负载紫杉醇的不对称树状大分子纳米载药系统及其制备方法与流程
本发明属于高分子靶向纳米载体的制备领域,具体涉及一种负载紫杉醇的不对称树状大分子纳米载药系统及其制备方法。
背景技术:
目前,化学治疗面临许多挑战,如药物在正常组织的高分布从而引起的毒副作用,以及在肿瘤组织的低富集而导致的令人不太满意的抗癌效果等。许多策略被研究应用来提高抗癌药的功效,这些策略合起来可以缩写为SAIR(Stability,Accumulation,Internalization and Release),即提高药物在循环过程中的稳定性(Stability)、通过主动或者被动靶向增强药物在肿瘤组织的富集累积(Accumulation)、促进药物快速进入肿瘤细胞实现内化(Internalization)以及加快药物在细胞内的释放速率(Release)。这些策略中有些达到了增强某些特殊药物特性的目的,然而并没有成功的实现抗癌效果全方位的提升。
紫杉醇(PTX)是一种具有细胞毒性的高效抗癌药物。然而,其在循环系统中会被快速地清除并且引起诸如骨髓毒性等毒副作用,这些不利因素限制了紫杉醇的应用。此外,一个很重要的方面是PTX自身酯键的断裂会导致其在循环系统中被降解。
肽类树状大分子是一类由氨基酸组成的大分子,其具有类树形的结构精确的枝化结构。他们具有诸多优良性能,例如结构精确可控、高支化的结构、高密度的末端官能团、具有水溶性以及良好的生物安全性,这些促使其能成为潜 在的药物递送载体。此外,将两种化学功能分别集中在其内核和表层的不对称树状大分子能够承担不同的任务和目标。由于将不同的功能集中在一个单独分子里,不对称树状大分子比起普通的扇形或球形树状大分子能提供更多的功能性以及更广泛的用途。然而,低代数的树状大分子的载体其粒径小于10nm,其粒径不够大,无法满足增强渗透及滞留效应(EPR效应)的标准。另外,尽管高代数的树状大分子具有相对较大的尺寸,其在循环过程中极不易被降解而增加了诱发细胞毒性的风险。
纳米粒子的静脉内注射(i.v.)通常会发生血浆蛋白吸附作用,该过程即大家所熟悉的调理素作用。目前降低或减少调理素作用的方法主要集中在引入亲水性基团或者对粒子进行改性修饰使其表面电荷成电中性或电负性。聚乙二醇(PEG)能增强材料的水溶性及血浆蛋白的稳定性,同时降低免疫原性。利用聚乙二醇来修饰改性纳米载体,从而降低单核巨噬细胞系统(MPS)对纳米载体的非特异细胞吞噬,与此同时也可以调节纳米粒子的循环半衰期。研究表明PEG修饰改性的树状大分子药物递送载体拥有有效的抗癌功效。然而,载体表面高密度的PEG干扰到了细胞对它的摄取。
组织蛋白酶B是一种溶酶体半胱氨酸蛋白酶,其在乳腺癌等大部分肿瘤细胞和肿瘤内皮细胞中过表达。这些为酶响应型药物递送载体的设计提供了更多的思路。甘氨酸-苯丙氨酸-亮氨酸-甘氨酸四肽片段(Gly-Phe-Leu-Gly;GFLG),是组织蛋白酶B的一种理想底物;其已被用于连接抗癌药到大分子上,并且这些大分子-药物偶联物已经表现出在血浆和血清运输过程中具有良好的稳定性的特性,同时在胞吞作用后能快速地实现胞内溶酶体中药物的释放。如发明专利CN201310511453.6,提供了一种基于GFLG的PEG化肽类树状大分子给药系统的制备方法。该给药系统为PEG化肽类树状大分子、靶向功能性因子GFLG与抗 肿瘤治疗因子的偶联物,通过GFLG将治疗因子和PEG化肽类树状大分子相连接后形成功能化树状大分子,在获得良好生物相容性的同时,使同等剂量或低剂量的药物达到明显的抗肿瘤疗效。但该给药系统为亲水性树状大分子,生物相容性较差。
技术实现要素:
基于以上技术问题,本发明提供了一种负载紫杉醇的不对称树状大分子纳米载药系统。该系统满足作为高效的药物递送系统的所有SAIR要求,通过结合不对称肽类树状大分子、PEG改性、纳米级系统以及酶敏感连接键的特征,纳米粒子在体内治疗中呈现出比游离PTX显著增强的抗肿瘤治疗指数和生物安全性。
为了实现上述发明目的,本发明采用如下技术方案:
负载紫杉醇的不对称树状大分子纳米载药系统,该载药系统为抗肿瘤药物紫杉醇、肿瘤胞内酶敏感短肽GFLG和PEG化不对称肽类树状大分子的偶联物;具体结构如下:
An-B
其中,A为:
B是以乙二胺为核的PEG化不对称赖氨酸肽类树状大分子,该大分子为 G1L-G2L、G2L-G3L或者G3L-G4L的不对称树状大分子,PEG修饰发生在该不对称树状大分子代数较小的一端,A取代了另一端的末端氨基,n=1-16。
本发明所述的不对称树状大分子为G2L-G3L树状大分子。最大化载药量和最恰当的亲水性的基团的修饰。
负载紫杉醇的不对称树状大分子纳米载药系统的制备方法,先分别合成叠氮化的GFLG-PTX和炔基化的PEG化树状大分子,再通过铜催化的点击反应合成终产物树状大分子-紫杉醇偶联物。
优选地,所述叠氮化GFLG-PTX的合成如下:
利用CH3ONa将N3-GFLG-OMe脱去甲酯保护,将反应产物N3-GFLG-OH置于氮气氛下,反应体系用DCM作为溶剂,加入DMAP和二环己基碳二亚胺(DCC),反应半小时后,加入PTX,反应48小时;过滤除去不溶物,旋干除去溶剂;再用乙酸乙酯溶解产物,将有机相分别用饱和NaHCO3溶液、稀盐酸与饱和NaCl溶液洗涤,再经干燥、过滤后,减压旋蒸浓缩,重结晶,即得。
优选地,所述不对称树状大分子纳米载药系统的合成如下:
A、炔基化不对称树状大分子的合成
以一端为Cbz基团保护,另一端为Boc基团保护的不对称树状大分子为原料,在氢气氛围下与Pb/C反应脱去Cbz保护基,反应结束后,在N2氛保护下,将反应产物、HBTU与HOBT溶于无水DMF中,在冰浴下与DIPEA和5-己炔酸反应;
B、PEG化树状大分子的合成
将A步骤的产物与叠氮化的PEG溶于含CuSO4·5H2O和抗坏血酸钠的水中,反应结束后,利用透析袋透析除去溶剂及杂质,将透析后留在透析袋中的溶液收集冻干得mPEG修饰的树状大分子;
C、PEG化树状大分子的炔基化
在N2氛保护下,将PEG化树状大分子溶于无水二氯甲烷中,然后滴加入三氟乙酸,搅拌4小时后,除去溶剂,真空干燥后收集样品;在N2氛保护下,将得到的样品、HATU、HOAt与5-己炔酸溶于无水DMF中,在冰浴下,加入DIPEA 反应;
D、树状大分子-紫杉醇偶联物的合成
在Ar氛保护下,将C步骤得到的产物与叠氮化的GFLG-PTX溶于含CuSO4·5H2O和抗坏血酸钠的水中,反应结束后,利用透析袋透析除去溶剂及杂质,将透析后留在透析袋中的溶液收集冻干即得树状大分子-紫杉醇偶联物。
进一步优选地,所述的A步骤和C步骤,在反应结束后,减压蒸馏移除溶剂,并加入EtOAc,将有机相分别用饱和NaHCO3溶液、稀盐酸与饱和NaCl溶液各洗涤三次;用无水硫酸镁干燥,过滤除去硫酸镁后将溶液减压旋蒸浓缩,利用乙酸乙酯/乙醚重结晶,即得。
进一步优选地,所述的B步骤和D步骤中,透析袋的截留分子量均为3500。
进一步优选地,所述的B步骤和D步骤中,透析的去离子水中加入乙二胺四乙酸二钠盐,帮助透析除去反应中的铜离子。
进一步优选地,所述的B步骤和D步骤,将透析后留在透析袋中的溶液收集冻干得到的固体溶解,利用排阻色谱法分离提纯产物,避光收集产物,再经纯水透析后冻干即得产物。
本发明的有益效果在于:
1、本发明成功地构筑了集SAIR功能于一体的PEGylated dendrimer-GFLG-PTX纳米粒子,对不对称PEGylated dendrimer-GFLF-PTX纳米粒子进行了全面的结构设计和优化,是一种安全高效的药物递送系统。将智能大分子载体用于疏水药物紫杉醇的运载,使其在循环过程中保持最大的稳定状态并且到达肿瘤后能实现胞内快速的药物释放。全面有效地提升了紫杉醇的抗肿瘤效果并具有良好的生物安全性。
2、采用两亲性不对称树状大分子的载药系统,针对高代数树状大分子在循环过程中极不易被降解而增加了诱发细胞毒性的风险,因此采用低代数树状大分子;同时针对低代数粒径不够大,无法满足增强渗透及滞留效应(EPR效应)的标准,采用PEG修饰以及自组装的手段来解决,具体为利用PEG修饰低代数的树状大分子形成两亲性的不对称树状大分子,疏水性基团集中在其内核,亲 水性基团集中在其表层,再利用其两亲性的性质实现材料的自组装,自组装成为尺寸合适的纳米粒子。
3、本发明的载药系统满足作为高效的药物递送系统的所有SAIR要求。由于药物是通过一个酶敏感短肽共价连接到载体上,并且不对称树状大分子利用PEG修饰优化过,该抗癌药紫杉醇能够在酶的存在下快速地从纳米粒子中释放出来,而且保持着原始的结构,该纳米粒子能有效地诱发乳腺癌细胞的凋亡,相比使用游离药物紫杉醇,其对正常细胞的毒性要低得多。通过结合不对称肽类树状大分子、PEG改性、纳米级系统以及酶敏感连接键的特征,纳米粒子在体内治疗中呈现出比游离PTX显著增强的抗肿瘤治疗指数和生物安全性。
4、本发明制备方法通过液相法合成了结构精确的炔基化的不对称树状大分子,利用两步“click”反应将mPEG与酶敏感短肽-药物偶联物高效精确地连接到树状大分子上。采用Cbz基团保护的树状大分子为原料,可根据需要选择性地脱保护,选择性地对不对称大分子的高代数端进行修饰,否则就得不到不对称的树状大分子,而是球形的树状大分子了。
附图说明
图1为不对称树状大分子PEG-G2L-G3L的质谱表征图。
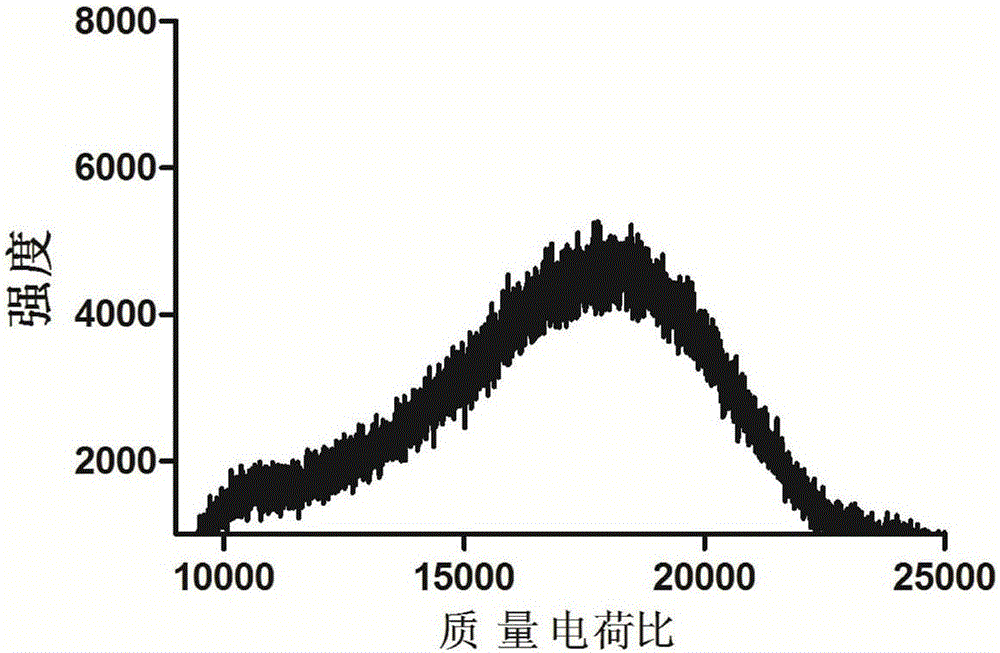
图2为PEGylated dendrimer-GFLF-PTX的质谱表征图。
图3为PEGylated dendrimer-GFLF-PTX纳米粒子的DLS水相粒径(A),AFM(B),和zeta电位(C)表征。
图4为通过HPLC检测药物释放。PTX注射液(A)和经过木瓜蛋白酶孵育48小时的PEGylated dendrimer-GFLF-PTX(B)的HPLC出峰图。
图5为PEGylated dendrimer-GFLF-PTX纳米粒子在pH为5.4的缓冲溶液中孵育24小时的水相粒径变化。
图6为PEGylated dendrimer-GFLF-PTX纳米粒子的粒径形貌变化。
图7为纳米粒子的体内抗肿瘤实验评价。荷瘤小鼠分别注射了生理盐水、紫杉 醇注射液和PEGylated dendrimer-GFLF-PTX纳米粒子。(A)相对肿瘤大小变化曲线。(B)小鼠体重变化曲线。(C)瘤重,(D)抑瘤率。
图8为19天治疗周期的小鼠体重变化曲线。
图9为19天的治疗周期后小鼠取血进行血常规检测分析。
具体实施方式
下面结合具体实施方式对本发明的实质性内容作进一步详细的描述。
实施例1
负载紫杉醇的不对称树状大分子纳米载药系统,该载药系统为抗肿瘤药物紫杉醇、肿瘤胞内酶敏感短肽GFLG和PEG化不对称肽类树状大分子的偶联物;
具体结构如下:
An-B
其中,A为:
B是以乙二胺为核的PEG化不对称赖氨酸肽类树状大分子,该大分子为G1L-G2L、G2L-G3L或者G3L-G4L的不对称树状大分子,PEG修饰发生在该不对称树状大分子代数较小的一端,A取代了另一端的末端氨基。
偶联体Ⅰ:n=1,B是PEG化的G1L-G2L代不对称树状大分子。
偶联体Ⅱ:n=4,B是PEG化的G1L-G2L代不对称树状大分子。
偶联体Ⅲ:n=5,B是PEG化的G2L-G3L代不对称树状大分子。
偶联体Ⅳ:n=8,B是PEG化的G2L-G3L代不对称树状大分子。
偶联体Ⅴ:n=16,B是PEG化的G3L-G4L代不对称树状大分子。
实施例2
负载紫杉醇的不对称树状大分子纳米载药系统的制备方法,先分别合成叠氮化的GFLG-PTX和炔基化的PEG化树状大分子,再通过铜催化的点击反应合成终产物树状大分子-紫杉醇偶联物。
实施例3
本实施例在实施例2的基础上:
所述叠氮化GFLG-PTX的合成如下:
利用CH3ONa将N3-GFLG-OMe脱去甲酯保护,将反应后的N3-GFLG-OH置于氮气氛下,反应体系用DCM作为溶剂;再加入溶于DCM的DMAP和DCC反应半小时后,加入PTX。低温搅拌反应48h后,停止反应。过滤除去不溶物,旋干除去溶剂;再用乙酸乙酯溶解产物,将有机相分别用饱和NaHCO3溶液、稀盐酸与饱和NaCl溶液各洗涤三次;用无水硫酸镁干燥,过滤除去硫酸镁后将溶液减压旋蒸浓缩,利用乙酸乙酯/乙醚重结晶,即得,MALDI-TOF MS测试结果为m/z:1733.8([M+H]+)。
实施例4
负载紫杉醇的不对称树状大分子纳米载药系统的制备方法,包括以下步骤:
A、炔基化不对称树状大分子的合成
以Cbz基团保护的树状大分子为原料,在氢气氛围下与Pb/C反应脱去Cbz保护基,反应结束后,在N2氛保护下,将反应产物、HBTU与HOBT溶于无水DMF中,在冰浴下与DIPEA和5-己炔酸反应;
B、PEG化树状大分子的合成
将A步骤的产物与叠氮化的PEG溶于含CuSO4·5H2O和抗坏血酸钠的水中,反应结束后,利用透析袋透析除去溶剂及杂质,将透析后留在透析袋中的溶液收集冻干得mPEG修饰的树状大分子;
C、PEG化树状大分子的炔基化
在N2氛保护下,将PEG化树状大分子溶于无水二氯甲烷中,然后滴加入三氟乙酸,搅拌4小时后,除去溶剂,真空干燥后收集样品;在N2氛保护下,将得到的样品、HATU、HOAt与5-己炔酸溶于无水DMF中,在冰浴下,加入DIPEA反应;
D、树状大分子-紫杉醇偶联物的合成
在Ar氛保护下,将C步骤得到的产物与叠氮化的GFLG-PTX溶于含CuSO4·5H2O和抗坏血酸钠的水中,反应结束后,利用透析袋透析除去溶剂及杂质,将透析后留在透析袋中的溶液收集冻干即得树状大分子-紫杉醇偶联物。
实施例5
本实施例在实施例4的基础上:
所述的B步骤和D步骤中,透析袋的截留分子量均为3500。
实施例6
本实施例在实施例4的基础上:
所述的B步骤和D步骤中,透析袋的截留分子量均为3500。
所述的B步骤和D步骤中,透析的去离子水中加入乙二胺四乙酸二钠盐。
实施例7
本实施例在实施例4的基础上:
所述的B步骤和D步骤中,透析袋的截留分子量均为3500。
所述的B步骤和D步骤中,透析的去离子水中加入乙二胺四乙酸二钠盐。
所述的B步骤和D步骤,将透析后留在透析袋中的溶液收集冻干得到的固体溶解,利用排阻色谱法分离提纯产物,避光收集产物,再经纯水透析后冻干即得产物。
实施例8
本实施例在实施例4的基础上:
所述的B步骤和D步骤中,透析袋的截留分子量均为3500。
所述的B步骤和D步骤中,透析的去离子水中加入乙二胺四乙酸二钠盐。
所述的B步骤和D步骤,将透析后留在透析袋中的溶液收集冻干得到的固体溶解,利用排阻色谱法分离提纯产物,避光收集产物,再经纯水透析后冻干 即得产物。
所述的A步骤和C步骤,在反应结束后,减压蒸馏移除溶剂,并加入EtOAc,将有机相分别用饱和NaHCO3溶液、稀盐酸与饱和NaCl溶液各洗涤三次;用无水硫酸镁干燥,过滤除去硫酸镁后将溶液减压旋蒸浓缩,利用乙酸乙酯/乙醚重结晶,即得。
实施例9
G2L-G3L不对称树状大分子纳米载药系统的合成路线如下:
化合物Alkyne-G2L-G3L的合成
在N2氛保护下,利用Pd/C在高压H2氛下脱去G2L-G3L(4.7g,2mmol)的Cbz保护基。在N2氛保护下,将脱去保护基得到的白色固体、HBTU(10.6g,12mmol)与HOBT(4.4g,12mmol)溶于100mL无水DMF中。在冰浴下,缓慢逐滴滴加入6mLDIPEA(39mmol)以及5-己炔酸(1.4mL,12mmol)。将其在冰浴下继续搅拌半小时,然后在室温下反应72小时。反应结束后,减压蒸馏移除溶 剂,并加入300mL EtOAc,将有机相分别用饱和NaHCO3溶液、稀盐酸(1M)与饱和NaCl溶液各洗涤三次。用无水硫酸镁干燥,过滤除去硫酸镁后将溶液减压旋蒸浓缩至15mL。利用乙酸乙酯/乙醚重结晶,得白色固体4.8g,产率96%。1H NMR(400MHz,CDCl3),δ=5.30(s,4H,CHCCH2),4.11(m,10H,COCH(NH)-CH2),3.07-3.73(t,32H,CH2CH2-NH and CHCCH2CH2CH2CO),2.04(t,8H,CHCCH2CH2),1.26-1.73(m,68H,CH2-Lys,CHCCH2CH2CH2CO and s,72H,CH3-Boc)。MALDI-TOF MS:m/z 2542[(M+Na)+,C126H216N22O30Na+]。
化合物mPEG-G2L-G3L的合成
将化合物alkyne-G2L-G3L(0.5g,0.2mmol)与叠氮化的PEG(2g,1mmol)溶于40mL水(含5mol%CuSO4·5H2O以及10mol%抗坏血酸钠)中。在50℃下搅拌反应24小时。反应结束后,利用透析袋(molecular weight cute off(MwCO=3500)透析除去溶剂及杂质,期间在透析的去离子水中加入大量的乙二胺四乙酸二钠盐(EDTA-Na2)帮助透析除去反应中的铜离子。将透析后留在透析袋中的溶液收集冻干得黄色固体。将该固体溶解后利用排阻色谱法,使用Superose 12HR/10/300GL色谱柱,通过FPLC系统(GE Healthcare)来分离提纯产物(利用pH为6.5的含30%乙腈的乙酸钠水溶液作为柱子的流动相)。避光条件下将收集到的产物部分利用纯水透析后冻干得黄色mPEG修饰的树状大分子产物(mPEG-G2L-G3L)2.0g,产率92.7%。MALDI-TOF MS:m/z 10710,[M+Na]+。化合物mPEG-G2L-G3L-alkyne的合成
在N2氛保护下,将化合物mPEG-G2L-G3L(480mg,0.05mmol)溶于2mL无水DCM中。将该溶液在冰浴下搅拌5分钟,然后滴加入2mL TFA。搅拌4小时后,除去溶剂并且真空干燥1小时收集样品。在N2氛保护下,将之前得到的样品、HATU(760mg,2mmol)、HOAt(272mg,2mmol)与5-己炔酸(0.22mL,2mmol)溶于30mL无水DMF中。在冰浴下,缓慢滴加入定量的DIPEA(1.3mL,8mmol)。将其在冰浴下继续搅拌半小时,然后在室温下反应72小时。反应结束后,减压蒸馏移除溶剂,并加入300mL EtOAc,将有机相分别用饱和NaHCO3溶液、稀盐酸(1M)与饱和NaCl溶液各洗涤三次。用无水硫酸镁干燥,过滤除去硫酸镁后将溶液减压旋蒸浓缩至15mL。利用乙酸乙酯/乙醚重结晶,得棕色固 体(mPEGylated G2L-G3L-alkyne)470mg,产率88.2%。MALDI-TOF MS:m/z 10651[M+Na]+。
化合物PEGylated Peptide Dendrimer-GFLG-PTX的合成
在Ar氛保护下,将化合物PEG-G2L-G3L-alkyne(540mg,50μmol)与叠氮化的GFLG-PTX(680mg,0.50mmol)溶于100mL水(含5mol%CuSO4·5H2O以及10mol%抗坏血酸钠)中。在50℃下搅拌反应24小时。反应结束后,利用透析袋(molecular weight cute off(MwCO=3500)透析除去溶剂及杂质,期间在透析的去离子水中加入大量的乙二胺四乙酸二钠盐(EDTA-Na2)帮助透析除去反应中的铜离子。将透析后留在透析袋中的溶液收集冻干得淡黄色固体。将该固体溶解后利用排阻色谱法,使用Superose 12 HR/10/300GL色谱柱,通过 FPLC系统(GE Healthcare)来分离提纯产物(利用pH为6.5的含30%乙腈的乙酸钠水溶液作为柱子的流动相)。将收集到的产物部分利用纯水透析后冻干得淡黄色产物846mg,产率91.2%。
在Ar氛保护下,将上面得到的淡黄色产物(500mg,28μmol)与叠氮化的Cy5.5(5mg,5μmol)溶于160mL水(含5mol%CuSO4·5H2O以及10mol%抗坏血酸钠)中。在50℃下搅拌反应24小时。反应结束后,利用透析袋(molecular weight cute off(MwCO=3500)透析除去溶剂及杂质,期间在透析的去离子水中加入大量的乙二胺四乙酸二钠盐(EDTA-2Na)帮助透析除去反应中的铜离子。将透析后留在透析袋中的溶液收集冻干得绿色固体。将该固体溶解后利用如前一个产物通过排阻色谱法提纯产物(利用pH为6.5的含30%乙腈的乙酸钠水溶液作为柱子的流动相)。避光条件下将收集到的产物部分利用纯水透析后冻干得绿色产物(PEGylated dendrimer-GFLG-PTX)420mg,产率83.3%。MALDI-TOF MS:m/z 18472,[M+H]+。通过荧光光谱检测,Cy5.5的含量接近0.5%,通过HPLC检测该偶联物中含有大约20.9%的药物(紫杉醇)。
由于上述合成中GFLG-PTX的接枝数量是不可控的过程,大分子的末端氨基接枝完全是最理想的状态,但实际上由于位阻效应及产率问题,会出现部分接枝的情况,实验结果得到平均接枝个数是5个。
为了将SAIR的这些必要功能整合进一个单一的树状支架中,本发明设计合成了两亲性的树状大分子-GFLG-PTX。为了高效地合成这种功能型的树状偶联物,采用点击反应来合成PEGylated dendrimer-GFLF-PTX。首先合成PEG修饰的树状大分子(dendrimer PEG-G2L-G3L-alkyne)。其通过MALDI-TOF质谱得出最大丰度峰为m/z=10,673,标记为[M+H]+(如图1所示),这表明有4个mPEG链段共价连接到树状支架上。其次,癌细胞内大量存在组织蛋白酶B,将对该酶敏感的N3-GFLG-PTX分子与聚乙二醇化修饰的树状大分子(PEG-G2L-G3L-alkyne)进行偶联。另外,Cy5.5通过点击反应也共价连接到了树状大分子上。利用稀的EDTA溶液通过透析的方法将反应中可能引起毒性的微量铜离子清除干净。最后,得到的初产物通过FPLC系统利用排阻色谱柱(Superose 12 HR/16/30)进行进一步提纯。将分离下来的液体进行透析、冻干,得到两亲性的PEGylated dendrimer-GFLF-PTX偶联物。通过MALDI-TOF飞行质谱检测其分子量,得到最大丰度峰为m/z=18,472,其为产物PEGylated dendrimer-GFLF-PTX,并标记为[M+H]+(如图2所示)。这表明大约一个PEGylated dendron单元平均连接有5个GFLG-PTX。这比理论的8个略少,该结果可能的原因是化学反应过程中存在着较大的空间位阻。通过HPLC检测得出载药量为20.9wt%,与MALDI-TOF质谱得到的结果相吻合。由于药物是共价连接到树状大分子上,该聚乙二醇化修饰的树状偶联物在PBS或者血液循环中是稳定的。
实施例10
偶联体Ⅰ和偶联体Ⅱ的合成方法参照实施例9,大分子原料为G1L-G2L代树状大分子。
偶联体Ⅴ的合成方法参照实施例10,大分子原料为G3L-G4L代树状大分子。
实施例11
不对称树状大分子的合成路线如下:
化合物G1L的合成
在N2氛保护下,Z-Lys(Z)-OH(14.5g,35mmol)和N-Boc-ethylenediamine(7.9g,42mmol)溶于100mL无水DMF中。将该溶液在冰浴下搅拌5分钟,然后加入DIPEA(29mL,175mmol)。再将HBTU(16.1g,47mmol)和HOBT(6.7g,47mmol)加入溶液体系中。将其在冰浴下搅拌半小时,然后在室温下反应24小时。反应结束后,减压蒸馏移除溶剂,并加入300mL EtOAc,将有机相分别用饱和NaHCO3溶液、稀盐酸(1M)与饱和NaCl溶液各洗涤三次。用无水硫酸镁干燥,过滤除去硫酸镁后将溶液减压旋蒸浓缩至15mL。利用乙酸乙酯/乙醚重结晶,得白色固体17.8g,产率91.4%。1H NMR(400MHz,CDCl3),δ=7.34(s,10H,NHCOOCH2Ph-H),5.09(s,4H,OCH2-Ph),4.08(m,1H,CH(NHCbz)-CH2),3.33-3.19(t,6H,CH2CH2-NH),1.25-1.83(m,6H,CH2-Lys and s,9H,CH3-Boc)。MALDI-TOF MS:m/z=559[(M+H)+,C29H41N4O7+]。
化合物G1L-G1L的合成
在N2氛保护下,G1L(14.4g,26mmol)溶于20mL无水DCM中。将该溶液在冰浴下搅拌5分钟,然后滴加入20mL TFA(260mmol)。搅拌4小时后,除去溶剂。加入无水乙醚,产生白色固体粉末。分离收集白色固体粉末即为G1L脱去Boc基团后产物。在N2氛保护下,将之前收集的白色固体、Boc-Lys(Boc)-OH (10.9g,31mmol)、HBTU(12.1g,36mmol)以及HOBT(5.0g,36mmol)溶于100mL DMF中。在冰浴下,缓慢滴加入22mL DIPEA(130mmol)。将其在冰浴下继续搅拌半小时,然后在室温下反应24小时。反应结束后,减压蒸馏移除溶剂,并加入300mL EtOAc,将有机相分别用饱和NaHCO3溶液、稀盐酸(1M)与饱和NaCl溶液各洗涤三次。用无水硫酸镁干燥,过滤除去硫酸镁后将溶液减压旋蒸浓缩至15mL。利用乙酸乙酯/乙醚重结晶,得白色固体17.7g,产率86.8%。1H NMR(400MHz,CDCl3),δ=7.34(s,10H,NHCOOCH2Ph-H),5.09(s,4H,OCH2-Ph),4.08(m,2H,CH(NHCbz)-CH2),3.33-3.19(t,8H,CH2CH2-NH),1.25-1.83(m,12H,CH2-Lys and s,18H,CH3-Boc)。MALDI-TOF MS:m/z 785[(M+H)+,C40H61N6O10+],807[(M+Na)+,C40H60N6O1ONa+]。
化合物G1L-G2L的合成
在N2氛保护下,参照“化合物G1L-G1L”合成中脱Boc保护基的方法脱去G1L-G1L(10.6g,14mmol)的Boc保护基。在N2氛保护下,将脱去保护基得到的白色固体、Boc-Lys(Boc)-OH(11.3g,32mmol)、HBTU(14.4g,38mmol)与HOBT(5.2g,38mmol)溶于100mL无水DMF中。在冰浴下,缓慢滴加入23mL DIPEA(135mmol)。将其在冰浴下继续搅拌半小时,然后在室温下反应24小时。反应结束后,减压蒸馏移除溶剂,并加入300mL EtOAc,将有机相分别用饱和NaHCO3溶液、稀盐酸(1M)与饱和NaCl溶液各洗涤三次。用无水硫酸镁干燥,过滤除去硫酸镁后将溶液减压旋蒸浓缩至15mL。利用乙酸乙酯/乙醚重结晶,得白色固体16.1g,产率92.4%。1H NMR(400MHz,CDCl3),δ=7.33(s,10H,NHCOOCH2Ph-H),5.08(s,4H,OCH2-Ph),4.08(m,4H,COCH(NH)-CH2),3.00-3.20(t,12H,CH2CH2-NH),1.25-1.80(m,24H,CH2-Lys and s,36H,CH3-Boc)。MALDI-TOF MS:m/z 1264[(M+Na)+,C62H100N10O16Na+]。
化合物G2L-G2L的合成
将G1L-G2L(14.5g,12mmol)溶于60mL甲醇(HPLC级)中;然后将Pd/C(6g)加入溶液中。在H2氛下,保持高压反应釜中压力为0.8MPa,搅拌反应48小时。反应结束后,过滤除去Pd/C,旋蒸除去溶剂得到脱去“Cbz保护基”的产物。在N2氛保护下,在支管瓶中加入脱去的“Cbz保护基”的产物、Z-Lys(Z)-OH (11.5g,28mmol)、HBTU(12.1g,32mmol)以及HOBT(4.4g,32mmol),并加入100mL无水DMF使其溶解。在冰浴下,缓慢滴加入19mL DIPEA(115mmol)。将其在冰浴下继续搅拌半小时,然后在室温下反应24小时。反应结束后,减压蒸馏移除溶剂,并加入300mL EtOAc,将有机相分别用饱和NaHCO3溶液、稀盐酸(1M)与饱和NaCl溶液各洗涤三次。用无水硫酸镁干燥,过滤除去硫酸镁后将溶液减压旋蒸浓缩至15mL。利用乙酸乙酯/乙醚重结晶,得白色固体16.6g,产率78.2%。1H NMR(400MHz,CDCl3),δ=7.33(s,20H,NHCOOCH2Ph-H),5.06(s,8H,OCH2-Ph),4.08(m,6H,COCH(NH)-CH2),2.95-3.25(t,16H,CH2CH2-NH),1.25-1.73(m,36H,CH2-Lys and s,36H,CH3-Boc)。MALDI-TOF MS:m/z 1789[(M+Na)+,C90H136N14O22Na+]。
化合物G2L-G3L的合成
在N2氛保护下,参照“化合物G1L-G1L”合成中脱Boc保护基的方法脱去G2L-G2L(13.2g,7.5mmol)的Boc保护基。在N2氛保护下,将脱去保护基得到的白色固体、Boc-Lys(Boc)-OH(13.0g,38mmol)、HBTU(16.0g,42mmol)与HOBT(5.7g,42mmol)溶于100mL无水DMF中。在冰浴下,缓慢滴加入25mL DIPEA(150mmol)。将其在冰浴下继续搅拌半小时,然后在室温下反应24小时。反应结束后,减压蒸馏移除溶剂,并加入300mL EtOAc,将有机相分别用饱和NaHCO3溶液、稀盐酸(1M)与饱和NaCl溶液各洗涤三次。用无水硫酸镁干燥,过滤除去硫酸镁后将溶液减压旋蒸浓缩至15mL。利用乙酸乙酯/乙醚重结晶,得白色固体17.6g,产率87.7%。1H NMR(400MHz,DMSO),δ=7.33(s,20H,NHCOOCH2Ph-H),5.07(s,8H,OCH2-Ph),4.08(m,10H,COCH(NH)-CH2),3.07-3.73(t,24H,CH2CH2-NH),1.26-1.73(m,60H,CH2-Lys and s,72H,CH3-Boc)。MALDI-TOF MS:m/z 2701[(M+Na)+,C134H216N22O34Na+]。
化合物G3L-G4L的合成
先参照化合物G2L-G2L的合成得到化合物G3L-G3L,再参照化合物G2L-G3L的合成得到化合物G3L-G4L。
本发明对G2L-G3L树状大分子-紫杉醇偶联物进行实验检测:
一、纳米粒子的粒径、形貌、zeta电位与稳定性的表征
利用DLS和扫描电子显微镜(SEM)来实时观测该偶联物在不同的条件下的粒径与形貌。首先,用去离子水制备好PBS缓冲液(pH 7.4)和McIlvaine’s缓冲液(pH 5.4)。将该偶联物分别溶于这两种缓冲溶液中,并形成纳米粒子。再取部分材料(1mg/mL)溶解于McIlvaine’s缓冲液中,37℃下与木瓜蛋白酶(2.0μM,pH=5.4)共孵育6小时。与此同时,在37℃无木瓜蛋白酶条件下,将纳米粒子分别孵育在两种不同的缓冲溶液中(1mg/mL;pH=7.4及pH=5.4)24小时。在不同的时间点,利用DLS对其粒径和zeta电位进行检测。在特定的时间点如0h,1h,4h和6h,取出部分溶液进行稀释制备SEM样品。
图3-A表明样品在水中聚集成纳米粒子,平均水相粒径大约为69nm,并且粒径分布狭窄,PDI=0.230。同时,利用AFM来对样品进行形貌分析,观察到粒径约74nm的均匀球形结构(图3-B),该结果与DLS检测结果非常接近。通过AFM进行粒径测量得出的结果也与DLS的结果一致。通过DLS与AFM得出的纳米级尺寸和狭窄的粒径分布表明我们制备的两亲性肽类树状大分子-PTX偶联物可以自组装成尺寸均一的纳米粒子。自组装等多种策略被设计来构筑纳米级载体。然而,树状大分子等亲水性材料无法单独进行化学或物理相互作用。在本研究中,自组装是由PEGylated dendrimer-GFLF-PTX自身介导的。自组装的驱动力可能归因于界面能的减少,其受到柔性PEG亲水作用与dendrimer-GFLF-PTX片段疏水作用之间的平衡影响。此外,其他诸如氢键,π-π堆叠和偶极作用等因素也可能影响到偶联物的自组装行为。此外,小的粒子比大的粒子能更快的渗透进肿瘤组织,然而极小的粒子会轻易被肝肾清除。在增强对肿瘤组织的渗透能力(通过降低纳米药物尺寸)与降低对正常组织的毒性以及MPS清除作用(通过增大纳米药物尺寸)之间寻找平衡是一个巨大的挑战。因此,本发明的纳米粒子尺寸大约为60-80nm,是具有长循环和更强肿瘤渗透性的载体。
理想的药物递送系统要求在经过血液长循环后能富集到肿瘤组织。纳米粒子的表面电荷在其经历血液循环过程中扮演着关键角色。利用DLS检测了PEGylated dendrimer-GFLF-PTX纳米粒子的zeta电位,显示表面电荷为电负性。 静脉注射正电荷纳米粒子会被巨噬细胞或者网状内皮系统(RES)从循环系统中快速清除。相反,电负性或者电中性的纳米粒子理论上将减少异常的蛋白结合,并且因此较低概率刺激到免疫系统,最终导致更长的循环半衰期。所以,该PEGylated dendrimer-GFLF-PTX偶联物具有合适的纳米级尺寸与弱表面负电荷,其作为一个药物递送系统能相应地具有机会通过EPR被动靶向效应富集到肿瘤组织,并且避免被快速清除,最终导致抗癌疗效增强。
二、药物释放试验
药物释放缓慢限制了药物递送系统的抗癌效率。一旦纳米粒子药物递送系统内化进入癌细胞后,本发明设计了一个智能的基于偶联物的纳米粒子,其在胞内能被癌细胞内过表达的组织蛋白酶B触发。因此,评价这些载体对刺激响应的能力就显得至关重要。木瓜蛋白酶具有与溶酶体内组织蛋白酶B相似的活性,其被选中来评价酶敏感药物释放情况。通过有酶/无酶(木瓜蛋白酶)不同条件的实验来检测其体外药物释放,并利用RP-HPLC来测试结果。
通过将mPEGylated dendrimer-GFLG-PTX分别置于含有木瓜蛋白酶和不含木瓜蛋白酶的溶液中进行药物释放研究。参照第三章3.2.4配置最终的缓冲溶液。将纳米粒子加入该缓冲溶液(酶浓度为2.0μM,pH=5.4)中并在37℃下孵育48个小时,其中纳米粒子浓度为3mg/mL。与此同时,相同条件下将该纳米粒子(3mg/mL)加入不含酶的缓冲溶液中(pH=7.4的PBS缓冲溶液与pH=5.4的McIlvaine缓冲溶液),同样在37℃下孵育48个小时。在特定的时间点,将样品取出并利用高效液相色谱仪分析(液相分析柱C8column 4.6×150mm),其中流动相选择为(流动相A:去离子水,流动相B:乙腈),以1.5mL/min的流速进行20分钟的洗脱(流动相B含量为50%),检测波长为227nm。将释放断裂下来的样品经过HPLC分离收集并利用ESI-MS表征分析。
图4表明PTX标准样品(如图4-A,peak:5.6min)与释放断裂下来的样品(如图4-B,peak:5.6min)具有同一的洗脱时间,这意味着从纳米粒子断裂释放下来的产物为原药紫杉醇。此外,将断裂下来的、峰值为5.6min的产物收集并用ESI-MS表征。这些发现表明在木瓜蛋白酶存在下的氨解反应没有明显影响到PTX的结构。
利用RP-HPLC检测不同时间点释放出来的PTX的浓度,结果表明PEGylated dendrimer-GFLF-PTX纳米粒子的药物释放存在酶响应性的特性。在木瓜蛋白酶存在的实验中,观测到快速的药物释放,与木瓜蛋白酶孵育8小时后大约有50%的PTX得到释放,并且在24小时后释放出了约90%的PTX。反之,无酶孵育的实验中,经过48小时后在pH=7.4或5.4的缓冲液中检测到仅有少量的PTX被断裂释放(如图4-C)。其间,胎牛血清的PBS(pH=7.4)缓冲液被选为一个模板来评价纳米粒子在血液中循环过程中药物释放活力。这些结果表明PEGylated dendrimer-GFLF-PTX存在高的木瓜蛋白酶选择性和高的稳定性。此外,尽管PTX分子的酯键的水解作用是不可避免的,偶联物为基础的纳米粒子表现出了高的稳定性并且PTX在循环系统中得到了载体的有效保护。这些更低的调理素作用率、高的稳定性以及循环过程中最少量的药物泄露有助于毒副作用的降低以及抗癌效率损失的最低化。
实验结果表明PEG保护的纳米粒子在循环系统中能保持稳定,并且酶敏感的纳米粒子可以在组织蛋白酶B的存在下释放出疏水药物PTX。DLS和SEM被用来实时观测纳米粒子在不同pH条件下以及有无酶孵育的情况下粒子的尺寸和形貌变化情况。疏水单元的断裂释放导致了自组装作用的减弱,这反过来导致纳米粒子结构不再紧密而在聚集过程中坍塌。无酶孵育下,粒子在缓冲液下24小时内保持大约60–140nm(如图5所示),表明没有明显的结构坍塌。相反,经过木瓜蛋白酶酶孵育的粒子被发现结构松散了,如DLS检测发现伴随有大而宽的PDIs(如图6所示)。而且通过SEM进行形貌学分析显示了相似的结果。孵育4小时后即观察到粒子的粒径增大并且发生坍塌。这些结果表明这些粒子在循环过程中能稳定。由组织蛋白酶B引起的粒径和形貌上的改变更进一步证明了酶敏感性能以及酶诱发的粒子坍塌有利于快速地药物释放。
三、体内抗肿瘤效果评价
在接种过4T1鼠源乳腺癌细胞的BALB/c小鼠上进行酶敏感的PEGylated dendrimer-GFLF-PTX纳米粒子与游离PTX的抗肿瘤效果及毒性研究。将荷瘤小鼠随机分配成三组(每组7只)并进行标记,然后通过尾静脉注射向小鼠注射药剂(200μL),分别是紫杉醇注射液、PEGylated dendrimer-GLFG-PTX形成 的纳米粒子以及作为对照组的生理盐水,其中每一次注射给药量统一为5mg/kg PTX,每天注射一次共注射10天。每两天检测一次小鼠的肿瘤体积与体重,并将第一天测定的肿瘤体积与小鼠体重分别设定为100%。在第19天的时候,对所有小鼠实施安乐死,将肿瘤进行剥离并称重,然后按照抑瘤率公式计算抑瘤率(TGI)。通过肿瘤组织的免疫组化分析进一步地研究肿瘤的新生血管、增殖与细胞凋亡情况。
每天检测小鼠的临床症状,将第一天测定的肿瘤体积和体重设定为100%并且每隔一天测量一次。图7-A显示了肿瘤体积随时间变化而增长,从第三次治疗开始所有药物治疗组小鼠的肿瘤相较于对照组呈现出有意义的增长阻滞。相比紫杉醇和生理盐水治疗组,纳米粒子治疗组有效地抑制了肿瘤的生长。图7-A显示在第19天的时候,各组小鼠相对肿瘤体积大小分别为972±134%(对照组)、902±152%(PTX治疗组)和383±71%(NPs治疗组)。特别是,经过19天的治疗后,如肿瘤生长曲线图(图7-A,p<0.001相对于生理盐水和p<0.002相对于临床注射药物PTX)和肉眼可见的实体瘤的大小,纳米粒子治疗小组比使用相同给药方案的紫杉醇治疗小组表现出显著增强的抗肿瘤功效。纳米粒子治疗组的肿瘤显著性地比其他治疗组的(对照组和PTX治疗组)肿瘤更小。治疗结束后,抑瘤率(TGI)和瘤重被测量并计算。图7-C和图7-D显示PTX注射液治疗组和纳米粒子治疗组的瘤重分别为994.2±158.1mg与465.3±84.8mg(p<0.0001相对于生理盐水组,p<0.001相对于游离药物紫杉醇注射液组)。此外,各自的TGIs分别为16.6%(PTX)和61.0%(NPs),这表明纳米粒子具有更高的抗肿瘤疗效。这些结果得益于旨在全面提高抗肿瘤效果(SAIR)方案的设计。具体可以归因于材料负电性的表面、合适的纳米级尺度(图7-A)以及更低的调理素作用率等特性提高了药物在循环过程的稳定性。同时这些特征导致了其通过EPR效应快速地富集累积到肿瘤组织,经癌细胞内吞以及摄取后在胞内溶酶体中实现紫杉醇的高效快速释放。体内抗肿瘤研究表明该PEG修饰的树状大分子纳米粒子满足SAIR的标准,因此全方位地提高了其抗肿瘤疗效。
四、体内毒性评价
将6到8周大的健康雌性BALB/c小鼠(体重约20±2g)随机分为三组,每组7只并对每只小鼠进行标记。然后通过尾静脉注射向小鼠注射药剂(200μL),分别是紫杉醇注射液、PEGylated dendrimer-GFLF-PTX形成的纳米粒子以及作为对照组的生理盐水,其中注射给药量统一为5mg/kg PTX,每天注射一次共注射10次。每两天记录一次小鼠的体重以及观察小鼠的行为并且将第一天的体重设定为100%。19天后,小鼠被实施安乐死,收集其血液进行分析。
在整个治疗的研究过程中,纳米粒子的重复注射普遍耐受良好,并且小鼠没有表现出任何显著的体重降低(图8)。治疗周期结束后,对小鼠实施安乐死,将其血液收集来进行常规分析。
为了进一步评定PTX注射液和PEGylated dendrimer-GFLF-PTX纳米粒子对血液的生物相容性,本发明进行了血液学分析和血常规检测。这些血液学标记物的水平大部分在正常范围内(图9)。其间,PEGylated dendrimer-GFLF-PTX纳米粒子治疗组小鼠的中性粒细胞(GR#)和血小板计数(PLT)与生理盐水组的小鼠相似。相反,临床药PTX注射液治疗组小鼠的这些标记物的水平发生显著的降低。观测到纳米粒子的血液学毒性(PTX剂量限制毒性)极低甚至没有毒性,而这有助于将来给药剂量的提升。这些结果表明通过静脉注射给药的PEGylated dendrimer-GFLF-PTX是一种具有高的生物相容性和低的血液毒性的有潜力的药物递送系统。
下表为本发明实验所用的主要药品。
试剂
N,N二甲基甲酰胺加入无水CaH2干燥24h后,重蒸,减压收集中间馏分。
二氯甲烷加入氢化钙过夜,常压蒸馏,弃去前馏分,收集中间馏分,密封置干燥器暂时保存。现蒸现用,存储时间不超过2周。
其他试剂未特别说明时为分析纯,未进一步纯化处理,直接使用。
仪器
磁力加热搅拌器(德国IKA公司)。
BP211D型电子天平(德国Sartorius公司)。
真空干燥箱(上海一恒科技有限公司,DZF-6050型)。
电热鼓风干燥箱(上海一恒科技有限公司,DHG-9140型)。
高速离心机(德国,eppendorf)
高分辨超导核磁共振波谱仪(瑞士Bruker公司,Bruker AV II-400MHz)。
飞行质谱仪(美国布鲁克·道尔顿公司,Autoflex MALDI-TOF/TOF)。
液相色谱质谱联用仪(美国ABI公司,API3000LC-MS/MS)。
质谱仪(美国Waters公司,Q-TOF Premier)。
傅立叶红外光谱仪(美国Thermo公司,FT-IR,Nicolet 6700)。
快速蛋白液相色谱(瑞典,GE Healthcare,FPLC system)。
紫外光谱仪(美国Perkin-Elmer)。
荧光分光光度计(日本HITACHI公司,F700)。
纳米粒度及电位分析仪(英国Malvern公司,DTS software version 3.32)。
原子力显微镜(美国Digital Instruments公司,Asylum Research MFP-3D-Bio)。
高效液相色谱仪(美国Agilent,1260LC,Zorbax C8column 4.6×150mm)。
激光共聚焦显微镜(德国Leica,TCS SP5)。
流式细胞仪(美国,BD公司)。
pH计(瑞士,梅特勒-托多利)。
活体成像仪(美国,Cri,Maestro In-Vivo Imaging System) 。
- 还没有人留言评论。精彩留言会获得点赞!