一种中药组合物的指纹图谱检测方法与流程
本发明涉及一种中药组合物的指纹图谱检测方法。
背景技术:
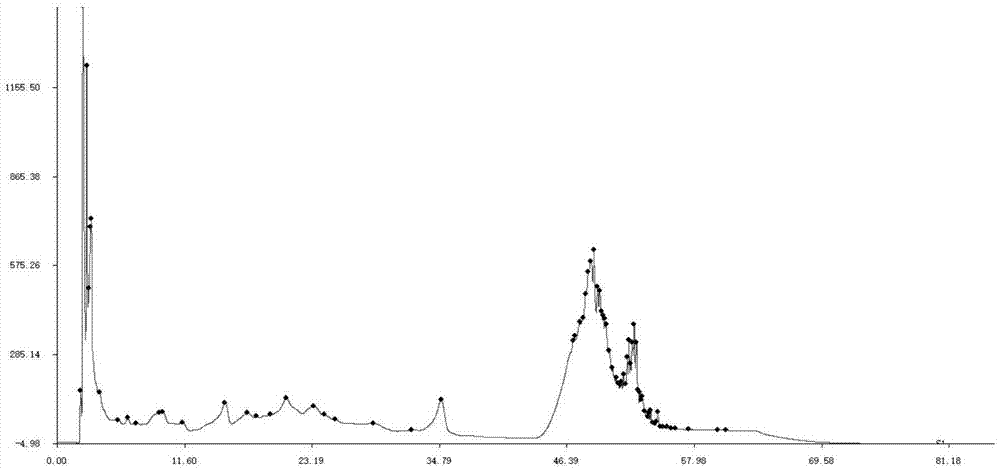
中药指纹图谱是指某些中药材或中药制剂经适当处理后,采用一定的分析手段,得到的能够标示其化学特征的色谱图或光谱图。中药指纹图谱是一种综合的,可量化的鉴定手段,它是建立在中药化学成分系统研究的基础上,主要用于评价中药材以及中药制剂半成品质量的真实性、优良性和稳定性。指纹图谱应该具备指纹性,即:(1)专属性强。指所制订的指纹图谱应该是该中药所独有的、能与其它中药相区别的,其反映的化学信息是具有高度的选择性的;(2)稳定性好。即中药的指纹图谱应该是从某中药的多批次中归纳出的共性,图谱中的共有峰或特征峰应相对稳定;(3)重现性好。所制订的指纹图谱在规定条件下应能再现指纹特征(如共有峰数目、大小、位置等),其误差应在允许的范围内。只有这样,所制订的指纹图谱才具有实用价值,才可以有效控制药品的质量。中药及其制剂均为多组分复杂体系,建立针对某一中药制剂的指纹图谱现有技术并没有直接的教导,需要大量的研究。中国专利申请201710327985.2公开了一种用于治疗儿童注意力缺陷多动障碍的中药组合物的制备方法。该中药组合物由太子参、熟地黄、枸杞子、五味子、远志、石菖蒲、茯苓药味组成。该专利并未提供其中药制剂的质量检测方法。本发明提供了该中药组合为的检测方法,有效控制该药品的质量。技术实现要素:本发明提供一种中药组合物的指纹图谱检测方法,该方法包括如下步骤:供试品溶液的制备:取中药组合物,制备供试品溶液;对照品溶液的制备:取五味子醇甲,制备对照品溶液;色谱条件:以乙腈-磷酸水溶液为流动相,梯度洗脱,所述磷酸水溶液浓度为0.05%~0.2%;测定法:分别吸取对照品溶液与供试品溶液,注入液相色谱仪,测定,即得。在具体的实施方式中,所述梯度洗脱程序为:优选的,所述梯度洗脱程序为:在具体的实施方式中,所述磷酸水溶液浓度为0.1%。在具体的实施方式中,所述供试品溶液的制备:取中药组合物,加有机溶剂提取,得供试品溶液。所述有机溶剂包括正丁醇、乙酸乙酯、丙酮、50%~100%的甲醇溶液或50%~100%的乙醇溶液;优选乙酸乙酯;所述提取方法包括超声提取、回流提取、浸渍提取、微波提取或渗漉提取;所述提取次数为1~3次;溶剂倍量为25~50倍量(g/ml)。在具体的实施方式中,所述对照品溶液的制备:取五味子醇甲,加有机溶剂溶解,得对照品溶液。所述有机溶剂包括50%~100%的甲醇溶液或0.1%磷酸水溶液-乙腈(40:60)。在具体的实施方式中,所述柱温为30℃~40℃,所述检测波长为210nm~230nm。在具体的实施方式中,所述检测方法包括如下步骤:供试品溶液的制备:取中药组合物,加30倍量乙酸乙酯超声提取1h,滤过,得滤液①;滤渣用乙酸乙酯洗涤,滤过,得滤液②,合并滤液①、②,回收乙酸乙酯;残渣用0.1%磷酸水溶液-乙腈(40:60)溶解,得供试品溶液;对照品溶液的制备:取五味子醇甲,加0.1%磷酸水溶液-乙腈(40:60)溶解,得供试品溶液;色谱条件:以乙腈-0.1%磷酸水溶液为流动相,梯度洗脱,洗脱程序为:测定法:分别吸取对照品溶液与供试品溶液,注入液相色谱仪,测定,即得。本发明所述中药组合物由如下重量份的原料药制成:太子参5~20重量份、熟地5~20重量份、枸杞子4~10重量份、五味子4~10重量份、远志4~10重量份、石菖蒲4~10重量份、茯苓5~20重量份;具体的,所述中药组合物由如下重量份的原料药制成:太子参10重量份、熟地10重量份、枸杞子6重量份、五味子6重量份、远志6重量份、石菖蒲6重量份、茯苓10重量份。所述中药组合物按如下方法制备:步骤a,五味子加溶剂提取,得提取物a;步骤b,石菖蒲提取挥发油,得挥发油b和水液c;步骤c,太子参、熟地、枸杞、茯苓、远志合并加溶剂提取,提取液与步骤b的水液c合并醇沉,取滤液浓缩,得提取物d;步骤d,将提取物a、挥发油b和提取物d混匀,即得。步骤a和c所述提取溶剂为水或50%~90%乙醇溶液;步骤b采用水蒸气蒸馏法提取挥发油,挥发油用倍他环糊精包合;步骤c所述醇沉浓度为50%~70%;所述中药组合物可进一步加入常规制剂辅料制成颗粒剂、片剂、胶囊剂、丸剂、口服液等常规剂型。采用本发明检测方法对11批样品进行检测,共指认出30个共有峰,其中t=21.876的信号峰为五味子醇甲的信号峰,各批样品色谱相似度均在0.99以上;经方法学考察,本发明检测方法稳定性、精密度、重复性均符合要求。附图说明图1为色谱条件一的色谱图;图2为色谱条件二的色谱图(分别为s1和s2两个批次样品的色谱图);图3为色谱条件三的色谱图(分别为210nm、220nm、228nm色谱图);图4为色谱条件四的色谱图(分别为s1、s2和s3三个批次样品的色谱图);图5为系统适应性考察色谱图(分别为中药组合物-五味子醇甲、中药组合物、五味子醇甲、空白组的色谱图);图6为11批本发明中药组合物色谱指纹图谱;图7为本发明中药组合物对照指纹图谱。具体实施方式实施例1原料:太子参10g、熟地10g、枸杞子6g、五味子6g、远志6g、石菖蒲6g、茯苓10g;制备方法:五味子加10倍量60%乙醇,加热回流提取3次,每次2小时,合并提取液,浓缩,减压干燥;石菖蒲加8倍量水,加热蒸馏8小时提取挥发油,所得挥发油用环糊精包合(包合工艺为:油:β-cd比例1:8,40℃,搅拌2小时);太子参、熟地、枸杞、茯苓、远志加12倍量水,回流提取3次,每次1.5小时,所得药液与石菖蒲水液合并,浓缩至相对密度1.15~1.20(60℃),加乙醇至含醇量为60%,醇沉,过滤,浓缩,减压干燥;取各提取物干膏粉均匀,即得中药组合物干膏。指纹图谱检测方法:1.仪器与试剂1.1主要仪器色谱系统:agilenttechnologies1200series(b1322adegasser,g1311aquartpump,g1320aals,g1316atcc,g1315ddad);超声波清洗仪:15b065,250w,宁波新芝生物科技股份有限公司;旋转蒸发仪:eyelan1100;1.2试药五味子醇甲:dst161117-010,纯度≥98%,esite;中药组合物干膏(按照本实施例制备);1.3试剂乙酸乙酯:20101112,纯度≥99.5%,天津市光复科技发展有限公司;正丁醇:20101112,纯度≥99.0%,北京化工厂;丙酮:20110104,纯度≥99.5%,北京化工厂;乙腈:lot175164,纯度≥99.9%,fisher;磷酸:20170616,纯度≥85%,北京化工厂;纯净水:20180208,娃哈哈。2.色谱条件2.1色谱条件一固定相:agilentzorboxsb-c18,4.6×250mm,5μm;流动相:甲醇-水系统,梯度洗脱程序如表1:表1色谱条件一t/mina(水)/%b(甲醇)/%09551095520802040802050595605957095580955供试品溶液的制备:取中药组合物干膏粉末5g,置100ml锥形瓶中,加50ml甲醇,超声提取20min,静置10min,0.45μm针孔滤膜过滤,取续滤液作为待测样品,冷藏保存备用,测定时放置至室温。检测波长:228nm;进样量10μl;流动性流速为1.0ml/min,柱温为35℃。取上述供试品,于色谱条件下分析,记录各色谱信息。结果见图1。由图1可见,在高水相流动相条件下对样品进行分析,采用甲醇提取时大极性物质含量很高,且信号峰丰富,采用agilentzorboxsb-c18色谱柱无法有效分离鉴别大极性成分,且对于sb-c18色谱柱不宜长期采用高水相流动相进行分析,因此不建议采用此色谱柱。2.2色谱条件二固定相:agilentzorboxsb-aqc18,4.6×250mm,5μm;流动相:甲醇-0.1%磷酸水溶液系统,梯度洗脱程序如表2;表2色谱条件二供试品溶液制备:取中药组合物干膏粉末5g,置100ml锥形瓶中,加50ml乙酸乙酯,超声提取60min,提取2次,0.45μm针孔滤膜过滤,取续滤液减压回收溶剂,加甲醇适量使溶解,并用甲醇稀释定容至10ml,0.45μm针孔滤膜过滤,取续滤液作为待测样品,冷藏保存备用,测定时放置至室温。检测波长:228nm;进样量10μl;流动性流速为1.0ml/min,柱温为35℃。取上述供试品两个批次(s1和s2),于色谱条件下分析,记录各色谱信息。结果见图2。由图2可见,更换色谱柱为agilentzorboxsb-aqc18,且将样品制备时的甲醇改为乙酸乙酯后,大极性物质(保留时间≤10min)的信号峰明显减少和降低,但甲醇的溶剂峰及杂峰信号较多,对样品分析干扰较大,因此拟采用乙腈作为流动相中的有机相,进一步考察分析条件。2.3色谱条件三固定相:agilentzorboxsb-aqc18,4.6×250mm,5μm;流动相:乙腈-0.1%磷酸水溶液系统,梯度洗脱程序如表3:表3色谱条件三供试品溶液的制备:取中药组合物干膏粉末1g,置100ml锥形瓶中,加50ml乙酸乙酯,超声提取60min,0.45μm针孔滤膜过滤,取续滤液减压回收溶剂,精密加0.1%磷酸-乙腈混合溶液(40-60)2.5ml使溶解,0.45μm针孔滤膜过滤,取续滤液作为待测样品,冷藏保存备用,测定时放置至室温。检测波长:210nm、220nm、228nm;进样量10μl;流动性流速为1.0ml/min,柱温为35℃。取上述供试品,于色谱条件下分析,记录各色谱信息。结果见图3。将流动相的有机相改为乙腈后,且比较210、220、228nm不同波长下的信号峰,发现228nm下溶剂基线平稳,样品信号峰丰富,分离度较好,溶剂峰及杂质影响较少,因此后期可以在此基础上进行分析条件优化。2.4色谱条件四固定相:zorbaxsb-aqc-184.6mmid×250mm(5μm);流动相:乙腈-0.1%磷酸系统,梯度程序如表4:表4色谱条件四t/min乙腈/%0.1%磷酸水溶液/%03070535656555457060607595578955833070903070;供试品溶液的制备:取中药组合物干膏粉末1g,置100ml锥形瓶中,加50ml乙酸乙酯,超声提取60min,0.45μm针孔滤膜过滤,取续滤液减压回收溶剂,精密加0.1%磷酸-乙腈混合溶液(40-60)2.5ml使溶解,0.45μm针孔滤膜过滤,取续滤液作为待测样品,冷藏保存备用,测定时放置至室温。检测波长:228nm;进样体积10μl;流动相流速:1ml/min;柱温:40℃。取上述供试品三个批次(分别为s1、s2、s3),于色谱条件下分析,记录各色谱信息。结果见图4。如图4可见,该色谱条件下分离效果最佳。选择该色谱条件进行检测。3.供试品溶液和对照品溶液的制备3.1提取溶剂考察分别以正丁醇、乙酸乙酯、丙酮为提取溶剂,考察提取效果,结果表明:提取溶媒为正丁醇时,峰信号最少,且信号强度弱;当提取溶媒为丙酮时,峰信号最多,且信号强度最大,但丙酮在减压回收过程中因沸点高不易回收;当溶媒为乙酸乙酯时,峰信号较丰富,信号强度适中,溶剂回收时容易回收(减压40-50℃),因此,将提取溶媒定为乙酸乙酯。3.2供试品溶液制备方法确定:称取干膏粉末0.3g,置100ml锥形瓶中,加乙酸乙酯9ml,提取1次,每次超声提取1h,静置至室温,过滤(玻璃注射器+0.45μm有机针孔滤膜),得滤液①保存备用;分别取乙酸乙酯9ml,分2-3次洗涤滤渣,过滤(玻璃注射器+0.45μm有机针孔滤膜),得滤液②,合并滤液①、②,于50℃水浴减压回收乙酸乙酯;残渣用10ml0.1%磷酸-乙腈(40-60)溶解,0.22μm滤膜过滤,取续滤液作为供试品,4℃保存待测,测定前取出放置至室温。3.3对照品溶液的制备:称取五味子醇甲10mg,加0.1%磷酸-乙腈(40-60)溶解,制成1mg/ml的五味子醇甲对照品溶液母液,精密移取母液0.1ml,置10ml容量瓶中,加0.1%磷酸-已经(40-60)溶解,稀释,定容,摇匀,0.22μm滤膜过滤,取续滤液作为供试品,4℃保存待测,测定前取出放置至室温。4方法学考察4.1系统适应性称取中药组合物干膏粉末0.3g,按“3.2”项下制备供试品溶液,4℃保存待测,测定前取出放置至室温;量取乙酸乙酯10ml,按“3.2”项下制备溶媒空白溶液,4℃保存待测,测定前取出放置至室温;称取五味子醇甲10mg,按“3.2”项下制备对照品溶液,4℃保存待测,测定前取出放置至室温;称取中药组合物干膏粉末0.3g,按“3.2”项下制备供试品溶液并精密加入五味子醇甲母液0.1ml,4℃保存待测,测定前取出放置至室温。取上述供试品,分别于“2.4色谱条件四”项下分析,观察色谱信息,并参考液相色谱参数分析色谱条件的系统适应性。色谱图见图5。通过比较五味子醇甲、中药组合物、中药组合物-五味子醇甲三组信号发现,t=21.848min的信号峰为五味子醇甲,该色谱峰与前后色谱峰的分离度为2.32,理论塔板数为40197,选择性为1.05,符合要求;综合上述信息表明,本分析方法系统适应性及专属性良好,符合指纹图谱研究的要求。4.2稳定性取供试品溶液和对照品溶液,每隔3h进样,样品与对照品中各色谱峰保留时间rsd值均小于1%;对照品峰面积rsd值均小于0.5%;供试品峰面积绝大部分rsd值均小于5%。表明供试品溶液和对照品溶液在24h内稳定。4.3精密度取供试品溶液,按确定的色谱条件连续进样6次,各色谱峰保留时间rsd均小于0.1%;主要色谱峰峰面积rsd小于2%,表明色谱分析条件精密度良好。4.4重复性取本发明中药组合物6份,按已确定的“供试品溶液制备方法”制备,注入液相色谱仪检测,测定各主要色谱峰峰面积的rsd均小于10%,表明方法重复性较好。5样品测定取11批本发明中药组合物样品,按“3.2”项下制备供试品溶液,在上述色谱条件下进样分析,得到11批本发明中药组合物的指纹图谱,导入到相似度软件拟合生成对照指纹图谱,结果详见图6和图7,相似度结果见表5。表511批中药组合物相似度评价相似度软件分析结果表明,11批样品中指认出30个共有峰,其中t=21.876的信号峰为五味子醇甲的信号峰,各批样品色谱相似度均在0.99以上。实施例2供试品溶液的制备:称取干膏粉末0.3g,置100ml锥形瓶中,加丙酮9ml,提取1次,每次超声提取1h,静置至室温,过滤(玻璃注射器+0.45μm有机针孔滤膜),得滤液①保存备用;分别取丙酮9ml,分2-3次洗涤滤渣,过滤(玻璃注射器+0.45μm有机针孔滤膜),得滤液②,合并滤液①、②,于50℃水浴减压回收乙酸乙酯;残渣用10ml0.1%磷酸-乙腈(40-60)溶解,0.22μm滤膜过滤,取续滤液作为供试品,4℃保存待测,测定前取出放置至室温。对照品溶液的制备:称取五味子醇甲10mg,加0.1%磷酸-乙腈(40-60)溶解,制成1mg/ml的五味子醇甲对照品溶液母液,精密移取母液0.1ml,置10ml容量瓶中,加0.1%磷酸-已经(40-60)溶解,稀释,定容,摇匀,0.22μm滤膜过滤,取续滤液作为供试品,4℃保存待测,测定前取出放置至室温。色谱条件固定相:zorbaxsb-aqc-184.6mmid×250mm(5μm)流动相:乙腈-0.2%磷酸系统,梯度程序如下:柱温:40℃;流动相流速:1ml/min;进样体积20μl。测定法:分别精密吸取对照品溶液与供试品溶液各20μl,注入超高液相色谱仪,测定,记录色谱图,即得。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1