一种液相自吸附SnO2/生物碳复合材料的制备方法及应用与流程
本发明涉及一种形貌可控的钠离子负极碳材料的制备方法,具体涉及一种液相自吸附SnO2/生物碳复合材料的制备方法及应用。
背景技术:
随着科技的发展和信息社会的到来,对化学电源的要求强度逐渐增大。锂离子电池具有高电压、高比能量的特点,因此在便携式电源应用中得到长足发展,但锂元素昂贵且地壳中含量少,随着其逐渐应用于电动汽车,锂的需求量将大大增加。而锂的储量有限且储藏分布不均匀,会制约长寿命储能电池的大规模发展[刘春娜.国外钠离子电池研究进展[J].电源技术,2014,38(1):12-13.]0。因此开发其他种类电池势在必行。
钠离子电池是目前最具研究价值的电池之一。与锂离子电池相比,其优势在于其密度高,这意味着它们质量更大可以储存更多能量,适合用于大规模储能。同时,其原料资源丰富易得,成本低廉;能用来分解电势更低的电解质溶剂及电解质盐,电解质的选择范围更宽;有相对稳定的电化学性能,使用更加安全。因此,它们能负担起可持续绿色能源开发的重任,具有强大的生命力和发展潜质[叶飞鹏,王莉,连芳等.钠离子电池研究进展[J].化工进展,2013,32(8):1789-1795.]。
但是,钠离子电池负极材料的筛选面临一些问题。由于钠离子半径大于锂离子半径,传统商品化的锂离子负极材料石墨层间距过小,并不适合钠离子的嵌入和脱出[苗艳丽,刘兴江.钠离子电池负极材料研究进展[J].电源技术,2015,39(2):23-25.],需要具有更大层间距或孔隙的碳材料及合金等其它储钠材料。钠离子电池的负极材料主要包括碳基负极材料、钛基负极材料和合金类负极材料等。在对负极材料的研究中,发现氧化锡基负极材料具有储钠容量高、与Na+反应电势低、能量密度高、安全性能好、低成本、无污染的特点。不仅如此,二氧化锡还具有丰富的资源,价格也便宜,是一类很好的钠离子电池负极材料。[杨旭.高比容量锡基负极材料的合成与电化学储能性质的研究[D].吉林大学,2016.]
然而,氧化锡基负极材料存在一些问题,(I)在Na+脱嵌的过程中电极存在着巨大的体积变化;(II)氧化锡作为钠离子电池负极材料,与Na+的第一步电化学反应不可逆,造成了比较大的不可逆容量,使这类材料库伦效率低下,这极大地影响了这类材料在实际中的应用。[方国清.锂离子电池锡基负极材料的合成及其性能表征[D].苏州大学,2013.]如果以碳材料作为基体,将氧化锡负极材料与各种类型的碳材料复合,从而提高复合材料的结构稳定性,改善体积膨胀的问题,并且增加材料的导电性,使电子迁移速度提高,更多的电极材料将参与到电化学反应中,提高库伦效率。[杨承昭.锂离子电池氧化锡/碳复合负极材料的制备性能研究[D].广东工业大学,2011.]
目前,适应于与氧化锡基负极材料复合的碳材料主要有石墨类材料和无定形材料等。在无定形材料中,利用天然存在的植物制备的生物碳材料具有制备材料天然、制备方法简单合理、性能优良、价格低廉、节能环保等优点,运用到电池当中,将会有更大的应用前景。[刘浩.铁和锡基氧化物及其碳复合材料作为锂离子电池负极材料的研究与应用[D].浙江大学,2016.]
技术实现要素:
为解决现有技术存在的问题,本发明提供了一种液相自吸附SnO2/生物碳复合材料的制备方法及应用。得到一种生物碳与SnO2复合粉体,该材料应用于钠离子电池具备良好的循环性能。
本发明目的是通过以下技术方案来实现的:
一种液相自吸附SnO2/生物碳复合材料的制备方法,包括以下步骤:
(1)将采集的绒毛类植物采用反应酸浸法进行羟基化处理后,去籽,在管式炉中煅烧,先按照1~10℃·min-1速率升温至500~1400℃,再保温煅烧3~8h,得到物质A;
(2)将物质A用蒸馏水冲洗至pH=7,抽滤、干燥后得到物质B;
(3)将SnCl2·2H2O与酸以质量比1:(1~10)混合均匀,得到物质C;
(4)将物质C和物质B按照质量比1:(1~10)混合,得到溶液D;
(5)采用微波-紫外-超声的合成模式,将微波、紫外灯和超声波同时作用于溶液D,使溶液D以5~10℃/min的升温速率由室温升温到100~320℃,微波功率为200~400W,保温至反应完全,然后冷却到室温,得到物质E;
(6)将物质E洗至pH=7,抽滤、干燥后得到生物碳/SnO2复合粉体。
步骤(1)中,反应酸浸法是指将采集的绒毛类植物经硫酸与水按照体积比1:(1~5)进行处理。
步骤(1)中,煅烧的保护气氛为氩气,氩气气流流速0.1~0.6sccm·min-1。
步骤(3)中,所述的酸为H2SO4、HCL、HNO3、H3PO4、HF、H2CO3中的任意一种。
所述的复合材料中,生物碳呈现出管状结构,管与管之间相互连通,SnO2颗粒均匀的包覆在碳管表面及内部,形成良好的生物碳包覆SnO2结构形状;SnO2颗粒粒径为100~200nm。
所述的绒毛类植物为枇杷、杜鹃花、菊花、蒲公英。
所述的制备方法得到的液相自吸附SnO2/生物碳复合材料在钠离子电池负极材料中的应用。
相对于现有技术,本发明具有以下优点:
本发明的目的是以绒毛类植物蒲公英为原料,将蒲公英进行硫酸浸泡,高温煅烧处理与不同类型的酸处理的SnCl2·2H2O混合均匀,通过微波-紫外-超声法,得到一种生物碳与SnO2复合粉体,该材料应用于钠离子电池具备良好的循环性能。具体的优点表现为:(1)采用生物碳的天然结构优势和二氧化锡的理论容量大的优点,采用微波-紫外-超声法,制备工艺简单。(2)通过控制酸的浓度和采用反应酸浸法等对生物碳进行羟基化处理,增加碳骨架上的羟基与羧基数目,得到一个易于承载二氧化锡的良好碳骨架。(3)通过大量的实验研究分析,采用控制温度、pH等方法优化了水热反应的步骤,简化了制备工艺,揭示了复合材料制备工艺、微观结构与产品的电化学性能之间的关系,实现了复合材料的可控制备,具有良好的理论性与指导意义。(4)利用复合物中氧化锡基负极材料具有较高的储钠容量,远大于石墨和钛基负极材料的理论容量,在一定程度上提高了电化学性能。(5)制备的SnO2与生物碳复合材料中,生物碳的结构上存在羧基和羟基,复合体表面及内部会产生吸附形成碳管包覆结构,在反应中会使钠与二氧化锡充分反应,有效缓解二氧化锡的膨胀现象。(6)将生物碳煅烧前进行预羟基化处理,生物绒毛类植物表面含有较多的纤维素、半纤维素、木质素,酸处理破坏了半纤维素和纤维素、木质素之间的连接,同时也破坏了部分的纤维素,通过控制酸处理的浓度可以较好的保持生物质骨架,在煅烧的过程中可以较好的保持这一骨架,使得生物碳成为一个好的负载二氧化锡的载体。(7)本发明中生物碳为微米结构,二氧化锡为纳米结构,具有较大的比表面积,且电化学反应过程中产生的氧化钠可作为基体阻止锡纳米颗粒的团聚,有利于钠与二氧化锡反应的进行,使得钠离子电池的反应更加充分,提高电池的电化学性能。
【附图说明】
图1为不同硝酸浓度下SnO2/生物碳复合产物的SEM图;图1a硝酸浓度为1mol;图1b硝酸浓度为1.5mol;图1c硝酸浓度为2mol;
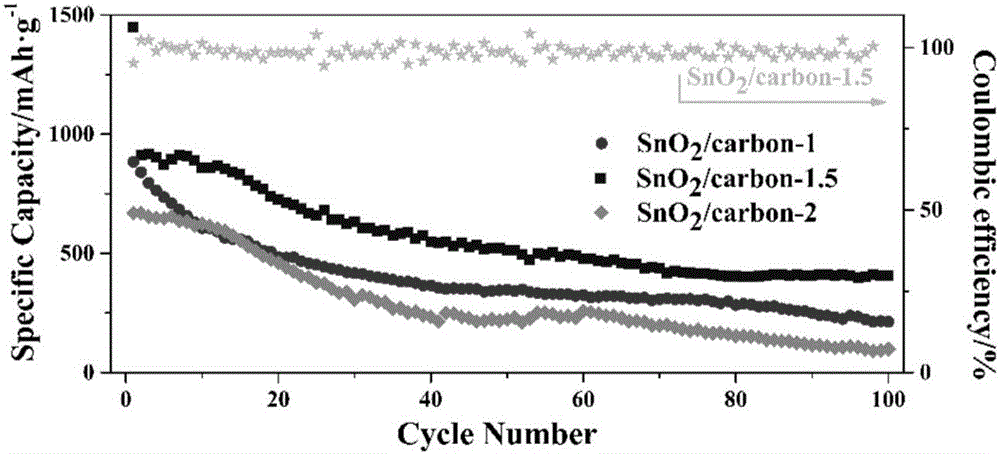
图2为电流密度为50mA·g-1时,所有产物的循环100圈稳定性能图;
图3为电流密度为50mA·g-1,100mA·g-1,200mA·g-1,500mA·g-1,1000mA·g-1再回到50mA·g-1时产物的倍率性能图。
【具体实施方式】
一种液相自吸附SnO2/生物碳复合材料的制备方法,其特征包括以下步骤:
(1)将采集的绒毛类植物经硫酸与水按照体积比1:(1~5)处理后,去籽,通过低温管式炉中煅烧,煅烧温度为500~1400℃,升温速率为1~10℃·min-1,保温时间为3~8h,采用氩气气体进行保护,气流流速0.1~0.6sccm·min-1,得到悬浮液A;
(2)将悬浮液A用蒸馏水冲洗至pH=7,抽滤、干燥后得到产物B;
(3)将SnCL2·2H2O与H2SO4、HCL、HNO3、H3PO4、HF、H2CO3中的任意一种以质量比1:1~1:10混合均匀,得到产物C;
(4)将产物C和产物B按照1:1~1:8混合,得到D溶液;
(5)采用微波-紫外-超声的合成模式,将微波、紫外灯和超声波同时作用于D溶液,使D溶液以10℃/min的升温速率由室温升温到100~320℃,保温10~100min,然后冷却到室温,得到产物E;
(6)将产物E洗至pH=7,抽滤、干燥后得到生物碳/SnO2复合粉体。
下面结合具体实施例,对本发明的具体实施方式进行详细阐述,但本发明不限于该实施例。为了使公众对本发明有彻底的了解,在以下本发明优选施例中详细说明具体的细节。
下面结合附图及具体实施例对本发明进行详细说明。
实施例1
(1)将采集的绒毛类植物(蒲公英)经硝酸与水按照体积比1:5处理后,去籽,通过低温管式炉中煅烧,煅烧温度为1000℃,升温速率为5℃·min-1,保温时间为5h,采用氩气气体进行保护,气流流速0.6sccm·min-1,得到悬浮液A;
(2)将悬浮液A用蒸馏水冲洗至pH=7,抽滤、干燥后得到物质B;
(3)将SnCl2·2H2O与H2SO4以质量比1:1混合均匀,得到物质C;
(4)将物质C和物质B按照质量比1:1混合,得到溶液D;
(5)采用微波-紫外-超声的合成模式,将微波、紫外灯和超声波同时作用于溶液D,使溶液D以5℃/min的升温速率由室温升温到200℃,保温60min,然后冷却到室温,得到物质E;
(6)将物质E洗至pH=7,抽滤、干燥后得到生物碳/SnO2复合粉体。
实施例2
(1)将采集的绒毛类植物经硝酸与水按照体积比1:5处理后,去籽,通过低温管式炉中煅烧,煅烧温度为1000℃,升温速率为5℃·min-1,保温时间为5h,采用氩气气体进行保护,气流流速0.6sccm·min-1,得到悬浮液A;
(2)将悬浮液A用蒸馏水冲洗至pH=7,抽滤、干燥后得到物质B;
(3)将SnCl2·2H2O与H2SO4以质量比1:1.5混合均匀,得到物质C;
(4)将物质C和物质B按照质量比1:1混合,得到溶液D;
(5)采用微波-紫外-超声的合成模式,将微波、紫外灯和超声波同时作用于溶液D,使溶液D以5℃/min的升温速率由室温升温到200℃,保温60min,然后冷却到室温,得到物质E;
(6)将物质E洗至pH=7,抽滤、干燥后得到生物碳/SnO2复合粉体。
实施例3
(1)将采集的绒毛类植物经硝酸与水按照体积比1:5处理后,去籽,通过低温管式炉中煅烧,煅烧温度为1000℃,升温速率为5℃·min-1,保温时间为5h,采用氩气气体进行保护,气流流速0.6sccm·min-1,得到悬浮液A;
(2)将悬浮液A用蒸馏水冲洗至pH=7,抽滤、干燥后得到物质B;
(3)将SnCl2·2H2O与H2SO4以质量比1:1.5混合均匀,得到物质C;
(4)将物质C和物质B按照质量比1:2混合,得到溶液D;
(5)采用微波-紫外-超声的合成模式,将微波、紫外灯和超声波同时作用于溶液D,使溶液D以5℃/min的升温速率由室温升温到200℃,保温60min,然后冷却到室温,得到物质E;
(6)将物质E洗至pH=7,抽滤、干燥后得到生物碳/SnO2复合粉体。
实施例4
(1)将采集的绒毛类植物经硫酸与水按照体积比1:5处理后,去籽,通过低温管式炉中煅烧,煅烧温度为1000℃,升温速率为5℃·min-1,保温时间为5h,采用氩气气体进行保护,气流流速0.6sccm·min-1,得到悬浮液A;
(2)将悬浮液A用蒸馏水冲洗至pH=7,抽滤、干燥后得到物质B;
(3)将SnCl2·2H2O与H2SO4以质量比1:5混合均匀,得到物质C;
(4)将物质C和物质B按照质量比1:5混合,得到溶液D;
(5)采用微波-紫外-超声的合成模式,将微波、紫外灯和超声波同时作用于溶液D,使溶液D以10℃/min的升温速率由室温升温到200℃,保温60min,然后冷却到室温,得到物质E;
(6)将物质E洗至pH=7,抽滤、干燥后得到生物碳/SnO2复合粉体。
实施例5
(1)将采集的绒毛类植物经硫酸与水按照体积比1:5处理后,去籽,通过低温管式炉中煅烧,煅烧温度为1000℃,升温速率为5℃·min-1,保温时间为5h,采用氩气气体进行保护,气流流速0.6sccm·min-1,得到悬浮液A;
(2)将悬浮液A用蒸馏水冲洗至pH=7,抽滤、干燥后得到物质B;
(3)将SnCL2·2H2O与HNO3以质量比1:5混合均匀,得到物质C;
(4)将物质C和物质B按照质量比1:5混合,得到溶液D;
(5)采用微波-紫外-超声的合成模式,将微波、紫外灯和超声波同时作用于溶液D,使溶液D以10℃/min的升温速率由室温升温到200℃,保温60min,然后冷却到室温,得到物质E;
(6)将物质E洗至pH=7,抽滤、干燥后得到生物碳/SnO2复合粉体。
实施例6
(1)将采集的绒毛类植物经硫酸与水按照体积比1:5处理后,去籽,通过低温管式炉中煅烧,煅烧温度为1000℃,升温速率为5℃·min-1,保温时间为6h,采用氩气气体进行保护,气流流速0.6sccm·min-1,得到悬浮液A;
(2)将悬浮液A用蒸馏水冲洗至pH=7,抽滤、干燥后得到物质B;
(3)将SnCL2·2H2O与H2SO4以质量比1:10混合均匀,得到物质C;
(4)将物质C和物质B质量比按照1:10混合,得到溶液D;
(5)采用微波-紫外-超声的合成模式,将微波、紫外灯和超声波同时作用于溶液D,使溶液D以8℃/min的升温速率由室温升温到200℃,保温60min,然后冷却到室温,得到物质E;
(6)将物质E洗至pH=7,抽滤、干燥后得到生物碳/SnO2复合粉体。
参见图3,从图3可以看出在200℃的煅烧温度下,控制物质C和物质B质量比1:10混合时,所得物质的低倍率SEM中,材料管状结构已被破坏,分成两个部分,这大大增大了材料的比表面积。
参见图3,从图3可以看出在200℃的煅烧温度下,控制物质C和物质B质量比1:10混合时,所得物质的高倍率SEM中,材料的外部和内部,分别有不同程度的剥落,表明酸对该材料的刻蚀程度。
实施例7
(1)将采集的绒毛类植物经硫酸与水按照体积比1:1处理后,去籽,通过低温管式炉中煅烧,煅烧温度为500℃,升温速率为1℃·min-1,保温时间为3h,采用氩气气体进行保护,气流流速0.1sccm·min-1,得到悬浮液A;
(2)将悬浮液A用蒸馏水冲洗至pH=7,抽滤、干燥后得到产物B;
(3)将SnCL2·2H2O与H2SO4以质量比1:1混合均匀,得到产物C;
(4)将产物C和产物B按照1:1混合,得到D溶液;
(5)采用微波-紫外-超声的合成模式,将微波、紫外灯和超声波同时作用于D溶液,使D溶液以10℃/min的升温速率由室温升温到100℃,保温10min,然后冷却到室温,得到产物E;
(6)将产物E洗至pH=7,抽滤、干燥后得到生物碳/SnO2复合粉体。
实施例8
(1)将采集的绒毛类植物经硫酸与水按照体积比1:5处理后,去籽,通过低温管式炉中煅烧,煅烧温度为1400℃,升温速率为10℃·min-1,保温时间为8h,采用氩气气体进行保护,气流流速0.6sccm·min-1,得到悬浮液A;
(2)将悬浮液A用蒸馏水冲洗至pH=7,抽滤、干燥后得到产物B;
(3)将SnCL2·2H2O与H2CO3以质量比1:10混合均匀,得到产物C;
(4)将产物C和产物B按照1:8混合,得到D溶液;
(5)采用微波-紫外-超声的合成模式,将微波、紫外灯和超声波同时作用于D溶液,使D溶液以10℃/min的升温速率由室温升温到320℃,保温100min,然后冷却到室温,得到产物E;
(6)将产物E洗至pH=7,抽滤、干燥后得到生物碳/SnO2复合粉体。
图1为不同硝酸浓度下SnO2/生物碳复合产物的SEM图,从图1a中可以看出,在硝酸浓度为1mol时,管状表面有少量氧化锡附着,这主要由于微波对生物碳管状表面和内部进行了处理,形成了较多的含氧官能团,它们极易与氧化锡形成化学键,促进氧化锡在表面的吸附;在图1b中,硝酸浓度为1.5mol时,氧化锡完全对碳管进行了包覆,表面和内部均有较多的氧化锡附着,说明此时产生的氧化还原反应最多;在图1c中,硝酸浓度为2mol时,硝酸浓度最大,氧化锡在碳管表面发生了剥离,使得它在碳管表面的附着量也大大减少。
图2为电流密度为50mA·g-1时,所有产物的循环100圈稳定性能图。从图中可以看出SnO2/carbon-1.5展示了较好的循环稳定性和初始库伦效率。前十圈可逆容量为800mAh·g-1左右,虽然随着循环次数的增加容量有衰减,但100圈后其可逆容量仍然有435mAh·g-1左右,显著高于水热产物的性能293mAh g-1。这表明了三合一法成功将SnO2颗粒吸附到生物碳管中,生物碳管的中空结构缓解了SnO2颗粒的体积膨胀,显示了较好的循环稳定性。而SnO2/carbon-1和SnO2/carbon-2较差的性能主要由于SnO2/carbon-1样品的SnO2颗粒负载较少和SnO2/carbon-2样品的SnO2附着力减弱,发生脱落。
图3为电流密度为50mA·g-1,100mA·g-1,200mA·g-1,500mA·g-1,1000mA·g-1再回到50mA·g-1时产物的倍率性能图。
从图中同样可以观察到SnO2/carbon-1.5展示了较好的倍率性能,SnO2/carbon-1其次,SnO2/carbon-2最差。这和循环性能图相对应。在50mA·g-1电流密度下,SnO2/carbon-1.5的可逆容量为798mAh·g-1左右;当电流密度增大为1000mA·g-1时,SnO2/carbon-1.5的可逆容量为250mAh·g-1左右;当电流密度回到50mA·g-1时,容量又回到了435mAh·g-1左右,展示了较好的倍率性能。这主要由于SnO2颗粒可以和生物碳较好的结合,减少了电流密度增大时对氧化锡颗粒的破坏,同时完整的碳管结构有利于电解液的进入,为钠离子的传输提供了通道,从而提升了其倍率性能。
以上,仅为本发明的较佳实施例,并非仅限于本发明的实施范围,凡依本发明专利范围的内容所做的等效变化和修饰,都应为本发明的技术范畴。
- 还没有人留言评论。精彩留言会获得点赞!