2/g,孔容0.475m3/g,平均孔径28nm),将此载体浸渍于上述溶液中,室温下随着搅拌采用超声震荡器进行超声处理1h(其中,超声输出功率为800w,工作频率为40khz),超声结束后继续搅拌12h,然后将得到的混合溶液在旋转蒸发仪(转速为150rpm)中于真空(负压-0.1mpa)、323k下蒸干溶剂,得到粉末状的固体混合物。将该固体粉末置于烘箱中于333k下干燥12h,然后在马弗炉中于673k下焙烧6h,得到co3o4/si3n4,将co3o4/si3n4在管式炉中于h2气氛、573k下还原12h,其中氢气流速为50ml/min,还原后的催化剂为半成品催化剂(即co/si3n4催化剂)。将6mg柠檬酸溶解于35mmol乙醇中,得到柠檬酸的乙醇溶液,将100mg的该半成品催化剂置于如上所述的柠檬酸的乙醇溶液中,于373k下搅拌2h,待柠檬酸充分包裹活性金属co后,即得到负载型钴基催化剂。制备例7称取0.247gco(no3)2·6h2o并溶于50g去离子水中,得到活性金属co含量为1.0wt%的co(no3)2溶液。取5.0gsic载体(比表面积35.6m2/g,孔容0.154m3/g,平均孔径17.43nm),将载体浸入上述溶液中,室温下随着搅拌采用超声震荡器进行超声处理2h(其中,超声输出功率为800w,工作频率为40khz),超声结束后继续搅拌12h,然后将得到的混合溶液在旋转蒸发仪(转速为150rpm)中于真空(负压-0.1mpa)、323k下蒸干溶剂,得到粉末状的固体混合物。将该固体粉末置于烘箱中于363k下干燥12h,然后在马弗炉中于573k下焙烧6h,得到co3o4/sic,将co3o4/sic在管式炉中于h2气氛、973k下还原2h,其中氢气流速为50ml/min,还原后的催化剂为半成品催化剂(即co/sic催化剂)。将2mg草酸溶解于30mmol四氢呋喃(thf)中,得到草酸的thf溶液,将150mg该半成品催化剂置于如上所述的草酸的thf溶液中,于373k下搅拌2h,待草酸充分包裹活性金属co后,即得到负载型钴基催化剂。实施例1将100mg的制备例1中得到的柠檬酸包裹的负载型钴基催化剂加入到间歇式反应釜中,向该间歇式反应釜中加入4mmol原料1-己烯,随后向其中充入5mpa合成气(co:h2=1:1),于373k下反应3h,反应后产物使用agilent-7890a气相色谱进行分析,该agilent-7890a气相色谱仪配有plot-al2o3柱(50m×530μm×1μm)、hp-innowax柱(30m×320μm×0.5μm),前后检测器均为fid检测器,采用联苯作为内标物对反应产物进行定量计算。初温30℃,保留时间4min,升温速率10℃/min,终温198℃,保留时间40min。分析结果见表1。实施例2将80mg的制备例3中得到的酒石酸包裹的负载型钴基催化剂加入到间歇式反应釜中,向该间歇式反应釜中加入4mmol原料2-己烯,随后向其中充入5mpa合成气(co:h2=1:1),于393k下反应15h,反应后产物使用agilent-7890a气相色谱进行分析,该agilent-7890a气相色谱仪配有plot-al2o3柱(50m×530μm×1μm)、hp-innowax柱(30m×320μm×0.5μm),前后检测器均为fid检测器,采用联苯作为内标物对反应产物进行定量计算。初温30℃,保留时间4min,升温速率10℃/min,终温198℃,保留时间40min。分析结果见表1。实施例3将50mg的制备例4中得到的甲酸包裹的负载型钴基催化剂加入到间歇式反应釜中,向该间歇式反应釜中加入4mmol原料1-辛烯,随后向其中充入5mpa合成气(co:h2=1:1),于393k下反应15h,反应后产物使用agilent-7890a气相色谱进行分析,该agilent-7890a气相色谱仪配有plot-al2o3柱(50m×530μm×1μm)、hp-innowax柱(30m×320μm×0.5μm),前后检测器均为fid检测器,采用联苯作为内标物对反应产物进行定量计算。初温30℃,保留时间4min,升温速率10℃/min,终温198℃,保留时间40min。分析结果见表1。实施例4将50mg的制备例2中得到的草酸包裹的负载型钴基催化剂加入到间歇式反应釜中,向该间歇式反应釜中加入4mmol原料2-辛烯,随后向其中充入5mpa合成气(co:h2=1:1),于393k下反应5h,反应后产物使用agilent-7890a气相色谱进行分析,该agilent-7890a气相色谱仪配有plot-al2o3柱(50m×530μm×1μm)、hp-innowax柱(30m×320μm×0.5μm),前后检测器均为fid检测器,采用联苯作为内标物对反应产物进行定量计算。初温30℃,保留时间4min,升温速率10℃/min,终温198℃,保留时间40min。分析结果见表1。实施例5将150mg的制备例7中得到的草酸包裹的负载型钴基催化剂加入到间歇式反应釜中,向该间歇式反应釜中加入2mmol原料1-己烯,随后向其中充入5mpa合成气(co:h2=1:1),于393k下反应5h,反应后产物使用agilent-7890a气相色谱进行分析,该agilent-7890a气相色谱仪配有plot-al2o3柱(50m×530μm×1μm)、hp-innowax柱(30m×320μm×0.5μm),前后检测器均为fid检测器,采用联苯作为内标物对反应产物进行定量计算。初温30℃,保留时间4min,升温速率10℃/min,终温198℃,保留时间40min。分析结果见表1。实施例6将100mg的实施例1中的负载型钴基催化剂回收利用,将催化剂从溶液中离心出来,随后进行烘干、焙烧、还原、柠檬酸的甲苯溶液包裹,然后重新加入到间歇式反应釜中,向该间歇式反应釜中加入4mmol原料1-己烯,随后向其中充入5mpa合成气(co:h2=1:1),于373k下反应5h,反应后产物使用agilent-7890a气相色谱进行分析,该agilent-7890a气相色谱仪配有plot-al2o3柱(50m×518μm×1μm)、hp-innowax柱(30m×320μm×0.5μm),前后检测器均为fid检测器,采用联苯作为内标物对反应产物进行定量计算。初温30℃,保留时间4min,升温速率10℃/min,终温198℃,保留时间40min。分析结果见表1。对比例1称取0.75gco(no3)2·6h2o并溶于100g去离子水中,得到活性金属co含量为3wt%的co(no3)2溶液。取5.0gsio2载体(比表面积150m2/g,孔容0.9m3/g,平均孔径23.4nm),将此载体浸渍于上述溶液中,室温下随着搅拌采用超声震荡器进行超声处理1h(其中,超声输出功率为800w,工作频率为40khz),超声结束后继续搅拌12h,然后将得到的混合溶液在旋转蒸发仪(转速为150rpm)中于真空条件、323k下蒸干溶剂,得到粉末状的固体混合物。将该固体粉末置于烘箱中于333k下干燥12h,然后在马弗炉中于673k下焙烧6h,得到co3o4/sio2,将co3o4/sio2在管式炉中于h2气氛、673k下还原6h,氢气流速为50ml/min,将还原后的催化剂用作对比的钴基催化剂。将100mg的上述钴基催化剂加入到间歇式反应釜中,然后向其中加入20mmol甲苯、4mmol原料1-己烯,并随后向其中充入5mpa合成气(co:h2=1:1),于373k下反应5h,反应后产物使用agilent-7890a气相色谱进行分析,该agilent-7890a气相色谱仪配有plot-al2o3柱(50m×518μm×1μm)、hp-innowax柱(30m×320μm×0.5μm),前后检测器均为fid检测器,采用联苯作为内标物对反应产物进行定量计算。初温30℃,保留时间4min,升温速率10℃/min,终温198℃,保留时间40min。分析结果见表1。对比例2称取0.75gco(no3)2·6h2o并溶于100g去离子水中,得到活性金属co含量为3wt%的co(no3)2溶液。取5.0gsi3n4载体(比表面积66m2/g,孔容0.475m3/g,平均孔径28nm),将此载体浸渍于上述溶液中,室温下随着搅拌采用超声震荡器进行超声处理2h(其中,超声输出功率为800w,工作频率为40khz),超声结束后继续搅拌12h,然后将得到的混合溶液在旋转蒸发仪(转速为150rpm)中于真空条件、323k下蒸干溶剂,得到粉末状的固体混合物。将该固体粉末置于烘箱中于333k下干燥12h,然后在马弗炉中于673k下焙烧6h,得到co3o4/si3n4,将co3o4/si3n4在管式炉中于h2气氛、673k下还原6h,氢气流速为50ml/min,将还原后的催化剂用作对比的钴基催化剂。将80mg的上述钴基催化剂加入到间歇式反应釜中,然后向其中加入20mmol甲苯、4mmol原料1-己烯,并随后向其中充入5mpa合成气(co:h2=1:1),于373k下反应5h,反应后产物使用agilent-7890a气相色谱进行分析,该agilent-7890a气相色谱仪配有plot-al2o3柱(50m×518μm×1μm)、hp-innowax柱(30m×320μm×0.5μm),前后检测器均为fid检测器,采用联苯作为内标物对反应产物进行定量计算。初温30℃,保留时间4min,升温速率10℃/min,终温198℃,保留时间40min。分析结果见表1。表1:实施例1-6和对比例1-2使用的催化剂的评价结果实施例钴流失量(%)转化率(%)总醛选择性(%)n:i实施例10.1122.1382.862.90实施例20.2733.7282.560.67实施例30.1142.3977.892.21实施例40.3025.380.202.65实施例50.0733.3282.322.56实施例61.1819.7483.523.16对比例140.0360.9890.463.06对比例238.1250.0080.123.02其中,n:i表示产物中的正构醛与异构醛的比例。由表1的上述结果可知,相比于对比例的钴基催化剂而言,本发明制备例1-7中制备的负载型钴基催化剂在烯烃氢甲酰化反应中具有较高的活性和选择性,且在反应过程中显著地抑制了活性金属钴的流失。上述具体实施方式,并不构成对本发明保护范围的限制。本领域技术人员应该明白的是,取决于设计要求和其他因素,可以发生各种各样的修改、组合、子组合和替代。任何在本发明的精神和原则之内所作的修改、等同替换和改进等,均应包含在本发明保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1