包含3,4-亚乙基二氧噻吩的电解质、包含其的电解电容器及电子材料原材料的制作方法
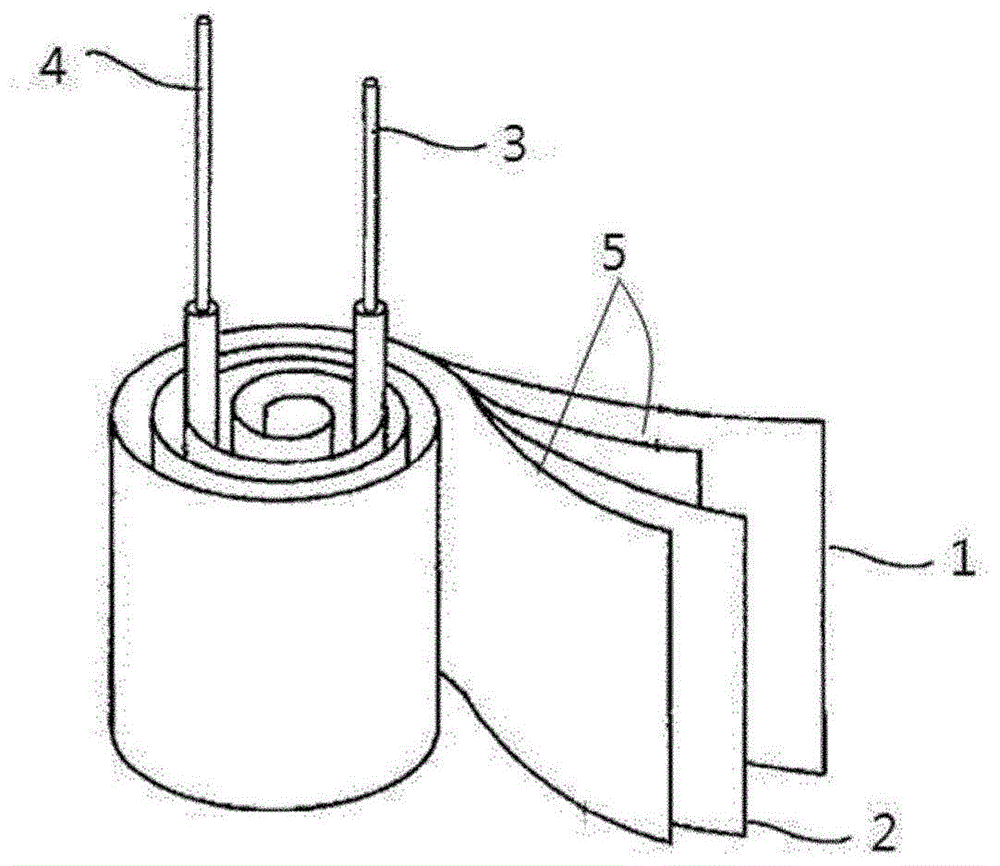
本发明涉及包含3,4-亚乙基二氧噻吩的电解质、包含其的电解电容器及电子材料原材料,更详细地,涉及具有高耐电压且低等效串联电阻的包含3,4-亚乙基二氧噻吩的电解质、包含其的电解电容器及电子材料用(导电性)原材料。
背景技术:
:最近,随着电子设备的性能提高且小型化,如移动设备那样不仅在固定的场所使用,还在移动的过程中使用电子设备的频率呈增加趋势。例如,在如电动车等具有较多的电子设备的苛刻的环境下,为了能够实现顺利的驱动,确保电子设备的长期可靠性尤为重要。在如上所述的用于电子设备的电解电容器中,使用铝及钽等作为正极侧对电极,在正极箔形成电介质,使间隔物介于正极箔及负极箔之间(位于正极箔与负极箔之间),之后,进行卷绕来形成电容器器件,在形成的电容器器件形成电解质层,从而具有在铝等的金属制壳体或合成树脂制的壳体密封电容器器件的结构。通常,作为用作所述电解质的导电性高分子,众所周知的是将3,4-亚乙基二氧噻吩等聚合性单体和氧化剂溶液分别混合并通过聚合制备的导电性高分子。以往合成的3,4-亚乙基二氧噻吩单体以根据仅将其作为单一的单体进行聚合或者在叔碳位单取代而成的结构而提高耐电压特性的形态形成(参照公开专利2012-0113701号)。但是,虽然可以通过这样的取代有烷基的3,4-亚乙基二氧噻吩的均聚来期待某程度的耐压特性得到提高,但是,具有难以制备具有高耐压特性的电解质的问题,可通过导入长烷基来制备具有高耐压特性的电解质,但在此情况下,具有等效串联电阻(esr)也变得过高的问题。技术实现要素:技术课题因此,本发明的目的在于,提供耐电压特性得到提高且等效串联电阻低、具有长期可靠性的包含3,4-亚乙基二氧噻吩的电解质、包含其的电解电容器及电子材料用(导电性)原材料。为了实现所述目的,提供包含一种以上的由以下化学式1表示的3,4-亚乙基二氧噻吩衍生物的电解质。化学式1在所述化学式1中,l及n各自独立地为0至3的整数,a不存在(a的左侧链通过单键与a的右侧链相连接)或为包含0个至5个氧原子(o)的具有1至20的碳原子数的烷基或亚烷基、具有6至20的碳原子数的亚芳基、具有4至15的碳原子数的环状或链状亚烷基,b为具有1至20的碳原子数的烷基或但是,l+n为3以下的整数。并且,本发明提供一种电解电容器,包括:具有氧化覆膜的正极电极层;负极电极层;以及位于所述正极电极层与负极电极层之间的间隔物和电解质,所述电解质包含一种以上的由所述化学式1表示的3,4-亚乙基二氧噻吩衍生物。发明效果本发明的电解质使用3,4-亚乙基二氧噻吩衍生物,具有耐电压特性优秀且等效串联电阻低的特性。并且,本发明的电容器具有耐电压特性优秀且等效串联电阻低的特性,并且长期可靠性优秀。进而,本发明的3,4-亚乙基二氧噻吩衍生物的导电性非常优秀。附图说明图1为本发明的电容器的一例的电解电容器的立体图。具体实施方式以下,参照附图对本发明进行更详细地说明。本发明的电解质包含一种以上的3,4-亚乙基二氧噻吩衍生物(3,4-ethylenedioxythiophenederivative,edot)。所述3,4-亚乙基二氧噻吩衍生物通过包含烷基和/或烷氧基来提高耐电压特性并使其具有低等效串联电阻(equivalentserialresistance,esr),其由以下化学式1表示。化学式1在所述化学式1中,l及n各自独立地为0至3的整数,具体为0至2的整数,a不存在(a的左侧链基团通过单键与a的右侧链基团相连接)或为包含0个至5个氧原子(o)的具有1至20的碳原子数的烷基或亚烷基(例如,链状等)或具有6至20的碳原子数的亚芳基,具体可为具有4至15的碳原子数的环状或链状亚烷基或具有6至15的碳原子数的亚苯基、联苯基或萘基,例如,可为并且,b可为具有1至20的碳原子数的烷基(例如,链状)或其中,意味着键合部,但是,l+n为3以下的整数。具体地,所述3,4-亚乙基二氧噻吩衍生物可由以下化学式2或3表示。化学式2化学式3在所述化学式2及3中,l、a与化学式1中的定义相同,p为1至19的整数,具体为1至10的整数,更具体为1至7的整数。并且,本发明的3,4-亚乙基二氧噻吩衍生物可单独使用或混合使用由所述化学式2及3表示的化合物,还可混合一种以上的由以下化学式4表示的化合物并使用,例如,可使用(a)一种以上的由化学式2表示的3,4-亚乙基二氧噻吩衍生物、(b)一种以上的由化学式3表示的3,4-亚乙基二氧噻吩衍生物、(c)一种以上的由化学式2表示的3,4-亚乙基二氧噻吩衍生物和一种以上的由化学式4表示的3,4-亚乙基二氧噻吩衍生物的混合物、(d)一种以上的由化学式3表示的3,4-亚乙基二氧噻吩衍生物和一种以上的由化学式4表示的3,4-亚乙基二氧噻吩衍生物的混合物、(e)一种以上的由化学式2表示的3,4-亚乙基二氧噻吩衍生物、一种以上的由化学式3表示的3,4-亚乙基二氧噻吩衍生物和一种以上的由化学式4表示的3,4-亚乙基二氧噻吩衍生物的混合物。在此情况下,耐电压特性更加优秀,可具有更低的等效串联电阻,因此有效。其中,混合物意味着单纯地混合或共聚的共聚物,具体意味着共聚物。化学式4在所述化学式4中,r为氢或具有1至20的碳原子数的烷基,具体为具有3至10的碳原子数的烷基(例如,链状烷基)。在本发明的电解质中,在混合使用由化学式2至4表示的化合物的情况下,相对于100重量份的电解质,由化学式3表示的3,4-亚乙基二氧噻吩衍生物的含量为0.1重量份至10重量份,具体为0.5重量份至5重量份,更具体为1重量份至3重量份,由化学式2和/或4表示的3,4-亚乙基二氧噻吩衍生物的含量各自为1摩尔%至99摩尔%,具体为20摩尔%至80摩尔%,更具体为50摩尔%至50摩尔%。若多个所述3,4-亚乙基二氧噻吩衍生物的含量超出所述范围,则由于3,4-亚乙基二氧噻吩聚合物之间的密集度低,耐电压特性及等效串联电阻不佳,或者,由于3,4-亚乙基二氧噻吩聚合物的密集度高,等效串联电阻可能变得过高。在本发明的电解质中,当由所述化学式3表示的化合物在电解质内形成聚合物时,如以下结构体1所示,可起到互不相同的聚合物之间的交联剂作用,在此情况下,使3,4-亚乙基二氧噻吩聚合物之间的结合更加坚固,从而提高耐电压特性并可具有低等效串联电阻(equivalentserialresistance,esr)。结构体1在所述结构体1中,r、l、p及a与在所述化学式1中的定义相同。在本发明的电解质中,只要是可起到作为导电性高分子电解质的作用,则由化学式1表示的3,4-亚乙基二氧噻吩衍生物的重均分子量没有特殊限制,通常为1000至500000,具体为5000至20000。若所述重均分子量的值过低,则电介质氧化覆膜层无法呈现充分的耐电压特性,漏电流可能变高,若过于大,则3,4-亚乙基二氧噻吩衍生物无法完全溶解,从而无法形成坚固且均匀的电解质层,耐电压特性及低等效串联电阻的效率可能降低。在由所述化学式2表示的3,4-亚乙基二氧噻吩衍生物的制备方法中,只要可制备,则没有特殊限制,例如,如以下反应式1及2所示,首先,在三乙胺中,使化合物e与甲苯磺酰氯进行反应来制备化合物f(化学式2的第一中间体)。接着,在对甲苯磺酸中,使3,4-二甲氧基噻吩与化合物g进行反应来制备化合物h(化学式2的第二中间体),之后,在二甲基亚砜(dimethylsulfoxide)中,使制备的化合物h与氯乙酰进行反应来制备化合物i。之后,使制备的化合物i与氢氧化钠进行反应来制备化合物j(化学式2的第三中间体),之后,在二甲基甲酰胺中,可使制备的化合物j、化合物f及氢化钠进行反应来制备。在以下反应式1及2中,l及p与在所述化学式2中定义的相同。反应式1反应式2如上所述,由所述化学式3表示的交联剂的制备方法没有特殊限制,例如,如以下反应式3及4所示,首先,在三乙胺(triethylamine,tea)及二氯甲烷(methylenechloride,mc)中,使化合物a与甲苯磺酰氯(toluensulfonylchloride)进行反应来制备化合物b(化学式3的第一中间体)。之后,在二甲基甲酰胺(dimethylformamide,dmf)中,可使制备的化合物b、所述化合物j及氢化钠(sodiumhydride,nah)进行反应来制备。反应式3由所述化学式4表示的3,4-亚乙基二氧噻吩衍生物的制备方法没有特殊限制,例如,如以下反应式4所示,可使3,4-二甲氧基噻吩(3,4-dimethoxythiphen)与由化合物d表示的二醇(diol)化合物进行反应来制备。反应式4本发明的电解质可通过使交联剂及3,4-亚乙基二氧噻吩衍生物聚合而形成,其方法没有特殊限制,例如,也可由化学氧化聚合及电分解氧化聚合中的任一种方法形成,具体地,可利用化学氧化聚合制备共聚物。例如,可以将所述交联剂和3,4-亚乙基二氧噻吩衍生物与溶解有氧化剂的溶液混合之后,加热来进行聚合。作为所述氧化剂,可以使用有机磺酸铁(ⅲ),具体地,可使用对甲苯磺酸铁盐、苯磺酸铁盐、萘磺酸铁盐等。并且,作为上述溶解有氧化剂的溶剂,可以使用正丁醇、乙醇、甲苯等,关于所述氧化剂,20重量%至90重量%、具体地30重量%至80重量%、更具体地40重量%至70重量%的氧化剂溶解于醇类的溶剂。接着,对本发明的电解电容器进行说明。图1为本发明的电容器的一例的电解电容器的立体图。如图1所示,本发明的电解电容器包括:具有氧化覆膜的正极电极层1;负极电极层2;以及位于所述正极电极层与负极电极层之间的间隔物(separator)5和电解质。所述电解质为包含一种以上的由所述化学式1表示的3,4-亚乙基二氧噻吩衍生物(例如,所述化学式2及4的单体与所述化学式3的交联剂)的单体的聚合物。所述间隔物5有含浸性优秀的纤维素类、耐压特性优秀的丙烯酸类,在利用本发明的交联剂及3,4-亚乙基二氧噻吩衍生物的情况下,由于密度低,优选使用无需碳化工序的尼龙类,但并不限定于尼龙类。为了制备本发明的电解电容器,首先,在具有氧化覆膜的正极电极层1与负极电极层2之间设置间隔物5。接着,使包含一种以上的由所述化学式1表示的3,4-亚乙基二氧噻吩衍生物的单体和氧化剂含浸在所述正极电极层1与负极电极层2之间,对所述单体进行加热来形成电解质,所述含浸可实施120秒钟。之后,对于形成的电容器器件,使用环氧树脂来制备所述电容器壳体,向正极施加4v的电压之后进行进行时效处理来制备电解电容器。本发明的电解电容器具有高耐电压及低等效串联电阻,因此,可用于各种电子部件,例如,中央处理电路及电源电路等使用电容器的电路,所述电路可用于计算机、服务器、摄像机、游戏机、dvd设备、av设备及移动电话等各种数字设备、各种电源等电子设备,本发明的3,4-亚乙基二氧噻吩衍生物具有优秀的导电性,可用作透明导电性液晶显示器、电致发光显示器、电致变色显示器、太阳能电池、触摸面板等的电极及电磁波屏蔽材料等的基材的原材料。发明实施方式以下,通过实施例对本发明进行更加详细的说明,但本发明并不局限于以下实施例。制备例1:制备化学式4的单体如以下反应式5所示,混合3,4-二甲氧基噻吩(3,4-dimethoxythiphene)1(30.00g,208.06mmol)、乙二醇(ethyleneglycol)2(25.83g,416.12mmol)、对甲苯磺酸(p-toluenesulfonicacid)(3.96g,20.81mmol)及300ml的甲苯,在100℃搅拌12小时并进行了反应。添加乙酸乙酯使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:乙酸乙酯=10:1(体积比)混合溶液展开已浓缩的有机层并进行柱纯化,从而获取了作为3,4-亚乙基二氧噻吩(3,4-ethylenedioxythiophene,edot)的化合物3(20.70g,70%的产率)。1h-nmr(cdcl3,varian400mhz):δ4.18(4h,s),6.31(2h,s)反应式5制备例2:制备化学式4的单体如以下反应式6所示,混合3,4-二甲氧基噻吩1(30.00g,208.06mmol)、1,2-戊二醇(1,2-pentanediol)4(43.34g,416.12mmol)、对甲苯磺酸(p-toluenesulfonicacid)(3.96g,20.81mmol)及300ml的甲苯,在110℃搅拌9小时并进行反应。通过添加乙酸乙酯完全溶解反应液,利用水提取之后利用硫酸钠对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅柱色谱法并利用己烷:乙酸乙酯=10:1(体积比)混合溶液展开已浓缩的有机层并进行柱纯化,从而获取了作为2-丙基-2,3-二氢噻吩并[3,4-b][1,4]二英(2-propyl-2,3-dihydrothieno[3,4-b][1,4]dioxine,propyl-edot)的化合物5(25.00g,70%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.88(3h,t,j=12hz),1.31-1.70(4h,m),3.86(1h,dd,j=8.0,3.2hz),4.10-4.16(2h,m),6.30(2h,s)。反应式6制备例3:制备化学式4的单体如以下反应式7所示,混合3,4-二甲氧基噻吩1(50.00g,346.76mmol)、1,2-癸二醇(1,2-octanediol)6(120.87g,693.52mmol)、对甲苯磺酸(p-toluenesulfonicacid)(6.60g,34.68mmol)及500ml的甲苯,在110℃搅拌9小时并进行反应。添加乙酸乙酯使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅柱色谱法并利用己烷:乙酸乙酯=10:1(体积比)混合溶液展开已浓缩的有机层并进行柱纯化,从而获取了作为2-辛基-2,3-二氢噻吩并[3,4-b][1,4]二英(2-octyl-2,3-dihydrothieno[3,4-b][1,4]dioxine,octyl-edot)的化合物7(57.33g,65%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.88(3h,t,j=12hz),1.27-1.71(14h,m),3.86(1h,dd,j=8.0,3.2hz),4.07-4.15(2h,m),6.29(2h,s)。反应式7制备例4:制备化学式4的单体如以下反应式8所示,混合3,4-二甲氧基噻吩1(30.00g,208.06mmol)、1,2-十二烷二醇(1,2-dodecanediol)8(42.07g,416.12mmol)、对甲苯磺酸(p-toluenesulfonicacid)(3.96g,20.81mmol)及300ml的甲苯,在110℃搅拌9小时并进行反应。添加乙酸乙酯使反应液完全溶解,利用水提取之后利用硫酸钠对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅柱色谱法并利用己烷:乙酸乙酯=10:1(体积比)混合溶液展开浓缩的有机层并进行柱纯化,从而获取了作为2-癸基-2,3-二氢噻吩并[3,4][1,4]二英(2-decyl-2,3-dihydrothieno[3,4-b][1,4]dioxine,decyl-edot)的化合物9(38.20g,65%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.88(3h,t,j=12hz),1.27-1.71(18h,m),3.86(1h,dd,j=8.0,3.2hz),4.07-4.15(2h,m),6.29(2h,s)。反应式8制备例5:制备化学式2的单体的第一中间体如以下反应式9-1所示,在二氯甲烷(dcm)(1000ml,10ml/g)中溶解正丙醇(n-propanol)10(100g,0.524mol)和三乙胺(tea)(108.95ml,0.786mol),之后,在0℃添加甲苯磺酰氯(tosylchloride)(100g,0.524mol)并在常温搅拌20小时。在反应结束之后,使用二氯甲烷(dcm)提取,利用无水na2so4进行干燥并浓缩有机层来获取了液体化合物10-1(105.5g,94%的产率)。1hnmr:δ0.87-0.90(3h,t,j=7.6hz),1.63-1.68(2h,m),2.43(3h,s),3.96-3.99(2h,t,j=6.8hz),7.32-7.34(2h,d,j=8.4hz),7.77-7.79(2h,d,j=8.0hz)。反应式9-1制备例6:制备化学式2的单体的第一中间体如以下反应式9-2所示,在二氯甲烷(dcm)(9000ml,10ml/g)中溶解正丁醇(n-butanol)11(130ml,1.416mol)和三乙胺(98.15ml,0.708mol),之后,在0℃添加甲苯磺酰氯(90g,0.472mol)并在常温搅拌了20小时。在反应结束之后,使用二氯甲烷提取,用无水na2so4进行干燥并浓缩有机层来获取了液体化合物11-1(89.9g,89%的产率)。1hnmr:δ0.82-0.86(3h,t,j=7.6hz),1.27-1.37(2h,m),1.57-1.64(2h,m)2.43(3h,s),3.99-4.2(2h,t,j=6.8hz),7.32-7.34(2h,d,j=8.4hz),7.76-7.78,d,j=8.0hz)。反应式9-2制备例7:制备化学式2的单体的第一中间体如以下反应式9-3所示,在二氯甲烷(200ml,10ml/g)中溶解二乙二醇单乙醚(diethyleneglycolmonoethylehther)12(20g,149.05mmol)和三乙胺(31.18ml,223.58mmol),之后,在0℃添加甲苯磺酰氯(25.57g,134.15mmol)并在常温搅拌3小时。在反应结束之后,通过使用二氯甲烷提取,利用无水na2so4进行干燥并浓缩有机层来获取了作为2-(2-乙氧基乙氧基)醚4-甲基苯磺酸酯(2-(2-ethoxyethoxy)ether4-methylbenzenesulfonate)的液体化合物12-1(35.0g,81%的产率)。1h-nmr(cdcl3,varian400mhz):δ1.19(3h,t,j=7.2hz),2.45(3h,s),3.47-3.53(4h,m),3.57-3.59(2h,m),3.70(2h,t,j=4.8hz)4.17(2h,t,j=5.2hz),7.34(2h,d,j=8.0hz),7.80(2h,d,j=8.4hz)。反应式9-3制备例8:制备化学式2的单体的第一中间体如以下反应式9-4所示,在二氯甲烷(200ml,10ml/g)中溶解1-辛醇(1-octanol)13(20g,153.57mmol)和三乙胺(32.13ml,230.36mmol)之后,在0℃添加甲苯磺酰氯(26.35g,138.21mmol)并在常温搅拌了3小时。在反应结束之后,使用二氯甲烷提取,利用无水na2so4进行干燥并浓缩有机层来获取了作为4-甲基苯磺酸辛酯(octyl4-methylbenzenesulfonate)的液体化合物13-1(34.0g,78%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.87(3h,t,j=7.2hz),1.21-1.28(10h,s),1.60-1.67(2h,m),2.45(3h,s),4.02(2h,t,j=6.4hz),7.34(2h,d,j=8.0hz),7.79(2h,d,j=8.0hz)。反应式9-4制备例9:制备化学式2的单体的第一中间体如以下反应式9-5所示,在二氯甲烷(200ml,10ml/g)中溶解乙二醇单己醚(ethyleneglycolmonohexylether)14(20g,136.77mmol)和三乙胺(28.61ml,205.16mmol),之后,在0℃添加甲苯磺酰氯(23.47g,123.09mmol)并在常温搅拌3小时。在反应结束之后,使用二氯甲烷(dcm)提取,利用无水na2so4进行干燥并浓缩有机层来获取作为2-(己氧基)乙基4-甲基苯磺酸酯(2-(hexyloxy)ethyl4-methylbenzenesulfonate)的液体化合物14-1(34.0g,92%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.88(3h,t,j=6.8hz),1.24-1.32(7h,m),1.47-1.50(2h,m),2.45(2h,s),3.37(2h,t,j=7.2hz),3.60(2h,t,j=4.8hz),4.16(2h,t,j=5.2hz),7.34(2h,d,j=8.0hz),7.81(2h,d,j=8.0hz)。反应式9-5制备例10:制备化学式2的单体的第一中间体如以下反应式9-6所示,在二氯甲烷(200ml,10ml/g)中溶解三乙二醇单丁醚(triethyleneglycolmonobutylether)15(20g,96.96mmol)和三乙胺(20.29ml,145.44mmol),之后,在0℃添加甲苯磺酰氯(16.64g,87.26mmol)并在常温搅拌3小时。在反应结束之后,使用二氯甲烷提取,用无水na2so4进行干燥并浓缩有机层而获取作为2-(2-(2-丁氧基乙氧基)乙氧基)乙基4-甲基苯磺酸酯(2-(2-(2-butoxyethoxy)ethoxy)ethyl4-methylbenzenesulfonate)的液体化合物15-1(24.90g,71%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.90(3h,t,j=7.2hz),1.30-1.39(2h,m),1.51-1.59(2h,m),2.44(3h,s),3.44(2h,t,j=6.4hz),3.54-3.61(8h,m),3.68(2h,t,j=4.8hz),4.15(2h,t,j=4.8hz),7.33(2h,d,j=8.0hz),7.79(2h,d,j=8.0hz)。反应式9-6制备例11:制备化学式2的单体的第一中间体如以下反应式9-7所示,在二氯甲烷(200ml,10ml/g)溶解2,5,8,11-四氧杂十三烷-13-醇(2,5,8,11-tetraoxatridecan-13-ol)16(20g,96.04mmol)和三乙胺(20.08ml,144.06mmol),之后,在0℃添加甲苯磺酰氯(16.48g,86.43mmol)并在常温搅拌3小时。在反应结束之后,使用二氯甲烷提取,利用无水na2so4进行干燥并浓缩有机层来获取作为2,5,8,11-四氧杂十三烷-13-基4-甲基苯磺酸酯(2,5,8,11-tetraoxatridecan-13-yl4-methylbenzenesulfonate)的液体化合物16-1(30.1g,86%的产率)。1h-nmr(cdcl3,varian400mhz):δ2.44(3h,s),3.30(3h,s),3.54-3.65(12h,m),3.70(2h,t,j=4.8hz),4.17(2h,t,j=5.2hz),7.33(2h,d,j=8.0hz),7.79(2h,d,j=8.0hz)。反应式9-7制备例12:制备化学式2的单体的第二中间体如以下反应式11所示,混合3,4-二甲氧基噻吩1(200.00g,1.387mol)、3-氯-1,2-丙二醇(3-chloro-1,2-propanediol)17(306.65g,2.774mol)对甲苯磺酸(26.38g,0.138mol)及2000ml的甲苯,在110℃搅拌15小时并进行反应。添加乙酸乙酯使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷溶液展开浓缩的有机层并进行柱纯化,从而获取了作为2-氯-2,3-二氢噻吩并[3,4-b][1,4]二英(2-chloro-2,3-dihydrothieno[3,4-b][1,4]dioxine)的化合物18(120.0g,45%的产率)。1h-nmr(cdcl3,varian400mhz):δ3.64-3.75(2h,m),4.11-4.18(1h,m),4.27-4.30(1h,m),4.35-4.44(1h,m),6.36(2h,s)。反应式11制备例13:制备化学式2的单体的第三中间体如以下反应式12所示,混合化合物18(60.00g,314.71mmol)、乙酸钠(sodiumacetate)(38.72g,472.07mmol)及900ml的二甲基亚砜,在110℃搅拌2小时并进行反应。添加二氯甲烷使反应液完全溶解,利用水提取之后利用硫酸钠对有机层进行干燥,并浓缩了剩余有机层。混合浓缩的化合物19(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)甲基乙酸酯(2,3-dihydrothieno[3,4-b][1,4]dioxin-2-yl)methylacetate))、氢氧化钠(sodiumhydroxide)(44.12g,1.102mol)及1200ml的水,在100℃搅拌4小时并进行反应。在反应结束之后,将温度降至常温,滴加盐酸(hydrochloricacid)直到ph3为止,利用水提取之后利用硫酸钠对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅柱色谱法并利用己烷:乙酸乙酯=1:3(体积比)混合溶液展开浓缩的有机层并进行柱纯化,从而获取了作为(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)甲醇(2,3-dihydrothieno[3,4-b][1,4]dioxin-2-yl)methanol)的化合物20(38.20g,70%的产率)。1h-nmr(cdcl3,varian400mhz):δ1.87(1h,s),3.83-3.88(2h,m),4.08-4.13(1h,m),4.22-4.26(1h,m),6.35(2h,s)。反应式12制备例14:制备化学式2的单体如以下反应式13所示,在70ml的n,n-二甲基甲酰胺中混合化合物20(7.00g,40.65mmol)并将温度降至0℃,之后,添加氢化钠(sodiumhydride,60%分散在矿物油中(dispersioninmineraloil))(1.63g,40.65mmol)将温度上升至常温并搅拌30分钟。再次将温度降至0℃之后,添加化合物10-1(9.58g,44.72mmol)来搅拌12小时并进行反应。添加乙酸乙酯使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:乙酸乙酯=1:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取了作为2-(丙氧基甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英(2-(propoxymethyl)-2,3-dihydrothieno[3,4-b][1,4]dioxine,5-edot-1)的化合物21(7.5g,86%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.89(3h,t,j=7.2hz),1.54(2h,t,j=8.8hz),3.45(2h,t,j=6.8hz),3.57-3.70(1h,m),4.00-4.18(1h,m),4.23-4.33(1h,m),6.25(2h,s)。反应式13制备例15:制备化学式2的单体如以下反应式14所示,在70ml的n,n-二甲基甲酰胺中混合化合物20(7.00g,40.65mmol)并将温度降至0℃,之后,添加氢化钠(sodiumhydride,60%分散在矿物油中)(1.63g,40.65mmol)将温度上升至常温并搅拌30分钟。再次将温度降至0℃之后,添加化合物11-1(10.21g,44.72mmol),搅拌12小时并进行反应。添加乙酸乙酯使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:乙酸乙酯=1:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取了作为2(丁氧基甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英(2-(butoxymethyl)-2,3-dihydrothieno[3,4-b][1,4]dioxine,6-edot-1)的化合物22(6.0g,65%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.90(3h,t,j=7.2hz),1.44-55(4h,m),3.45(2h,t,j=6.8hz),3.58(2h,t,j=6.8hz),3.57-3.70(1h,m),4.00-4.18(1h,m),4.23-4.33(1h,m),6.27(2h,s)。反应式14制备例16:制备化学式2的单体如以下反应式15所示,在70ml的n,n-二甲基甲酰胺中混合化合物20(7.00g,40.65mmol)并将温度降至0℃,之后,添加氢化钠(sodiumhydride,60%分散在矿物油中)(1.63g,40.65mmol),将温度上升至常温并搅拌30分钟。再次将温度降至0℃之后,添加化合物12-1(12.89g,44.72mmol),搅拌12小时并进行反应。添加乙酸乙酯使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:乙酸乙酯=1:1(体积比)溶液展开浓缩的有机层,并进行柱纯化,从而获取作为2((2-(2-乙氧基乙氧基)乙氧基)甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英(2((2-(2-ethoxyethoxy)ethoxy)methyl)-2,3-dihydrothieno[3,4-b][1,4]dioxine,10-edot-3)的化合物23(11.50g,98%的产率)。1h-nmr(cdcl3,varian400mhz):δ1.21(3h,t,j=6.8hz),3.53(2h,dd,j=7.2hz),3.58-3.60(2h,m),3.64-3.79(8h,m),4.04-4.15(1h,m),4.24-4.35(2h,m),6.32(2h,s)。反应式15制备例17:制备化学式2的单体如以下反应式16所示,在50ml的n,n-二甲基甲酰胺中混合化合物20(5.00g,29.04mmol)并将温度降至0℃,之后,添加氢化钠(sodiumhydride)(1.70g,29.04mmol,60%分散在矿物油中)将温度上升至常温并搅拌30分钟。再次将温度降至0℃之后,添加化合物13-1(9.08g,31.94mmol)搅拌12小时并进行反应。添加乙酸乙酯使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:乙酸乙酯=5:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取作为2-(辛氧甲基)2,3-二氢噻吩并[3,4-b][1,4]二英(2-(octyloxymethyl)-2,3-dihydrothieno[3,4-b][1,4]dioxine,10-edot-1)的化合物24(5.30g,64%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.88(3h,t,j=7.2hz),1.22-1.32(12h,m),1.63(2h,t,j=8.8hz),3.49(2h,t,j=6.8hz),3.57-3.70(1h,m),4.00-4.15(1h,m),4.23-4.33(1h,m),6.32(2h,s)。反应式16制备例18:制备化学式2的单体如以下反应式17所示,在60ml的n,n-二甲基甲酰胺中混合化合物20(6.00g,34.84mmol)并将温度降至0℃,之后,添加氢化钠(1.39g,34.84mmol,60%分散在矿物油中)将温度上升至常温并搅拌30分钟。再次将温度降至0℃之后,添加化合物14-1(9.42g,31.36mmol)搅拌12小时并进行反应。通过添加乙酸乙酯使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:乙酸乙酯=5:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取作为2-((2-(己氧基)乙氧基)甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英(2-((2-(hexyloxy)ethoxy)methyl)-2,3-dihydrothieno[3,4-b][1,4]dioxine,11-edot-2)的化合物25(5.30g,54%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.89(3h,t,j=6.8hz),1.24-1.35(6h,m),1.54-1.60(2h,m),3.45(2h,t,j=7.2hz),3.57-3.60(2h,m),3.66-3.80(4h,m),4.04-4.13(1h,m),4.24-4.36(2h,m),6.32(2h,t,j=4.4hz)。反应式17制备例19:制备化学式2的单体如以下反应式18,在70ml的n,n-二甲基甲酰胺中混合化合物20(6.85g,39.76mmol)并将温度降至0℃,之后,添加氢化钠(1.59g,39.76mmol,60%分散在矿物油中)将温度上升至常温并搅拌30分钟。再次将温度降至0℃之后,添加化合物15-1(12.90g,35.79mmol)搅拌12小时并进行反应。添加乙酸乙酯使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:乙酸乙酯=2:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取作为2,2,5,8,11-四氧杂十五烷基-2,3-二氢噻吩并[3,4-b][1,4]二英(2,2,5,8,11-tetraoxapentadecyl-2,3-dihydrothieno[3,4-b][1,4]dioxine,15-edot-4)的化合物26(7.10g,50%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.91(3h,t,j=7.6hz),1.31-1.40(2h,m),1.52-1.58(2h,m),3.45(2h,t,j=6.8hz),3.57(2h,d,j=4.8hz),3.63-3.79(12h,m),4.04-4.13(1h,m),4.24-4.34(2h,m),6.32(2h,s)。反应式18制备例20:制备化学式2的单体如以下反应式19所示,在60ml的n,n-二甲基甲酰胺中混合化合物20(6.00g,34.84mmol)并将温度降至0℃,之后,添加氢化钠(1.39g,34.84mmol,60%分散在矿物油中)来将温度上升至常温并搅拌30分钟。再次将温度降至0℃之后,添加化合物16-1(11.37g,31.36mmol)搅拌12小时并进行反应。添加乙酸乙酯使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:乙酸乙酯=5:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取作为2-2,5,8,11,14-五氧杂十五烷基-2,3-二氢噻吩并[3,4-b][1,4]二英(2-2,5,8,11,14-pentaoxapentadecyl-2,3-dihydrothieno[3,4-b][1,4]dioxine,15-edot-5)的化合物27(6.50g,52%的产率)。1h-nmr(cdcl3,varian400mhz):δ3.30(3h,t,j=7.6hz),3.54-3.65(18h,m),4.04-4.13(1h,m),4.24-4.34(2h,m),6.32(2h,s)。反应式19制备例21:制备化学式3的第一中间体如以下反应式20所示,在二氯甲烷(300ml,10ml/g)中溶解三乙二醇(triethyleneglycol)28(30g,199.77mmol)和三乙胺(84ml,599.32mol),之后,在0℃添加甲苯磺酰氯(114.26g,599.31mmol)并在常温搅拌6小时。使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:二氯甲烷=1:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取作为2,2'-(乙烷-1,2-二基双(氧基))双(乙烷-2,1-二基)双(4-甲基苯磺酸酯)(2,2'-(ethane-1,2-diylbis(oxy))bis(ethane-2,1-diyl)bis(4-methylbenzenesulfonate))的化合物28-1(54.20g,59%的产率)。1h-nmr(cdcl3,varian400mhz):δ2.45(6h,s),3.53(4h,s),3.66(4h,t,j=4.8hz),4.14(4h,t,j=4.8hz),7.34(4h,d,j=8.0hz),7.79(4h,d,j=8.0hz)。反应式20制备例22:制备化学式3的第一中间体如以下反应式21,在二氯甲烷(300ml,10ml/g)中溶解1,4-环己烷二甲醇(1,4-cyclohexanedimethanol)29(30g,208.03mmol)和三乙胺(87ml,624.09mol),之后,在0℃添加甲苯磺酰氯(118.98g,624.09mmol)并在常温搅拌6小时。使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:二氯甲烷=1:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取作为环己烷-1,4-二基双(亚甲基)双(4-甲基苯磺酸酯)(cyclohexane-1,4-diylbis(methylene)bis(4-methylbenzenesulfonate))的化合物29-1(38.50g,41%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.90(4h,t,j=9.2hz),1.58(2h,m),1.73(4h,d,j=7.2hz),2.44(6h,s),3.80(4h,d,j=6.4hz),7.34(4h,d,j=8.0hz),7.77(4h,d,j=8.0hz)。反应式21制备例23:制备化学式3的第一中间体如以下反应式22所示,在二氯甲烷(300ml,10ml/g)中溶解1,6-己二醇(1,6-hexanediol)30(30g,253.85mmol)和三乙胺(106ml,761.55mmol),之后,在0℃添加甲苯磺酰氯(118.98g,624.09mmol)并在常温搅拌6小时。使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:二氯甲烷=1:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取作为己烷-1,6-二基双(4-甲基苯磺酸酯)(hexane-1,6-diylbis(4-methylbenzenesulfonate))的化合物30-1(50.65g,57%的产率)。1h-nmr(cdcl3,varian400mhz):δ1.27(4h,s),1.58-1.62(4h,m),2.44(6h,s),3.98(4h,t,j=7.6hz),7.35(4h,d,j=8.4hz),7.78(4h,d,j=8.4hz)。反应式22制备例24:制备化学式3的第一聚合物如以下反应式23所示,在二氯甲烷(100ml,10ml/g)中溶解二甘醇31(10g,94.23mmol)和三乙胺(39ml,282.70mol),之后,在0℃添加甲苯磺酰氯(53.89g,282.70mmol)之后,在常温搅拌6小时。使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:二氯甲烷=1:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取作为2,2'-氧基双(乙烷-2,1-二基)双(4-甲基苯磺酸酯)(2,2'-oxybis(ethane-2,1-diyl)bis(4-methylbenzenesulfonate))的化合物31-1(28.0g,72%的产率)。1h-nmr(cdcl3,varian400mhz):δ2.34(6h,s),3.56(4h,t,j=4.8hz),3.70(4h,t,j=4.8hz),7.34(4h,d,j=8.0hz),7.79(4h,d,j=8.0hz)。反应式23制备例25:制备化学式1的交联剂(a-1)如以下反应式24所示,在400ml的n,n-二甲基甲酰胺中混合化合物20(4.09g,23.75mmol)并将温度降至0℃,之后,添加氢化钠(0.95g,23.75mmol,60%分散在矿物油中)将温度上升至常温并搅拌30分钟。再次将温度降至0℃之后,添加化合物28-1(5.11g,11.16mmol)搅拌12小时并进行反应。添加二氯甲烷(dcm)使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:乙酸乙酯=1:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取作为1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷(1,12-bis(2,3-dihydrothieno[3,4-b][1,4]dioxin-2-yl)-2,5,8,11-tetraoxadodecane)的化合物32(3.10g,61%的产率)。1h-nmr(cdcl3,varian400mhz):δ3.65-3.78(16h,m),4.03-4.06(2h,dd,j=11.6hz),4.23-4.33(4h,m)。反应式24制备例26:制备化学式1的交联剂(a-2)如以下反应式25所示,在100ml的n,n-二甲基甲酰胺中混合化合物20(9.8g,56.91mmol)并将温度降至0℃,之后,添加氢化钠(2.28g,56.91mmol,60%分散在矿物油中)将温度上升至常温并搅拌30分钟。再次将温度降至0℃之后,添加化合物29-1(12.11g,26.75mmol)搅拌12小时并进行反应。添加乙酸乙酯使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:乙酸乙酯=1:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取作为1,4-双(((2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)甲氧基)甲基环己烷(1,4-bis(((2,3-dihydrothieno[3,4-b][1,4]dioxin-2-yl)methoxy)methyl)cyclohexane)的化合物33(6.50g,54%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.96(4h,t,j=6.0hz),1.80(4h,d,j=7.6hz),3.36(4h,d,j=7.6hz),3.56-3.70(4h,m),4.03-4.08(2h,m),4.23-4.32(4h,m),6.33(4h,s)。反应式25制备例27:制备化学式1的交联剂(a-3)如以下反应式26所示,在150ml的n,n-二甲基甲酰胺中混合化合物20(15.0g,87.11mmol)并将温度降至0℃,之后,通过添加氢化钠(3.48g,87.11mmol,60%分散在矿物油中)将温度上升至常温并搅拌30分钟。再次将温度降至0℃之后,添加化合物30-1(17.46g,40.94mmol)搅拌12小时并进行反应。添加二氯甲烷使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:乙酸乙酯=3:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取作为1,6-双((2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)甲氧基)己烷(1,6-bis((2,3-dihydrothieno[3,4-b][1,4]dioxin-2-yl)methoxy)hexane)的化合物34(4.90g,28%的产率)。1h-nmr(cdcl3,varian400mhz):δ1.35-1.38(4h,m),1.57-1.61(4h,m),3.48-3.52(4h,m),3.57-3.70(4h,m),4.05(2h,dd,j=11.6hz),4.22-4.32(4h,m),6.33(4h,s)。反应式26制备例28:制备化学式1的交联剂(a-4)如以下反应式27所示,在20ml的n,n-二甲基甲酰胺中混合化合物20(2.0g,11.62mmol)并将温度降至0℃,之后,添加氢化钠(0.46g,11.62mmol,60%分散在矿物油中)将温度上升至常温并搅拌30分钟。再次将温度降至0℃之后,添加化合物31-1(2.26g,5.46mmol)搅拌12小时并进行反应。添加二氯甲烷(dcm)使反应液完全溶解,利用水提取之后利用硫酸钠(na2so4)对有机层进行干燥,并浓缩了剩余有机层。通过二氧化硅(sio2)柱色谱法并利用己烷:乙酸乙酯=1:1(体积比)溶液展开浓缩的有机层并进行柱纯化,从而获取作为2,2'-(2,2'-氧基双(乙烷-2,1-二基)双(氧基))双(亚甲基)双(2,3-二氢噻吩并[3,4-b][1,4]二英(2,2'-(2,2'-oxybis(ethane-2,1-diyl)bis(oxy))bis(methylene)bis(2,3-dihydrothieno[3,4-b][1,4]dioxine))的化合物35(2.05g,88%的产率)。1h-nmr(cdcl3,varian400mhz):δ3.65-3.79(12h,m),4.06(2h,dd,j=7.2hz),4.23-4.34(4h,m),6.32(4h,s)。反应式27制备例29:制备化学式2的单体的第一中间体使用2-(2-(庚氧基)乙氧基)乙醇(2-(2-(heptyloxy)ethoxy)ethanol)36(20.0g,97.89mmol)代替所述制备例11中的2,5,8,11-四氧杂十三烷-13-醇(2,5,8,11-tetraoxatridecan-13-ol)16,除此之外,以与制备例11相同的方法合成,获取了作为2-(2-(2-(庚氧基)乙氧基)乙基4-甲基苯磺酸酯(2-(2-(heptyloxy)ethoxy)ethyl4-methylbenzenesulfonate)的液体化合物36-1(28.50g,81%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.90(3h,t,j=7.2hz),1.30-1.43(8h,m),1.51-1.59(2h,m),2.44(3h,s),3.44(2h,t,j=6.4hz),3.54-3.61(8h,m),7.33(2h,d,j=8.0hz),7.79(2h,d,j=8.0hz)。反应式27-1制备例30:制备化学式2的单体使用所述制备例29的化合物36-1代替所述制备例20中的化合物16-1,除此之外,以与制备例20相同的方法合成,获取了作为2-((2-2-(庚氧基)乙氧基)乙氧基)甲基)-2-3-二氢噻吩并[3,4-b][1,4]二英(2-((2-(2-(heptyloxy)ethoxy)ethoxy)methyl)-2,3-dihydrothieno[3,4-b][1,4]dioxide,15-edot-3)的液体化合物37(6.10g,50%的产率)。1h-nmr(cdcl3,varian400mhz):δ0.89(3h,t,j=6.8hz),1.29-1.35(8h,m),1.54-1.62(2h,m),3.45(2h,t,j=7.2hz),3.57-3.60(6h,m),3.66-3.80(4h,m),4.04-4.13(1h,m),4.24-4.36(2h,m),6.32(2h,t,j=4.4hz)。反应式28比较例1:制备铝卷绕型电解电容器(聚单-丙基edot)首先,将铝箔用作正极侧对电极,对正极箔的表面进行蚀刻处理,之后,实施化学转化处理,在正极附着引线端子,所述正极在铝箔的表面形成了由氧化覆膜形成的电介质层。接着,在由铝箔形成的负极附着引线端子,通过尼龙类间隔物卷绕附着有所述引线端子的正极和负极来制备电容器器件。之后,准备(2-丙基2,3-二氢噻吩并)二英)单体,之后,利用1比2.5的重量比的氧化剂50%对甲苯磺酸铁盐/正丁醇溶液制备导电性高分子电解质溶液,将所述电容器器件浸渍于所述导电性高分子电解质溶液并提取,在45℃加热2小时、在105℃加热35分钟、在125℃加热1小时,进行氧化聚合来形成了由导电性高分子形成的电解质层。最后,利用外置材料对所述电解质层进行外包装,进行对正极施加4v的电压的时效处理来制备铝卷绕型电解电容器。比较例2:制备铝卷绕型电解电容器(聚单辛基-edot)使用(2-辛基2,3-二氢噻吩并)二英)单体代替(2-丙基2,3-二氢噻吩并)二英)单体,除此之外,以与比较例1相同的方法制备电容器器件及铝卷绕型电解电容器。比较例3:制备铝卷绕型电解电容器(聚单癸基-edot)使用(2-癸基2,3-二氢噻吩并)二英)单体代替(2-丙基2,3-二氢噻吩并)二英)单体,除此之外,以与比较例1相同的方法制备电容器器件及铝卷绕型电解电容器。比较例4:制备铝卷绕型电解电容器(聚单-(15-edot-5))使用2-2,5,8,11,14-五氧杂十五烷基-2,3-二氢噻吩并[3,4-b][1,4]二英单体代替(2-丙基2,3-二氢噻吩并)二英)单体,除此之外,以与比较例1相同的方法制备电容器器件及铝卷绕型电解电容器。比较例5:制备铝卷绕型电解电容器(聚单-(5-edot-1))使用2-(丙氧基甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英单体代替(2-丙基2,3-二氢噻吩并)二英)单体,除此之外,以与比较例1相同的方法制备电容器器件及铝卷绕型电解电容器。实施例1:制备铝卷绕型电解电容器(聚单-(6-edot-1))使用2(丁氧基甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英单体代替(2-丙基2,3-二氢噻吩并)二英)单体,除此之外,以与比较例1相同的方法制备电容器器件及铝卷绕型电解电容器。实施例2:制备铝卷绕型电解电容器(聚单-(10-edot-3))使用(2-((2-(2-乙氧基乙氧基)乙氧基)甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体代替(2-丙基2,3-二氢噻吩并)二英)单体,除此之外,以与比较例1相同的方法制备电容器器件及铝卷绕型电解电容器。实施例3:制备铝卷绕型电解电容器(聚单-(10-edot-1))使用(2-(辛氧甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体代替(2-丙基2,3-二氢噻吩并)二英)单体,除此之外,以与比较例1相同的方法制备电容器器件及铝卷绕型电解电容器。实施例4:制备铝卷绕型电解电容器(聚单-(11-edot-2))使用2-((2-(己氧基)乙氧基)甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英单体代替(2-丙基2,3-二氢噻吩并)二英)单体,除此之外,以与比较例1相同的方法制备电容器器件及铝卷绕型电解电容器。实施例5:制备铝卷绕型电解电容器(聚单-(15-edot-4))使用2,2,5,8,11-四氧杂十五烷基-2,3-二氢噻吩并[3,4-b][1,4]二英单体代替(2-丙基2,3-二氢噻吩并)二英)单体,除此之外,以与比较例1相同的方法制备电容器器件及铝卷绕型电解电容器。实施例6:制备铝卷绕型电解电容器(聚edot:10-edot-1)以与比较例1相同的方法制备电容器器件,聚合用化合物由以下方法制备。首先,以50%(1:1)摩尔比分别混合3,4-亚乙基二氧噻吩单体和(2-(辛氧甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体并准备,之后,利用1比2.5的重量比的氧化剂50%对甲苯磺酸铁盐/正丁醇溶液制备导电性高分子电解质溶液。接着,将所述电容器器件浸渍于所述导电性高分子电解质溶液并提取,在45℃加热2小时、在105℃加热35分钟、在125℃加热1小时,进行氧化聚合来形成由导电性高分子形成的电解质层。利用外置材料对所述导体电解质层进行外包装,进行向正极施加4v的电压的时效处理来制备铝卷绕型电解电容器。实施例7:制备铝卷绕型电解电容器(聚edot:15-edot-4)首先,以与比较例1相同的方法制备电容器器件。接着,使用3,4-亚乙基二氧噻吩单体及(2,2,5,8,11-四氧杂十五烷基-2,3-二氢噻吩并[3,4-b][1,4]二英单体代替3,4-亚乙基二氧噻吩单体及(2-(辛氧甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体,除此之外,以与实施例6相同的方法制备铝卷绕型电解电容器。实施例8:制备铝卷绕型电解电容器(聚丙基-edot:15-edot-4)首先,以与比较例1相同的方法制备电容器器件。接着,使用2-丙基-2,3-二氢噻吩并[3,4-b][1,4]二英单体及(2,2,5,8,11-四氧杂十五烷基-2,3-二氢噻吩并[3,4-b][1,4]二英单体代替3,4-亚乙基二氧噻吩单体及(2-(辛氧甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体,除此之外,以与实施例6相同的方法制备铝卷绕型电解电容器。实施例9:制备铝卷绕型电解电容器(聚edot:10-edot-3)首先,以与比较例1相同的方法制备电容器器件。接着,使用3,4-亚乙基二氧噻吩单体及2((2-(2-乙氧基乙氧基)乙氧基)甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英单体代替3,4-亚乙基二氧噻吩单体及(2-(辛氧甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体,除此之外,以与实施例6相同的方法制备铝卷绕型电解电容器。实施例10:制备铝卷绕型电解电容器(聚edot:11-edot-2)首先,以与比较例1相同的方法制备电容器器件。接着,使用3,4-亚乙基二氧噻吩单体及(2-((2-(己氧基)乙氧基)甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体代替3,4-亚乙基二氧噻吩单体及(2-(辛氧甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体,除此之外,以与实施例6相同的方法制备铝卷绕型电解电容器。实施例11:制备铝卷绕型电解电容器(聚辛基-edot:a-1)首先,以与比较例1相同的方法制备电容器器件。接着,单独准备(2-辛基2,3-二氢噻吩并)二英)单体,以100比2的重量比混合(mixing)作为交联剂的(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷)并准备,之后,利用1比2.5的重量比的氧化剂50%对甲苯磺酸铁盐/正丁醇溶液制备导电性高分子电解质溶液。接着,将所述电容器器件浸渍于所述导电性高分子电解质溶液并提取,在45℃加热2小时、在105℃加热35分钟、在125℃加热1小时,进行氧化聚合来形成由导电性高分子形成的电解质层。利用外置材料对所述电解质层进行外包装,进行向正极施加4v的电压的时效处理来制备铝卷绕型电解电容器。实施例12:制备铝卷绕型电解电容器(聚辛基-edot:a-2)首先,以与比较例1相同的方法制备电容器器件。接着,作为交联剂,使用(1,4-双(((2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)甲氧基)甲基)环己烷)代替(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷),除此之外,以与实施例11相同的方法制备氧化剂铝卷绕型电解电容器。实施例13:制备铝卷绕型电解电容器(聚辛基-edot:a-3)首先,以与比较例1相同的方法制备电容器器件。接着,作为交联剂,使用(1,6-双((2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)甲氧基)己烯)代替(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷),除此之外,以与实施例11相同的方法制备铝卷绕型电解电容器。实施例14:制备铝卷绕型电解电容器(聚辛基-edot:a-4)首先,以与比较例1相同的方法制备电容器器件。接着,作为交联剂,使用(2,2'-(2,2'-氧基双(乙烯-2,1-二基)双(氧基))双(亚甲基)双(2,3-二氢噻吩并)[3,4-b][1,4]二英)代替(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷),除此之外,以与实施例11相同的方法制备铝卷绕型电解电容器。实施例15:制备铝卷绕型电解电容器(聚10-edot-3:a-1)首先,以与比较例1相同的方法制备电容器器件。接着,使用(2-((2-(2-乙氧基乙氧基)乙氧基)甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体代替(2-辛基2,3-二氢噻吩并)二英单体,并且作为交联剂,使用(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷)代替(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷),除此之外,以与实施例11相同的方法制备铝卷绕型电解电容器。实施例16:制备铝卷绕型电解电容器(聚10-edot-3:a-4)首先,以与比较例1相同的方法制备电容器器件。接着,使用(2-((2-(2-乙氧基乙氧基)乙氧基)甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体代替(2-辛基2,3-二氢噻吩并)二英单体,并且作为交联剂,使用(2,2'-(2,2'-氧基双(乙烯-2,1-二基)双(氧基))双(亚甲基)双(2,3-二氢噻吩并)[3,4-b][1,4]二英)代替(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷),除此之外,以与实施例11相同的方法制备铝卷绕型电解电容器。实施例17:制备铝卷绕型电解电容器(聚11-edot-2:a-1)首先,以与比较例1相同的方法制备电容器器件。接着,使用(2-((2-(己氧基)乙氧基)甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体代替(2-辛基2,3-二氢噻吩并)二英单体,并且作为交联剂,使用(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷)代替(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷)之外,以与实施例11相同的方法制备铝卷绕型电解电容器。实施例18:制备铝卷绕型电解电容器(聚11-edot-2:a-4)首先,以与比较例1相同的方法制备电容器器件。接着,使用(2-((2-(己氧基)乙氧基)甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体代替(2-辛基2,3-二氢噻吩并)二英单体,并且作为交联剂,使用(2,2'-(2,2'-氧基双(乙烯-2,1-二基)双(氧基))双(亚甲基)双(2,3-二氢噻吩并)[3,4-b][1,4]二英)代替(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷),除此之外,以与实施例11相同的方法制备铝卷绕型电解电容器。实施例19:制备铝卷绕型电解电容器(聚-edot:10-edot-1:a-1)首先,以与比较例1相同的方法制备电容器器件。接着,以50%(1:1)摩尔比分别使用3,4-亚乙基二氧噻吩单体及(2-(辛氧甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体代替(2-辛基2,3-二氢噻吩并)二英单体,并且作为交联剂,使用(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷)代替(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷),除此之外,以与实施例11相同的方法制备铝卷绕型电解电容器。实施例20:制备铝卷绕型电解电容器(聚edot:11-edot-2:a-1)首先,以与比较例1相同的方法制备电容器器件。接着,以20%:80%摩尔比分别使用3,4-亚乙基二氧噻吩单体及(2-((2-(己氧基)乙氧基)甲基)-2,3-二氢噻吩并[3,4-b][1,4]二英)单体代替(2-辛基2,3-二氢噻吩并)二英单体,并且作为交联剂,使用(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷)代替(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷)之外,以与实施例11相同的方法制备铝卷绕型电解电容器。实施例21:制备铝卷绕型电解电容器(聚edot:15-edot-4:a-1)首先,以与比较例1相同的方法制备电容器器件。接着,以20%:80%摩尔比分别使用3,4-亚乙基二氧噻吩单体及(2,2,5,8,11-四氧杂十五烷基-2,3-二氢噻吩并[3,4-b][1,4]二英单体代替(2-辛基2,3-二氢噻吩并)二英单体,并且作为交联剂,使用(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷)代替(1,12-双(2,3-二氢噻吩并[3,4-b][1,4]二英-2-基)-2,5,8,11-四氧杂十二烷),除此之外,以与实施例11相同的方法制备铝卷绕型电解电容器。实施例22:制备铝卷绕型电解电容器(聚单-15-edot-3))使用2-((2-2-(庚氧基)乙氧基)乙氧基)甲基)-2-3-二氢噻吩并[3,4-b][1,4]二英单体代替(2-丙基2,3-二氢噻吩并)二英)单体,除此之外,以与比较例1相同的方法制备电容器器件及铝卷绕型电解电容器。整理用于所述比较例1至5及实施例1至22的3,4-亚乙基二氧噻吩衍生物、其组成比及交联剂并示于以下表1及2。表1表2实验例1:铝卷绕型电解电容器的评价1对于由所述比较例1至5及实施例1至22制备的铝卷绕型电解电容器,使用惠普(hewlettpackard)公司的lcr测试仪(4284a),在25℃的温度条件下以100khz测定等效串联电阻(esr),以120hz测定静电容量,通过使用matsusadaprecision公司制造的prk650-2.5,在25℃的条件下使电压以1v/分钟的速度上升并测定击穿电压,并在以下表1至8示出其结果。在以下表3至10中,等效串联电阻及静电容量分别求得10个平均值,并对小数点第二位进行四舍五入来示出,击穿电压值对小数点以下进行四舍五入来示出(6.3×6的器件尺寸,工作电压:50v,电容:10μf)。表3规定链数静电容量(μf)esr(mω)漏电流(μa)击穿电压(v)比较例1311.0240.0540比较例2810.4380.1550比较例31010.02850.1568如所述表3所示,在比较例1至3中,仅以在3,4-亚乙基二氧噻吩单体的叔碳位单取代的结构的取代有单一烷基的单体进行聚合来制备卷绕型铝电解电容器,关于击穿电压的说明如下,即,在比较例1至3中,由于具有碳数为3~10的长烷基链的卷绕型铝电解电容器,比较例3具有较高的击穿电压,这是因为烷基链越增加击穿电压越高。但是,若链数超过10个,无法具有高击穿电压,而是,如比较例3所示,可确认esr快速提高。并且,在所述表3中呈现随着链数的增加具有高击穿电压的特性,但是可知,esr也增加。为了解决所述问题,通过向烷基内导入氧(o)原子来导入了溶解度比仅由碳(c)形成的烷基优秀的烷基烷氧基。表4规定链数静电容量(μf)esr(mω)漏电流(μa)击穿电压(v)比较例5510.9280.0340实施例1611.0340.0454实施例21010.0600.0358实施例3109.71590.0472实施例41110.51060.0572实施例5159.91520.0580如所述表4所示,在实施例1至5中,仅利用在3,4-亚乙基二氧噻吩的叔碳位单取代的单体取代有烷基烷氧基的单体来制备卷绕型铝电解电容器。在所述实施例1至5中的击穿电压的说明如下,即,与所述表2同样地,可知,随着链数的增加呈现高击穿电压特性。并且,相比于烷基仅由碳(c)形成的比较例3,在具有烷基链包含氧(o)基团的烷基烷氧基的实施例2及实施例3中,具有相同的链数时可呈现更低的esr特性。如所述表4,关于比较例5的说明如下,即,当链数为5个时,击穿电压为40v,较低,因此,难以呈现有效的耐电压特性,当链数为6个以上时,击穿电压为50v以上,呈现优秀的耐电压特性。并且,若链数为16个以上,则存在相比于高耐电压特性,esr值显著变高的缺点。根据所述结果,为了在相同的链数中比较具有氧(o)原子时和没有氧(o)原子时的情况,在表5中对具有相同的链数的比较例3、实施例2及实施例3的情况进行比较。设计烷基链仅由碳(c)形成的比较例3、烷基链的碳(c)之间具有3个氧(o)的实施例2及碳(c)之间具有1个氧(o)的实施例3,比较了在相同的烷基链中具有氧(o)原子时的情况的特性。结果可确认,相比于比较例3,实施例2的击穿电压略微降低,但esr值显著降低。在实施例3的情况下,确认到与比较例1相比击穿电压反而更高且esr值显著降低。表5当利用所述结果向烷基链导入氧(o)原子时,耐压特性不会大大降低并且显著改善esr特性。但是,根据表6,在向烷基导入氧(o)原子的结构中,对具有15个链数的烷基烷氧基进行比较。在比较例4中,具有15个链数且具有5个氧(o)原子,在实施例5中,具有15个链数且具有4个氧(o)原子,在实施例22中,具有15个链数且具有3个氧(o)原子,关于击穿电压的说明如下,即,相比于实施例5和实施例22,具有5个氧(o)原子的比较例4具有低击穿电压。可确认,相比于实施例4及实施例22,esr特性也具有显著高的值。在这样的长链内,若氧(o)原子大于5个,则击穿电压特性反而降低且esr特性反而提高。表6若导入烷基烷氧基,则相比于烷基链仅由碳(c)形成的3,4-亚乙基二氧噻吩衍生物,击穿电压及esr特性得到显著改善,但是,为了进一步改善esr特性,如表6的实施例6至10那样,将在3,4-亚乙基二氧噻吩单体的叔碳位取代有烷基烷氧基的单体与由化学式4衍生的单体各自一种以上混合来制备卷绕型铝电解电容器。表7根据所述表7的结果,在实施例6至10中,击穿电压为54v至68v左右,这与表4的实施例3及实施例5的将烷基烷氧基均聚的电解电容器相比,具有低击穿电压,但是esr特性得到大的改善。虽然esr特性大大提高,但是击穿电压产生了某种程度的损失,为了使这种损失最小化,添加由化学式3衍生的交联剂来进行了用于弥补耐电压特性的实验。在下表7中,通过实施例11至18以规定重量比添加多种由化学式3表示的化合物(交联剂)来改善耐电压特性。表8所述表8所示,在实施例11至14中,使用(2-辛基2,3-二氢噻吩并)二英)作为单体,添加在所述制备例25至28中制备的a-1、a-2、a-3及a-4型各个交联剂,并在维持esr特性的状态下进行了用于提高耐电压特性的实验。在实施例11至14的情况下,可确认相比于比较例2,esr特性得到维持并且击穿电压特性显著提高。因此,在之前的实验中,向esr特性及击穿电压特性较优秀的具有烷基烷氧基的实施例2及实施例4中分别添加a-1及a-4的交联剂并观察esr特性及击穿电压特性。表9如所述表9所示,在实施例15至18的情况下,可确认相比于未向实施例2及实施例4添加交联剂并聚合时,在较高的电压具有击穿电压。通过添加这样的交联剂使由化学式3衍生的交联剂位于高分子与高分子之间进行交联,从而使结合更加坚固(参照所述结构体1)。根据所述表9的结果,在实施例19至21的情况下,在通过非单一单体的混合组成进行均聚时,向作为击穿电压特性优秀的实施例2至4的单体的10-edot-1、11-edot-2、15-edot-4中分别混合不同组成的3,4-亚乙基二氧噻吩单体(5:5、8:2及8:2的组成)并添加交联剂a-1来进行聚合(表9)。表10在相同组成时,相比于实施例6,添加交联剂a-1的实施例19的击穿电压特性得到提高,同样地,实施例20至实施例21也在较高的击穿电压时具有较低的esr值。尤其,在实施例20中,esr的值为目标值(60mω)以下的58mω,击穿电压为72v,具有高击穿电压。如所述表3至10所示可知,在使用本发明的3,4-亚乙基二氧噻吩衍生物的情况下,耐电压特性优秀且具有低等效串联电阻,而且导电性非常优秀。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1