IL-13拮抗剂用于治疗特应性皮炎的用途的制作方法
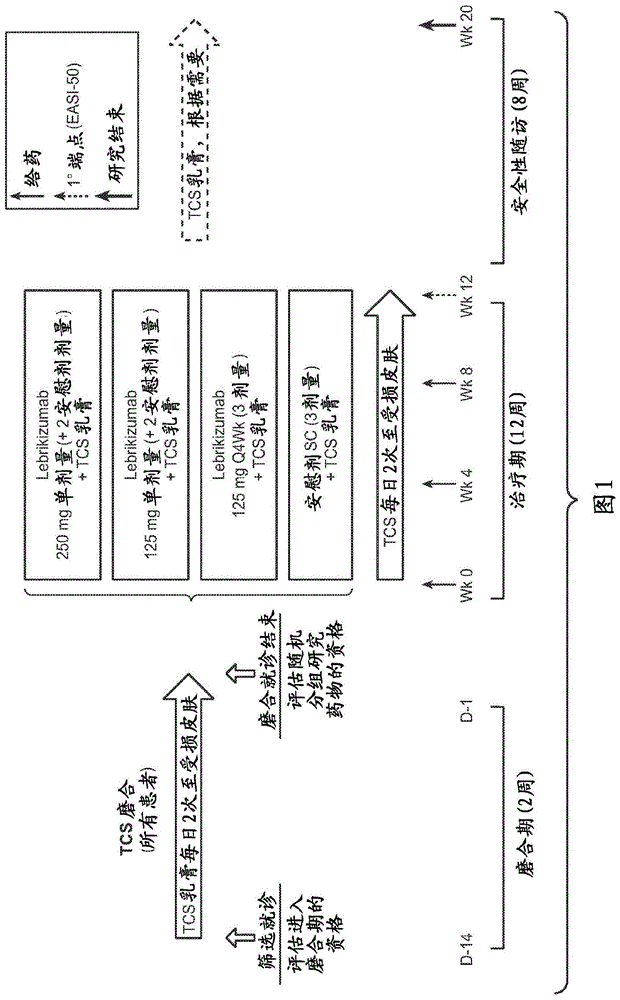
本申请要求2016年9月23日提交的美国临时申请号62/398,713、2017年6月30日提交的美国临时申请号62/527,204、2017年7月10日提交的美国临时申请号62/530,683和2017年7月31日提交的美国临时申请号62/539,037的优先权,其全部内容通过引用并入本文。序列表本申请包含已经通过efs-web提交的序列表,并且其全部内容通过引用并入本文。2017年8月30日创建的所述ascii拷贝命名为p33854-wo.sl.txt,大小为12,532字节。本发明提供了il-13拮抗剂用于治疗特应性皮炎的用途。还提供了治疗特应性皮炎的方法和通过施用il-13拮抗剂降低特应性皮炎严重性的方法。
背景技术:
:特应性皮炎(ad)是影响所有年龄组的慢性复发和缓解性炎性皮肤病。临床上,ad的特征在于干燥、红斑性结痂皮疹、苔藓化、皮肤屏障受损和强烈瘙痒(biebert.,nengljmed2008;358:1483-94)。患有ad的患者具有高度的疾病负担,并且其生活质量(qol)受到显著影响。在一项研究中,ad显示出对患者心理健康的负面影响大于糖尿病和高血压(zuberbiert等人,jallergyclinimmunol2006;118:226-32)。患有中度至重度ad的患者具有较高的社交功能障碍和睡眠障碍的患病率,其与疾病的严重性直接相关(williamsh等人,jallergyclinimmunol2008;121:947-54.e15)。抑郁、焦虑和社交功能障碍不仅影响ad患者,还影响其看护者(zuberbiert等人,jallergyclinimmunol2006;118:226-32)。与银屑病(另一种常见和使人衰弱的皮肤病)相比,ad患者具有较低的生理职能(rolephysical)、活力、社交功能、角色情绪和心理健康亚量评分(kiebertg等人,intjdermatol2002;41:151-8)。白介素(il)-13被认为是2型t辅助细胞(th2)炎症的关键介质,升高的il-13水平不仅与特应性皮炎相关,而且与许多其他疾病相关,包括但不限于哮喘、炎性肠疾病、特发性肺纤维化(ipf)和慢性阻塞性肺疾病(copd)(ohck等人,eurrespirrev19:46–54(2010);fahyjv等人,natrevimmunol15:57-65[2015])。il-13由许多细胞类型产生,包括th2细胞、嗜碱性粒细胞、嗜酸性粒细胞和肥大细胞、以及气道上皮细胞和2型先天淋巴样细胞。il-13与异二聚体受体il-4rα/il-13rα1结合(与il-4共享该异二聚体受体),并激活stat-6信号传导通路(hersheygk,jallergyclinimmunol111(4):677-90[2003])。因为th2炎症涉及th2细胞以外的几种细胞类型的活性,包括2型先天淋巴样细胞(ilc2s),所以“th2炎症”最近在科学文献中被称为“2型炎症”。除th2细胞外,已经将ilc2鉴定为细胞因子(如il-5和il-13)的重要来源。因此,先前已被鉴定为th2细胞因子的细胞因子如il-13和il-5现在在科学文献中也称为2型细胞因子。同样,与这些细胞因子相关的疾病状态,包括特应性皮炎,现在也被称为2型驱动的疾病或2型相关疾病。参见,例如,noonan等人,j.allergyclinimmunol.,132(3):567-574(2013);hanania等人,thorax70(8):748-56(2015);和cai等人,bioanalysis8(4):323-332(2016)。嗜酸性粒细胞性炎症与多种疾病相关,包括过敏性和非过敏性疾病(gonlugur(2006)immunol.invest.35(1):29-45)。炎症是活组织对损伤的恢复反应。炎性反应的特征是由组织本身产生的某些化学物质导致的白细胞在受损组织中的累积。嗜酸性粒细胞白细胞在多种病症中累积,如过敏性疾病、蠕虫感染和肿瘤性疾病(kudlacz等人,(2002)inflammation26:111–119)。嗜酸性粒细胞白细胞是免疫系统的组分,其是粘膜表面的防御元素。它们不仅对抗原、还对寄生虫、化学物质和创伤反应。组织嗜酸性粒细胞增多发生在皮肤病中,如湿疹、天疱疮、急性荨麻疹和毒性表皮坏死松解症以及特应性皮炎(rzany等人,br.j.dermatol.135:6-11(1996))。在ige介导的过敏性皮肤反应中嗜酸性粒细胞累积在组织和空颗粒蛋白中(nielsen等人,ann.allergyasthmaimmunol.,85:489-494(2001))。嗜酸性粒细胞与肥大细胞组合可能引起关节炎症(miossec,j.clin.rheumatol.3:81-83(1997))。嗜酸性粒细胞性炎症有时伴有关节创伤。滑液嗜酸性粒细胞增多可与以下疾病相关:如类风湿性关节炎、寄生虫病、嗜酸性粒细胞增多症、莱姆病和过敏性过程以及关节积血和关节造影(atanes等人,scand.j.rheumatol.,25:183-185(1996))。嗜酸性粒细胞性炎症也可以影响骨骼(yetiser等人,int.j.pediatr.otorhinolaryngol.,62:169-173(2002))。嗜酸性粒细胞性肌病的实例包括嗜酸性粒细胞性肌周炎、嗜酸性粒细胞性多发性肌炎和局灶性嗜酸性粒细胞性肌炎(lakhanpal等人,semin.arthritisrheum.,17:331-231(1988))。影响骨骼肌的嗜酸性粒细胞性炎症可能与寄生虫感染或药物或嗜酸性粒细胞增多症的某些全身性疾患(如特发性嗜酸性粒细胞增多症和嗜酸性粒细胞增多-肌痛综合征)的特征有关。嗜酸性粒细胞参与对自身免疫抗体识别的表位的炎性反应(engineer等人,cytokine,13:32-38(2001))。结缔组织疾病可导致中性粒细胞、嗜酸性粒细胞或淋巴细胞性血管炎症(chen等人,j.am.acad.dermatol.,35:173-182(1996))。组织和外周血嗜酸性粒细胞增多可发生在活动性风湿性疾病中。强直性脊柱炎(一种结缔组织病)中血清ecp水平的升高表明嗜酸性粒细胞也参与潜在的过程(feltelius等人,ann.rheum.dis.,46:403-407(1987))。韦格纳肉芽肿病可罕见地出现肺结节、胸腔积液和外周血嗜酸性粒细胞增多(krupsky等人,chest,104:1290-1292(1993))。至少400/mm3的外周血嗜酸性粒细胞增多可发生在系统性硬化的7%的病例中、局限性硬皮病的31%的病例中和嗜酸性粒细胞筋膜炎的61%的病例中(falanga等人,j.am.acad.dermatol.,17:648-656(1987))。硬皮病产生的炎症过程与迈斯纳氏和奥尔巴赫氏神经丛的非常相似,其由胃肠系统中的肥大细胞和嗜酸性粒细胞白细胞组成。源自嗜酸性粒细胞的神经毒素可导致胃肠运动功能障碍,如发生在硬皮病中的(deschryver-kecskemeti等人arch.pathol.labmed.,113:394-398(1989))。嗜酸性粒细胞可伴随局部(varga等人,curr.opin.rheumatol.,9:562-570(1997))或全身(bouros等人,am.j.respir.crit.caremed.,165:1581-1586(2002))结缔组织增生。它们可以通过抑制成纤维细胞中的蛋白多糖降解来诱导纤维化(hernnas等人,eur.j.cellbiol.,59:352-363(1992)),成纤维细胞通过分泌gm-csf介导嗜酸性粒细胞存活(vancheri等人,am.j.respir.cellmol.biol.,1:289-214(1989))。嗜酸性粒细胞可以在鼻中(bacherct等人,j.allergyclin.immunol.,107:607-614(2001))、支气管中(arguelles等人,arch.intern.med.,143:570-571(1983))和胃肠息肉组织中(assarian等人,hum.pathol.,16:311-312(1985))发现。同样,嗜酸性粒细胞可定位于炎性假瘤(肌成纤维细胞瘤)中。嗜酸性粒细胞通常伴随眼眶区域的炎性假瘤,在这种情况下,病症可以模拟血管性水肿或过敏性鼻结膜炎(li等人,ann.allergy,69:101-105(1992))。嗜酸性粒细胞性炎症可以在组织创伤中发现(例如,由于手术或损伤所致)。嗜酸性粒细胞性炎症也可与心血管疾病(例如,嗜酸性粒细胞性心肌炎、嗜酸性粒细胞性冠状动脉炎、缺血性心脏病、急性心肌梗塞、心脏破裂)相关。坏死性炎症过程还可涉及嗜酸性粒细胞性炎症(多发性肌炎、冠状动脉夹层、神经-白塞病的坏死性病灶、痴呆、脑梗塞)。几个证据表明il-13是特应性皮炎(ad)中的关键致病成分。报道在ad皮肤中il-13的表达增加一致(hamidq等人,jallergyclinimmunol98:225-31[1996];jeongcw等人,clinexpallergy33:1717-24[2003];tazawat等人,archdermatolres295:459-64[2004];neismm等人,jallergyclinimmunol118:930-7[2006];m等人,jallergyclinimmunol132:361-70[2013];choydf等人,jallergyclinimmunol.130:1335-43[2012]),一些报道提示il-13表达与疾病严重性之间的关系(lagruttas等人,allergy60:391-5[2005])。在ad患者的血清中也报道了il-13的增加(novakn等人,jinvestdermatol2002;119:870-5;国际专利申请号pct/us2016/022481[公开号wo2016149276])。一些研究报道了ad患者血液中表达il-13的t细胞增加(akdism等人,jimmunol1997;159:4611-9;alekszam等人,brjdermatol2002;147:1135-41;lagruttas等人,allergy2005;60:391-5)。因此,il-13及其受体已成为治疗各种2型炎症相关疾病(包括哮喘、ipf和ad)的治疗靶点(correnj等人,nengjmed365:1088-98[2011];scheerensh等人,clinexpallergy44:38–46[2014];beckla等人,nengjmed371:130-9[2014];thacid等人,lancet2016;387:40-52)。讨论特应性皮炎中il-13或lebrikizumab的其他出版物包括hejq等人,genesimmun2003;4:385–89;kimbe等人clinimmunology2008;126,332–7;bhogalrk&bonacaintrevimmunol2008;27:472–96;kimst等人jgenemed2009;11:26–37;biebert等人jallergyclinimmunol2014;133:ab404;thacid等人jallergyclinimmunol2014;133:ab192;2.ultschm等人jmolbiol2013;425:1330–1339。此外,研究具有广泛抗炎活性作用的试剂的几项临床研究已经证明il-13表达的降低与改善的临床疾病相关。例如,19名患有中度至重度ad的成年人患者用环孢菌素a治疗12周,表明皮肤中il-13的表达减少(khattris等人,jallergyclinimmunol2014;133(6):1626-34),用环孢菌素a微乳液治疗的10名儿科患者表现出血液中表达il-13的cd3+t细胞减少(bunikowskir等人,pediatrallergyimmunol2001;12:216-23),12名患有中度至重度ad的成年人用窄带紫外线b处理显示il-13皮肤表达的显著降低(tintles等人,jallergyclinimmunol2011;128:583-93.e1-4)。已经描述了许多il-13拮抗剂并在各种2型炎症相关疾病(包括哮喘、copd和ipf)中进行临床测试。这些il-13拮抗剂包括ima-026、ima-638(也称为anrukinzumab,innno.910649-32-0;qax-576);tralokinumab(也称为cat-354,cas号1044515-88-9);和aer-001、abt-308(也称为人源化13c5.5抗体)。此外,已经开发了某些il-4受体α拮抗剂,它们是il-13和il-4的拮抗剂。il-4受体α拮抗剂的实例包括amg-317、air-645、dupilumab(其已在临床上在特应性皮炎以及哮喘中进行了测试(参见,例如beckla等人,nengjmed371:130-9[2014]))和aer-001(il4/il-13捕获)。另一种il-13拮抗剂是lebrikizumab。lebrikizumab是人源化单克隆免疫球蛋白(ig)g4抗体(huigg4),其在铰链区具有突变以增加稳定性。lebrikizumab以高亲和力特异性结合可溶性人il-13,并以高效力中和其功能活性。lebrikizumab通过il-4rα/il-13rα1受体抑制il-13信号传导。它阻断il-13与il-4rα的结合,但不阻断il-13与il-13rα1或il-13rα2的结合。lebrikizumab已在各种出版物中描述并在若干哮喘研究中进行测试(参见,例如,corren等人(2011)nengljmed365:1088-98;scheerens等人(2012)amjrespircritcaremed185:a3960;jia等人(2012)jallergyclinimmunol130:647-654e10;hanania等人,thorax2015;70:748–756;hanania等人,lancetrespirmed2016,提供于dx(dot)doi(dot)org(slash)s2213-2600(16)30265-x,线上公开于2016年9月5日;wo2012/083132)。ad的治疗方法主要包括避免触发、通过沐浴使皮肤水合以及使用润肤剂和主要由局部皮质类固醇(tcs)组成的抗炎疗法。在许多患者中,使用tcs治疗可提供一些症状缓解措施,但不能充分控制其疾病。此外,tcs的使用与许多合并症相关,并具有局限性,包括高的患者负担。全身免疫抑制剂治疗和口服糖皮质激素治疗中度至重度ad的随机、对照、盲法临床试验的文献综述(包括pubmed、nankervis系统综述、湿疹试验的全球资源[great]数据库)突出了常规全身免疫抑制剂治疗的使用通常受到显著副作用的限制;另外,它们的使用主要是药品的标示外使用。这些发现表明在开发靶向潜在ad特异性通路的药物(如最近批准的抗il-4rα抗体dupilumab)中存在大量未满足的需求以及其重要性。在难治性ad中进行生物治疗的前景是提供更有效的治疗,这样的治疗减少对全身免疫抑制治疗的需求,并最终减少对集中的tcs治疗的需求。dupilumab是第一个也是唯一一个批准用于治疗中度至重度ad成年人的生物制剂,其以初始剂量600mg(两次300mg皮下注射)、然后每隔一周300mg施用(simpson等人,nengljmed.2016;375(24):2335-2348)。在那些对tcs没有充分反应的持续中度至重度疾病的患者中,最近的指南概述了许多升级治疗选项(ringj等人,jeuracaddermatolvenereol2012;26:1176-93;schneiderl等人,jallergyclinimmunol2013;131:295-9.e1-27)。升级选项包括局部钙依赖磷酸酶抑制剂(tci)、光疗和免疫抑制剂,如口服皮质类固醇、环孢菌素、硫唑嘌呤、甲氨蝶呤和霉酚酸酯。其中,只有环孢菌素被批准用于治疗中度至重度ad(在许多欧洲国家获得国家许可,但在美国没有),其使用仅限于16岁及以上的患者(最多8周)。尽管环孢菌素是最广泛研究的全身性试剂,但试验结果的解释和对临床实践的适用性受到试验设计的限制(schmittj等人,jeuracaddermatolvenereol2007;21:606-619)。对其他免疫抑制剂如硫唑嘌呤和甲氨蝶呤的研究主要包括病例报告,其中很少有报道的随机对照试验(haeckim等人,jamacaddermatol2011;64:1074-84;schramme等人,jallergyclinimmunol2011;128:353-9)。通过最近欧洲皮肤科医生调查的处方实践表明显著的国家特定差异,该调查报告了没有明显优势试剂:43%使用环孢菌素、31%口服皮质类固醇、22%硫唑嘌呤(proudfootle等人,brjdermatol2013;169:901-9)。尽管用于治疗患有中度至重度ad的患者的升级疗法(包括全身免疫抑制剂)显示出适度至良好功效的证据,但由于副作用导致的差的耐受性限制了它们的长期使用。即使在环孢菌素已显示出显著功效的情况下,约50%的患者在2周内复发,并且在停止治疗后6周内80%复发(amorkt等人,jamacaddermatol2010;63:925-46)。尽管有害的副作用和限制,这些试剂的继续使用表明对更安全和更有效治疗的高度未满足的医疗需求。为了寻找具有可接受的收益风险特征的中度至重度ad的新疗法,已经测试了许多特异性靶向炎性细胞和介质的生物制剂(a等人,jdtschdermatolges2012;10:174-8;guttman-yasskye等人,expertopinbiolther2013;13:549-61)。然而,这些研究和病例报告中的一些仅在少数患者(少至一个或两个)中进行,并且显示出不一致的功效和/或安全性信号。因此,需要新的治疗方法和治疗方案,其将降低特应性皮炎疾病症状的严重性并使功效最大化。此外,需要新的治疗方法和治疗方案,其与现有治疗相比提供具有有限毒性的改善的安全性特征或为患者提供更多的耐受性或便利性,从而改善患者对治疗剂使用的依从性和对治疗方案的遵守。本文描述的发明满足某些上述需求并提供其他益处。本文引用的所有参考文献,包括专利申请和出版物,出于任何目的通过引用整体并入。技术实现要素:本发明至少部分基于惊人和意想不到的发现,即,如通过若干功效结果测量所评估的,当使用本文提供的给药方案施用于特应性皮炎患者(包括同时使用局部皮质类固醇的患者)时,抗il-13拮抗剂单克隆抗体lebrikizumab提供治疗益处。因此,本文提供il-13拮抗剂(包括抗il-13抗体,如lebrikizumab)在治疗特应性皮炎中的用途和用il-13拮抗剂(包括抗il-13抗体,如lebrikizumab)治疗特应性皮炎的方法。因此,在一个方面,提供了治疗患者的特应性皮炎的方法,所述方法包括向患者施用包含治疗有效量的il-13拮抗剂的药物组合物,其中所述药物组合物降低患者的疾病严重性并且其中通过特应性皮炎疾病严重性结果测量(addsom)评估疾病严重性。在一些实施方案中,特应性皮炎根据rajka/langeland标准评分确定为中度至重度,并且其中rajka/langeland标准评分被确定为4.5至9。在一些实施方案中,该方法还包括施用一种或多种局部皮质类固醇。在一些实施方案中,一种或多种局部皮质类固醇在施用il-13拮抗剂之前施用、与施用il-13拮抗剂同时施用、或施用il-13拮抗剂之后施用。在一些实施方案中,一种或多种局部皮质类固醇选自曲安奈德、氢化可的松、以及曲安奈德和氢化可的松的组合。在一些实施方案中,患者年龄为12岁及以上。在一些实施方案中,患者对局部皮质类固醇的控制不充分。在一些实施方案中,il-13拮抗剂是单克隆抗il-13抗体。在一些实施方案中,抗il-13抗体是包含vh和vl的抗体,所述vh包含hvr-h1、hvr-h2和hvr-h3,其中各vhhvr具有seqidno:5、seqidno:6和seqidno:7的氨基酸序列,所述vl包含hvr-l1、hvr-l2和hvr-l3,其中各vlhvr具有seqidno:8、seqidno:9和seqidno:10的氨基酸序列。在某些实施方案中,抗il-13抗体是igg1、igg2、igg3或igg4。在某些实施方案中,抗il-13抗体是人、人源化或嵌合抗体。在一些实施方案中,抗il-13抗体是包含vh和vl的抗体,所述vh包含选自seqidno:1和seqidno:3的序列,所述vl包含选自seqidno:2和seqidno:4的序列。在某些实施方案中,抗il-13抗体是结合人il-13的全长抗体或其片段。在一些实施方案中,抗il-13抗体是igg4。在一个实施方案中,抗il-13抗体是lebrikizumab。在一个实施方案中,抗il-13抗体包含具有seqidno:11的氨基酸序列的重链和具有seqidno:12的氨基酸序列的轻链。在一些实施方案中,使用皮下施用装置向患者施用il-13拮抗剂。在某些这样的实施方案中,皮下施用装置选自预填充注射器、一次性笔注射装置、微针装置、微输注装置、无针注射装置和自动注射装置。在一个实施方案中,il-13拮抗剂是lebrikizumab,并且使用预填充注射器向患者施用lebrikizumab。在一个实施方案中,il-13拮抗剂是lebrikizumab,并且使用自动注射装置向患者施用lebrikizumab。另一方面,用于治疗特应性皮炎的药物组合物包含125mg或250mg或500mg或约125mg或约250mg或约500mg的抗il-13抗体。在一些实施方案中,药物组合物包含110mg至140mg的抗il-13抗体或120mg至130mg的抗il-13抗体。在一些实施方案中,药物组合物包含225mg至275mg的抗il-13抗体或240mg至260mg的抗il-13抗体。在一些实施方案中,药物组合物包含450mg至550mg的抗il-13抗体或475mg至525mg的抗il-13抗体或490mg至510mg的抗il-13抗体。在一些实施方案中,药物组合物包含125mg、或约125mg、或110mg至140mg、或120mg至130mg的抗il-13抗体,并且组合物每四周皮下施用一次。在一些实施方案中,药物组合物施用12周的期间或20周的期间或24周的期间。在一些实施方案中,药物组合物包含250mg、或约250mg、或225mg至275mg、或240mg至260mg的抗il-13抗体,并且组合物每四周或每八周皮下施用一次。在一些实施方案中,药物组合物施用24周或更长的期间或施用24周。在一些实施方案中,特应性皮炎根据rajka/langeland标准评分确定为中度至重度,并且其中rajka/langeland标准评分被确定为4.5至9。在一些实施方案中,il-13拮抗剂是单克隆抗il-13抗体。在一些实施方案中,抗il-13抗体是包含vh和vl的抗体,所述vh包含hvr-h1、hvr-h2和hvr-h3,其中各vhhvr具有seqidno:5、seqidno:6和seqidno:7的氨基酸序列,并且所述vl包含hvr-l1、hvr-l2和hvr-l3,其中各vlhvr具有seqidno:8、seqidno:9和seqidno:10的氨基酸序列。在某些实施方案中,抗il-13抗体是igg1、igg2、igg3或igg4。在某些实施方案中,抗il-13抗体是人、人源化或嵌合抗体。在一些实施方案中,抗il-13抗体是包含vh和vl的抗体,所述vh包含选自seqidno:1和seqidno:3的序列,所述vl包含选自seqidno:2和seqidno:4的序列。在某些实施方案中,抗il-13抗体是结合人il-13的全长抗体或其片段。在一些实施方案中,抗il-13抗体是igg4。在一个实施方案中,抗il-13抗体是lebrikizumab。在一个实施方案中,抗il-13抗体包含具有seqidno:11的氨基酸序列的重链和具有seqidno:12的氨基酸序列的轻链。在一些实施方案中,使用皮下施用装置向患者施用il-13拮抗剂。在某些这样的实施方案中,皮下施用装置选自预填充注射器、一次性笔注射装置、微针装置、微输注装置、无针注射装置和自动注射装置。在一个实施方案中,il-13拮抗剂是lebrikizumab,并且使用预填充注射器向患者施用lebrikizumab。在一个实施方案中,il-13拮抗剂是lebrikizumab,并且使用自动注射装置向患者施用lebrikizumab。在一些实施方案中,该方法还包括施用一种或多种局部皮质类固醇。在一些实施方案中,一种或多种局部皮质类固醇在施用il-13拮抗剂之前施用、与施用il-13拮抗剂同时施用、或在施用il-13拮抗剂之后施用。在一些实施方案中,一种或多种局部皮质类固醇选自曲安奈德、氢化可的松、以及曲安奈德和氢化可的松的组合。在一些实施方案中,患者年龄为12岁及以上。在一些实施方案中,患者对局部皮质类固醇的控制不充分。在另一个方面,提供了治疗患者特应性皮炎的方法,所述方法包括向患者施用包含治疗有效量的il-13拮抗剂的药物组合物,其中所述药物组合物降低患者疾病的严重性并且其中疾病的严重性通过特应性皮炎疾病严重性结果测量(addsom)评估,其中特应性皮炎疾病严重性结果测量是湿疹面积和严重性指数(easi)、或特应性皮炎的严重性评分(scorad)、或研究者全面评估(iga)、或患者报告的结果(pro)。在一些实施方案中,特应性皮炎根据rajka/langeland标准评分确定为中度至重度,并且其中rajka/langeland标准评分被确定为4.5至9。在一些实施方案中,该方法还包括施用一种或多种局部皮质类固醇。在一些实施方案中,一种或多种局部皮质类固醇在施用il-13拮抗剂之前施用、与施用il-13拮抗剂同时施用、或在施用il-13拮抗剂之后施用。在一些实施方案中,一种或多种局部皮质类固醇选自曲安奈德、氢化可的松、以及曲安奈德和氢化可的松的组合。在一些实施方案中,患者年龄为12岁及以上。在一些实施方案中,患者对局部皮质类固醇的控制不充分。在一些实施方案中,addsom是easi,并且与施用第一剂量的药物组合物之前测定的easi相比,药物组合物使easi降低50%或75%或90%。在一些实施方案中,在施用第一剂量12周后或在施用第一剂量20周后或在施用第一剂量24周后测定easi。在一些实施方案中,addsom是scorad,并且与施用第一剂量的药物组合物之前测定的scorad相比,药物组合物使scorad降低50%。在一些实施方案中,addsom是scorad,并且与施用第一剂量的药物组合物之前测定的scorad相比,药物组合物使scorad降低75%。在一些实施方案中,在施用第一剂量12周后测定scorad。在一些实施方案中,addsom是iga并且药物组合物将iga降低至零或一。在一些实施方案中,在施用第一剂量的药物组合物12周后测定iga。在一些实施方案中,addsom是pro并且pro是瘙痒直观模拟标度(pruritusvisualanaloguescale,vas)、睡眠丧失vas或特应性皮炎影响问卷(adiq)评分。在一些实施方案中,在施用第一剂量的药物组合物12周后测定pro。在一些实施方案中,pro是瘙痒vas,并且药物组合物将瘙痒vas降低40%至55%。在一些实施方案中,pro是睡眠丧失vas,并且药物组合物将睡眠丧失vas降低53%至61%。在一些实施方案中,pro是adiq,并且药物组合物将adiq评分降低54%至65%。在一些实施方案中,il-13拮抗剂是单克隆抗il-13抗体。在一些实施方案中,抗il-13抗体是包含vh和vl的抗体,所述vh包含hvr-h1、hvr-h2和hvr-h3,其中各vhhvr具有seqidno:5、seqidno:6和seqidno:7的氨基酸序列,所述vl包含hvr-l1、hvr-l2和hvr-l3,其中各vlhvr具有seqidno:8、seqidno:9和seqidno:10的氨基酸序列。在某些实施方案中,抗il-13抗体是igg1、igg2、igg3或igg4。在某些实施方案中,抗il-13抗体是人、人源化或嵌合抗体。在一些实施方案中,抗il-13抗体是包含vh和vl的抗体,所述vh包含选自seqidno:1和seqidno:3的序列,所述vl包含选自seqidno:2和seqidno:4的序列。在某些实施方案中,抗il-13抗体是结合人il-13的全长抗体或其片段。在一些实施方案中,抗il-13抗体是igg4。在一个实施方案中,抗il-13抗体是lebrikizumab。在一个实施方案中,抗il-13抗体包含具有seqidno:11的氨基酸序列的重链和具有seqidno:12的氨基酸序列的轻链。在一些实施方案中,使用皮下施用装置向患者施用il-13拮抗剂。在某些这样的实施方案中,皮下施用装置选自预填充注射器、一次性笔注射装置、微针装置、微输注装置、无针注射装置和自动注射装置。在一个实施方案中,il-13拮抗剂是lebrikizumab,并且使用预填充注射器向患者施用lebrikizumab。在一个实施方案中,il-13拮抗剂是lebrikizumab,并且使用自动注射装置向患者施用lebrikizumab。在另一个方面,提供了方法,所述方法包括向患者施用治疗有效量的il-13拮抗剂,其中治疗有效量选自125mg和250mg,并且其中每四周一次皮下施用il-13拮抗剂。在一些实施方案中,治疗有效量为约125mg或110mg至140mg的抗il-13抗体或120mg至130mg的抗il-13抗体。在一些实施方案中,治疗有效量为约250mg或225mg至275mg之间的抗il-13抗体或240mg至260mg之间的抗il-13抗体。在一些实施方案中,il-13拮抗剂是单克隆抗il-13抗体。在一些实施方案中,抗il-13抗体是包含vh和vl的抗体,所述vh包含hvr-h1、hvr-h2和hvr-h3,其中各vhhvr具有seqidno:5、seqidno:6和seqidno:7的氨基酸序列,并且所述vl包含hvr-l1、hvr-l2和hvr-l3,其中各vlhvr具有seqidno:8、seqidno:9和seqidno:10的氨基酸序列。在某些实施方案中,抗il-13抗体是igg1、igg2、igg3或igg4。在某些实施方案中,抗il-13抗体是人、人源化或嵌合抗体。在一些实施方案中,抗il-13抗体是包含vh和vl的抗体,所述vh包含选自seqidno:1和seqidno:3的序列,所述vl包含选自seqidno:2和seqidno:4的序列。在某些实施方案中,抗il-13抗体是结合人il-13的全长抗体或其片段。在一些实施方案中,抗il-13抗体是igg4。在一个实施方案中,抗il-13抗体是lebrikizumab。在一个实施方案中,抗il-13抗体包含具有seqidno:11的氨基酸序列的重链和具有seqidno:12的氨基酸序列的轻链。在一些实施方案中,使用皮下施用装置向患者施用il-13拮抗剂。在某些这样的实施方案中,皮下施用装置选自预填充注射器、一次性笔注射装置、微针装置、微输注装置、无针注射装置和自动注射装置。在一个实施方案中,il-13拮抗剂是lebrikizumab,并且使用预填充注射器向患者施用lebrikizumab。在一个实施方案中,il-13拮抗剂是lebrikizumab,并且使用自动注射装置向患者施用lebrikizumab。在一些实施方案中,如rajka/langeland标准评分所测定,特应性皮炎是中度至重度,并且其中rajka/langeland标准评分被测定为4.5至9。在一些实施方案中,该方法还包括施用一种或多种局部皮质类固醇。在一些实施方案中,一种或多种局部皮质类固醇在施用il-13拮抗剂之前施用、与施用il-13拮抗剂同时施用、或在施用il-13拮抗剂之后施用。在一些实施方案中,一种或多种局部皮质类固醇选自曲安奈德、氢化可的松、以及曲安奈德和氢化可的松的组合。在一些实施方案中,患者年龄为12岁及以上。在一些实施方案中,患者对局部皮质类固醇的控制不充分。在一些实施方案中,治疗有效量降低患者疾病的严重性,并且通过特应性皮炎疾病严重性结果测量(addsom)评估疾病的严重性。在一些实施方案中,addsom是湿疹面积和严重性指数(easi)、或特应性皮炎的严重性评分(scorad)、或研究者全面评估(iga)、或患者报告的结果(pro)。在一些实施方案中,addsom是easi,并且与施用第一剂量的il-13拮抗剂之前测定的easi相比,治疗有效量使easi降低50%或75%或90%。在一些实施方案中,在施用第一剂量12周后或在施用第一剂量20周后或在施用第一剂量24周后测定easi。在一些实施方案中,addsom是scorad,并且与施用第一剂量的il-13拮抗剂之前测定的scorad相比,治疗有效量使scorad降低50%。在一些实施方案中,addsom是scorad,并且与施用第一剂量的il-13拮抗剂之前测定的scorad相比,治疗有效量使scorad降低75%。在一些实施方案中,在施用第一剂量12周后测定scorad。在一些实施方案中,addsom是iga并且治疗有效量将iga降低至零或一。在一些实施方案中,在施用第一剂量的il-13拮抗剂12周后测定iga。在一些实施方案中,addsom是pro,并且pro是瘙痒直观模拟标度(vas)、睡眠丧失vas或特应性皮炎影响问卷(adiq)评分。在一些实施方案中,在施用第一剂量的il-13拮抗剂12周后测定pro。在一些实施方案中,pro是瘙痒vas,并且治疗有效量使瘙痒vas降低40%至55%。在一些实施方案中,pro是睡眠丧失vas,并且治疗有效量使睡眠丧失vas降低53%至61%。在一些实施方案中,pro是adiq,并且治疗有效量使adiq评分降低54%至65%。另一方面,提供了通过向患者施用治疗有效量的il-13拮抗剂来治疗患者的特应性皮炎的方法,其中所述施用包括施用至少一种负荷剂量和施用至少一种后续维持剂量,并且其中至少一种负荷剂量中的每一种和至少一种维持剂量中的每一种以均一剂量(flatdose)皮下施用。在一些实施方案中,如通过rajka/langeland标准评分确定的,特应性皮炎是中度至重度,并且其中rajka/langeland标准评分被确定为4.5至9。在一些实施方案中,该方法还包括施用一种或多种局部皮质类固醇。在一些实施方案中,一种或多种局部皮质类固醇在施用il-13拮抗剂之前施用、与施用il-13拮抗剂同时施用、或在施用il-13拮抗剂之后施用。在一些实施方案中,一种或多种局部皮质类固醇选自曲安奈德、氢化可的松、以及曲安奈德和氢化可的松的组合。在一些实施方案中,患者年龄为12岁及以上。在一些实施方案中,患者对局部皮质类固醇的控制不充分。在一些实施方案中,il-13拮抗剂是单克隆抗il-13抗体。在一些实施方案中,抗il-13抗体是包含vh和vl的抗体,所述vh包含hvr-h1、hvr-h2和hvr-h3,其中各vhhvr具有seqidno:5、seqidno:6和seqidno:7的氨基酸序列,所述vl包含hvr-l1、hvr-l2和hvr-l3,其中各vlhvr具有seqidno:8、seqidno:9和seqidno:10的氨基酸序列。在某些实施方案中,抗il-13抗体是igg1、igg2、igg3或igg4。在某些实施方案中,抗il-13抗体是人、人源化或嵌合抗体。在一些实施方案中,抗il-13抗体是包含vh和vl的抗体,所述vh包含选自seqidno:1和seqidno:3的序列,并且所述vl包含选自seqidno:2和seqidno:4的序列。在某些实施方案中,抗il-13抗体是结合人il-13的全长抗体或其片段。在一些实施方案中,抗il-13抗体是igg4。在一个实施方案中,抗il-13抗体是lebrikizumab。在一个实施方案中,抗il-13抗体包含具有seqidno:11的氨基酸序列的重链和具有seqidno:12的氨基酸序列的轻链。在一些实施方案中,负荷剂量为250mg或500mg,维持剂量为125mg。在一些实施方案中,负荷剂量为250mg,维持剂量为125mg。在一些实施方案中,负荷剂量为500mg,维持剂量为125mg。在一些实施方案中,在施用负荷剂量4周后施用维持剂量并且其后每四周施用一次维持剂量持续整个治疗期间。在一些实施方案中,负荷剂量为250mg,维持剂量为125mg,其中在负荷剂量4周后施用维持剂量,并且其后每四周一次持续整个治疗期间。在一些实施方案中,负荷剂量为250mg,施用负荷剂量,接着在15天后施用第二负荷剂量,维持剂量为125mg。在一些实施方案中,在施用第二负荷剂量两周后施用维持剂量,并且其后每四周施用一次维持剂量持续整个治疗期间。在一些实施方案中,负荷剂量为250mg,施用负荷剂量,接着在29天后施用第二负荷剂量,并且维持剂量为125mg。在一些实施方案中,在施用第二负荷剂量四周后施用维持剂量,并且其后每四周施用一次维持剂量持续整个治疗期间。在一些实施方案中,负荷剂量为500mg,维持剂量为250mg。在一些实施方案中,在施用负荷剂量四周后施用维持剂量,并且其后每四周施用一次维持剂量持续整个治疗期间。在一些实施方案中,在施用负荷剂量四周后施用维持剂量,并且其后每八周施用一次维持剂量持续整个治疗期间。在一些实施方案中,负荷剂量为500mg,维持剂量为250mg,其中在负荷剂量4周后施用维持剂量,并且其后每四周一次维持剂量持续整个治疗期间。在一些实施方案中,整个治疗期间为24周或更长。在一些实施方案中,整个治疗期间为24周。在一些实施方案中,治疗有效量降低患者的疾病严重性,通过特应性皮炎疾病严重性结果测量(addsom)评估疾病严重性。在一些实施方案中,addsom是湿疹面积和严重性指数(easi)、或特应性皮炎的严重性评分(scorad)、或研究者全面评估(iga)、或患者报告的结果(pro)。在一些实施方案中,addsom是easi,并且与施用第一剂量的il-13拮抗剂之前测定的easi相比,治疗有效量使easi降低50%或75%或90%。在一些实施方案中,在施用第一剂量12周后或在施用第一剂量20周后或在施用第一剂量24周后测定easi。在一些实施方案中,addsom是scorad,并且与施用第一剂量的il-13拮抗剂之前测定的scorad相比,治疗有效量使scorad降低50%。在一些实施方案中,addsom是scorad,并且与施用第一剂量的il-13拮抗剂之前测定的scorad相比,治疗有效量使scorad降低75%。在一些实施方案中,在施用第一剂量12周后测定scorad。在一些实施方案中,addsom是iga并且治疗有效量使iga降低至零或一。在一些实施方案中,在施用第一剂量的il-13拮抗剂12周后测定iga。在一些实施方案中,addsom是pro并且pro是瘙痒直观模拟标度(vas)、睡眠丧失vas或特应性皮炎影响问卷(adiq)评分。在一些实施方案中,在施用第一剂量的il-13拮抗剂12周后测定pro。在一些实施方案中,pro是瘙痒vas,并且治疗有效量使瘙痒vas降低40%至55%。在一些实施方案中,pro是睡眠丧失vas,并且治疗有效量使睡眠丧失vas降低53%至61%。在一些实施方案中,pro是adiq,并且治疗有效量使adiq评分降低54%至65%。在一些实施方案中,使用皮下施用装置向患者施用il-13拮抗剂。在某些这样的实施方案中,皮下施用装置选自预填充注射器、一次性笔注射装置、微针装置、微输注装置、无针注射装置和自动注射装置。在一个实施方案中,il-13拮抗剂是lebrikizumab,并且使用预填充注射器向患者施用lebrikizumab。在一个实施方案中,il-13拮抗剂是lebrikizumab,并且使用自动注射装置向患者施用lebrikizumab。附图的简要说明图1显示了如实施例2中所述的研究i方案。缩写如下:d=天;pbo=安慰剂;sc=皮下;tcs=局部皮质类固醇;wk=周。图2a显示如实施例2中所述的在经过12周实现easi-50的患者的比例。带有空心圆圈的短划线,lebrikizumab250mg单剂量加局部皮质类固醇(tcs)每天两次(bid);带有封闭三角形的虚线,lebrikizumab125mg单剂量加tcs每天两次;带有空心方块的实线,lebrikizumab125mg每4周一次(q4w)加tcs每天两次;带有加号的实线,安慰剂加tcs每天两次。图2b显示如实施例2中所述经过12周实现scorad-50的患者的比例。带有空心圆圈的短划线,lebrikizumab250mg单剂量加局部皮质类固醇(tcs)每天两次(bid);带有封闭三角形的虚线,lebrikizumab125mg单剂量加tcs每天两次;带有空心方块的实线,lebrikizumab125mg每4周一次(q4w)加tcs每天两次;带有加号的实线,安慰剂加tcs每天两次。图2c显示如实施例2中所述经过12周实现iga0/1的患者的比例。带有空心圆圈的短划线,lebrikizumab250mg单剂量加局部皮质类固醇(tcs)每天两次(bid);带有封闭三角形的虚线,lebrikizumab125mg单剂量加tcs每天两次;带有空心方块的实线,lebrikizumab125mg每4周一次(q4w)加tcs每天两次;带有加号的实线,安慰剂加tcs每天两次。图3a-3c显示如实施例2中所述在改良的意向治疗群体中经过20周实现easi-50(图3a)、easi-75(图3b)和easi-90(图3c)的患者的比例。带有空心圆圈的短划线,lebrikizumab250mg单剂量加局部皮质类固醇(tcs)每天两次(bid);带有封闭三角形的虚线,lebrikizumab125mg单剂量加tcs每天两次;带有空心方块的实线,lebrikizumab125mg每4周一次(q4w)加tcs每天两次;带有加号的实线,安慰剂加tcs每天两次。图4显示如实施例2中所述在改良的意向治疗群体中经过12周easi从基线的变化百分比。带有空心圆圈的短划线,lebrikizumab250mg单剂量加局部皮质类固醇(tcs)每天两次(bid);带有封闭三角形的虚线,lebrikizumab125mg单剂量加tcs每天两次;带有空心方块的实线,lebrikizumab125mg每4周一次(q4w)加tcs每天两次;带有加号的实线,安慰剂加tcs每天两次。显示的数据点是用标准误差棒调整的平均值。星号(*)表示安慰剂校正的变化为17.4%,p=0.025。图5显示如实施例3中所述的lebrikizumab的特应性皮炎纵向pk-pd模型。在图5中,r(t)是easi评分;kin0是基线疾病进展速度常数;kin是使用lebrikizumab的疾病进展速率常数,包括安慰剂/tcs作用;edrug是lebrikizumab对疾病进展抑制的药物作用;econ是安慰剂/tcs作用(随时间常数(constantovertime));emax是对疾病进展抑制的最大lebrikizumab药物作用;ec50是导致50%emax的lebrikizumab浓度;kout是组织修复速度常数;r(t=0)是基线easi评分;iiv是个体间变异性;ωkout是kout的个体间变异性;ωecon是econ的个体间变异性;ρkoutxecon是kout和econ之间的相关性;σ是残差。图6显示了如实施例3中所述的第1组(参见表5)lebrikizumab给药方案随时间变化的模型预期的中值easi-75反应。粗实线是安慰剂治疗的中值模拟反应(安慰剂治疗中改善的easi-75代表局部皮质类固醇对整体功效的可能贡献)。中等实线是每四周施用一次125mglebrikizumab(lebri125mgq4w)给药方案的中值模拟反应。点划线是在第1天给予250mg负荷剂量、接着从第4周开始每四周施用一次125mg维持剂量的lebrikizumab(lebri250mg负荷,125mgq4w)的中值模拟反应。细实线是在第1天给予500mg负荷剂量、接着从第4周开始每四周施用一次125mg维持剂量的lebrikizumab(lebri500mg负荷,125mgq4w)的中值模拟反应。粗短划线是在第1天和第29天施用250mg剂量的lebrikizumab、接着从第8周开始每四周施用一次125mg维持剂量的lebrikizumab(lebri250mg第1天和第29天,125mgq4w)的中值模拟反应。细短划线是在第1天和第15天施用250mg剂量的lebrikizumab、接着从第4周(第29天)开始每四周施用一次125mg维持剂量的lebrikizumab(lebri250mg第1天和第15天,125mgq4w)的中值模拟反应。为清楚起见,从图中删除了置信区间。图7显示了如实施例3中所述的组2(参见表5)lebrikizumab给药方案随时间变化的模型预期的中值easi-75反应。粗实线是安慰剂治疗的中值模拟反应(安慰剂治疗中改善的easi-75代表局部皮质类固醇对整体功效的可能贡献)。中等实线是每四周施用一次125mglebrikizumab(lebri125mgq4w)给药方案的中值模拟反应。细短划线是每四周施用一次250mglebrikizumab(lebri250mgq4w)给药方案的中值模拟反应。粗短划线是在第1天施用500mg负荷剂量、接着是从第4周开始每四周施用一次250mg维持剂量的lebrikizumab(lebri500mg负荷,250mgq4w)。为清楚起见,从图中删除了置信区间。图8显示了如实施例3中所述的组3(参见表5)给药方案随时间变化的模型预期的中值easi-75反应。粗实线是安慰剂治疗的中值模拟反应(安慰剂治疗中改善的easi-75代表局部皮质类固醇对整体功效的可能贡献)。中等实线是每四周施用一次125mglebrikizumab(lebri125mgq4w)给药方案的中值模拟反应。细短划线是每八周施用一次250mglebrikizumab的中值模拟反应(lebri250q8w)。粗短划线是500mg负荷剂量、接着从第4周开始每8周施用一次250mglebrikizumab的维持剂量(lebri500mg负荷,250mgq8w)的中值模拟反应。为清楚起见,从图中删除了置信区间。图9显示了如实施例3中所述的第4组(参见表5)lebrikizumab给药方案随时间变化的模型预期的中值easi-75反应。粗实线是安慰剂治疗的中值模拟反应(安慰剂治疗中改善的easi-75代表局部皮质类固醇对整体功效的可能贡献)。中等实线是每四周施用一次125mglebrikizumab(lebri125mgq4w)给药方案的中值模拟反应。粗短划线是每四周施用一次37.5mglebrikizumab给药方案(lebri37.5mgq4w)的中值模拟反应。细短划线是125mg负荷剂量、接着从第4周开始每四周施用一次37.5mglebrikizumab的维持剂量(lebri125mg负荷,37.5mgq4w)的中值模拟反应。为清楚起见,从图中删除了置信区间。图10显示如实施例4中所述的研究ii方案。缩写如下:d=天;easi=湿疹面积和严重性指数;q4wk=每4周一次;tcs=局部皮质类固醇;wk=周。图11a-11d显示了如实施例2中所述在第12周实现easi-50(图11a)、easi-75(图11b)、iga0/1(图11c)和scorad-50(图11d)的患者的比例以及在研究的每个组中与安慰剂相比分别实现easi-50、easi-75、iga0/1和scorad-50的患者比例的变化,在图11a-11d每个图中,该变化由虚线和与每个柱相邻的箭头表示。缩写如下:easi=湿疹面积严重性指数;iga=研究者全面评估;q4w=每4周一次;scorad=评分特应性皮炎;sd=单剂量。斑点柱=lebrikizumab125mg单剂量加局部皮质类固醇(tcs)每天两次(bid);阴影线柱=lebrikizumab250mg单剂量加tcs每天两次;点刻柱=lebrikizumab125mg每4周一次(q4w)加tcs每天两次;空心柱=安慰剂加tcs每天两次。图12a-12d显示了如实施例2中所述的瘙痒vas(图12a)、adiq(图12b)、睡眠丧失vas(图12c)和dlqi(图12d)随时间从基线的变化。带有空心圆圈的点划线,lebrikizumab250mg单剂量加局部皮质类固醇(tcs)每天两次(bid);带有封闭三角形的虚线,lebrikizumab125mg单剂量加tcs每天两次;带有空心方块的实线,lebrikizumab125mg每4周一次(q4w)加tcs每天两次;带有加号的实线,安慰剂加tcs每天两次。缩写如下:vas=直观模拟评分;adiq=特应性皮炎影响问卷;dlqi=皮肤病生活质量指数。详细说明本文引用的所有参考文献,包括专利申请和出版物,出于任何目的通过引用整体并入。除非另外定义,否则本文使用的技术和科学术语具有与本发明所属领域的普通技术人员通常理解的含义相同的含义。singleton等人,dictionaryofmicrobiologyandmolecularbiology第2版,j.wiley&sons(newyork,n.y.1994),和march,advancedorganicchemistryreactions,mechanismsandstructure第4版,johnwiley&sons(newyork,n.y.1992),为本领域技术人员提供本申请中使用的许多术语的一般指南。某些定义出于解释本说明书的目的适用以下定义,并且在适当时,以单数使用的术语也将包括复数,反之亦然。如果下面列出的任何定义与通过引用并入本文的任何文件冲突,则以以下列出的定义为准。如在本说明书和所附权利要求中所使用的,单数形式“一(a)”、“一个(an)”和“该(the)”包括复数指代物,除非上下文另有明确说明。因此,例如,提及“蛋白质”或“抗体”分别包括多个蛋白质或抗体;提及“细胞”包括细胞的混合物等。说明书和所附权利要求中提供的范围包括端点和端点之间的所有点。因此,例如,2.0到3.0的范围包括2.0、3.0、以及2.0和3.0之间的所有点。术语“标记物”和“生物标记物”可互换使用,指分子,包括基因、蛋白质、碳水化合物结构或糖脂、代谢物、mrna、mirna、蛋白质、dna(cdna或基因组dna)、dna拷贝数或后生变化,例如增加、减少或改变的dna甲基化(例如,胞嘧啶甲基化、或cpg甲基化、非cpg甲基化);组蛋白修饰(例如,(去)乙酰化、(去)甲基化、(去)磷酸化、泛素化、sumo化、adp-核糖基化);改变的核小体定位、其在哺乳动物组织或细胞中或上的表达或存在可通过标准方法(或本文公开的方法)检测,并且可预期、诊断和/或预后哺乳动物细胞或组织对基于2型炎症通路抑制的治疗方案的敏感性,该治疗方案使用例如2型炎症通路抑制剂,如本文所述的抗il-13抗体。生物标记物也可以是在从受试者获得的生物样品中测量的生物学或临床属性,例如但不限于血细胞计数。术语“生物样品”包括但不限于血液、血清、血浆、外周血单个核细胞(pbmc)、痰、组织活检(例如肺样品)和鼻样品,包括鼻拭子或鼻息肉。可以在治疗前、治疗期间或治疗后采集样品。术语“特应性皮炎”或“ad”是指慢性复发和缓解的炎性皮肤疾患,其特征在于强烈瘙痒(例如,严重发痒)、干燥(例如,异常干燥的皮肤)、红斑性结痂皮疹、苔藓化、皮肤屏障受损和鳞状和干性湿疹病灶。术语“特应性皮炎”包括但不限于由表皮屏障功能障碍、过敏(例如,对某些食物、花粉、霉菌、尘螨、动物等过敏)、辐射暴露和/或哮喘引起或与之相关的ad。在许多情况下,慢性ad病灶包括皮肤增厚的斑块、苔藓化和纤维性丘疹。本文提供的治疗剂包括可以与靶细胞因子,白介素(il)-13结合的试剂,如多肽(例如,抗体、免疫粘附素或肽体(peptibody))、适体或可以结合蛋白质的小分子或可以结合编码本文鉴定的靶标的核酸分子的核酸分子(即sirna)。“抗il-13结合剂”是指结合人il-13的试剂。此类结合剂可包括小分子、适体或多肽。此类多肽可包括但不限于选自免疫粘附素、抗体、肽体和肽的多肽。根据一个实施方案,结合剂以1μm-1pm的亲和力结合人il-13序列。抗il-13结合剂的具体实例可包括抗il-13抗体、与人fc融合的可溶性il-13受体α2、与人fc融合的可溶性il4受体α、与人fc融合的可溶性il-13受体α。示例性的抗il-13抗体包括但不限于ima-026、ima-638(也称为anrukinzumab,innno.910649-32-0;qax-576)、tralokinumab(也称为cat-354,casno.1044515-88-9);aer-001、abt-308(也称为人源化13c5.5抗体和lebrikizumab。根据一个实施方案,抗il-13抗体包含vh和vl,所述vh包含选自seqidno:1、3和和24的序列,所述vl包含选自seqidno:2、4和25的序列。在一个实施方案中,抗il-13抗体包含hvrh1、hvrh2、hvrh3、hvrl1、hvrl2和hvrl3,其中各hvr具有seqidno:5、seqidno:6、seqidno:7、seqidno:8、seqidno:9和seqidno:10的氨基酸序列。在一个实施方案中,抗il-13抗体是lebrikizumab。根据一个实施方案,抗体是igg1抗体。根据另一个实施方案,抗体是igg4抗体。根据一个实施方案,igg4抗体在其恒定结构域中包含s228p突变。在一个实施方案中,抗il-13抗体在其可变重链区中包含q1e突变。在一个实施方案中,抗il-13抗体在其可变轻链区中包含m4l突变。术语“小分子”是指分子量在50道尔顿至2500道尔顿之间的有机分子。术语“抗体”以最广泛的含义使用,并且具体涵盖例如单克隆抗体、多克隆抗体、具有多表位特异性的抗体、单链抗体、包括双特异性抗体的多特异性抗体、以及抗体片段,只要其表现出所需的抗原结合活性。此类抗体可以是嵌合的、人源化的、人的和合成的抗体。术语“不受控制的”或“不可控制的”是指治疗方案不足以使疾病的症状最小化。如本文所用,术语“不受控制的”和“不充分控制的”可以互换使用,并且意指相同的状态。患者的控制状态可以由主治医师基于多种因素来确定,包括患者的临床病史、对治疗的反应性和规定的当前治疗水平。术语“治疗剂”是指用于治疗疾病的任何试剂。术语“节约皮质类固醇(corticosteroidsparing)”或“cs”意指由于施用了其他治疗剂,降低了应用皮质类固醇治疗疾病的患者中用于治疗疾病的皮质类固醇的频率和/或量,或消除了用于治疗疾病的皮质类固醇。“cs剂”是指可以在应用皮质类固醇的患者中引起cs的治疗剂。术语“皮质类固醇”包括但不限于局部皮质类固醇。示例性局部皮质类固醇包括曲安奈德(通常在乳膏中以0.1%的浓度配制)和氢化可的松(通常在乳膏中以1%或2.5%的浓度配制)。某些局部皮质类固醇被认为具有非常高的疗效,例如,二丙酸倍他米松、丙酸氯倍他索、二乙酸二氟拉松、醋酸氟轻松和丙酸乌倍他索。某些局部皮质类固醇被认为是高疗效的,例如,安西奈德、去羟米松、哈西奈德和曲安奈德。某些局部皮质类固醇被认为是中等疗效,例如,戊酸倍他米松、氯可托龙戊酸酯、氟轻松、氟氢缩松、醋酸氟轻松、丙酸氟替卡松、丁酸氢化可的松、戊酸氢化可的松、糠酸莫米松和泼尼卡酯。某些局部皮质类固醇被认为是低疗效,例如,二丙酸阿氯米松、地奈德和氢化可的松。“可吸入的皮质类固醇”是指适于通过吸入递送的皮质类固醇。示例性的可吸入皮质类固醇是氟替卡松、二丙酸倍氯米松、布地奈德、糠酸莫米松、环索奈德、氟尼缩松、曲安奈德和目前可获得或将来可用的任何其他皮质类固醇。可被吸入并与长效β2-激动剂组合的皮质类固醇的实例包括但不限于:布地奈德/福莫特罗和氟替卡松/沙美特罗。术语“负荷剂量”是指在治疗过程开始时给予的药物剂量,其高于随后给予的剂量和治疗剩余部分所给予的每个剂量(被称为“维持剂量”)。通常,负荷剂量施用一次或两次。在施用一次或多次负荷剂量后,施用维持剂量,并且此后通常以规则的间隔施用维持剂量,用于治疗过程的剩余部分。术语“均一剂量”是指用于所有患者的单剂量,不考虑体重或与体重或身体质量相关的任何患者特异性因素。例如,施用100mg均一剂量的抗体意味着无论体重如何,每个患者都将接受100mg的剂量。均一剂量有时被称为固定剂量。术语“基于体重的剂量”意指基于患者体重计算的剂量。因此,施用剂量取决于患者的体重。例如,1mg/kg抗体的剂量意味着体重50kg的患者将接受50mg的剂量,而体重80kg的患者将接受80mg的剂量。可以使用指示对患者有益的任何端点来评估对治疗剂的“患者反应”或“反应”(及其语法变体),包括但不限于:(1)在一定程度上抑制疾病进展,包括减慢和完全阻止;(2)减少疾病发作次数和/或症状;(3)减少受损的尺寸;(4)抑制(即减少、减慢或完全停止)免疫或炎性细胞浸润到邻近的外周器官和/或组织中;(5)抑制(即减少、减慢或完全停止)疾病的传播;(6)减少自身免疫反应,可能但非必须导致疾病病变的消退或消融;(7)在某种程度上缓解与该病患相关的一种或多种症状;和/或(8)增加治疗后无病表现的长度。当患者的反应性在治疗过程中不随时间降低时,“患者对治疗维持反应性”。当患者的反应性在治疗过程中随时间降低时,患者是“不充分的反应者”。例如,特应性皮炎患者的症状在治疗开始时被局部皮质类固醇(tcs)控制,但在治疗过程中后期tcs施用后症状无缓解,则该患者对治疗的反应性丧失,被认为是不充分的tcs反应者。“皮下施用装置”是指适于或设计成通过皮下途径施用药物(例如治疗性抗体或药物制剂)的装置。示例性皮下施用装置包括但不限于注射器,包括预填充注射器、注射装置、输液泵、注射笔、无针装置和贴剂递送系统。皮下施用装置施用一定体积的药物制剂,例如约1.0ml、约1.25ml、约1.5ml、约1.75ml或约2.0ml。“亲和力”是指分子(例如抗体)的单个结合位点与其结合配偶体(例如抗原)之间的非共价相互作用的总和的强度。除非另有说明,否则如本文所用,“结合亲和力”是指内在结合亲和力,其反映结合对的各成员(例如,抗体和抗原结合臂)之间的1:1相互作用。分子x对其配偶体y的亲和力通常可由解离常数(kd)表示。可以通过本领域已知的常规方法测量亲和力,包括本文所述的那些。用于测量结合亲和力的具体说明性和示例性实施方案描述如下。“亲和力成熟的”抗体是指在一个或多个高变区(hvr)中具有一个或多个改变的抗体,与不具有这种改变的亲本抗体相比,这种改变导致抗体对抗原的亲和力改善。术语“抗靶抗体”和“与靶标结合的抗体”是指能够以足够的亲和力结合靶标的抗体,使得该抗体可用作靶向靶标的诊断和/或治疗剂。在一个实施方案中,抗靶抗体与不相关非靶蛋白的结合程度小于抗体与靶结合的约10%,如例如通过放射免疫测定法(ria)或biacore测定法所测量的。在某些实施方案中,结合靶标的抗体具有≤1μm、≤100nm、≤10nm、≤1nm、≤0.1nm、≤0.01nm或≤0.001nm的解离常数(kd)(例如10-8m或更小,例如10-8m至10-13m,例如10-9m至10-13m)。在某些实施方案中,抗靶抗体结合在不同物种中是保守的靶标的表位。“抗体片段”是指完整抗体以外的分子,其包含完整抗体的一部分并结合完整抗体所结合的抗原。抗体片段的实例包括但不限于单链fv、fab、fab'、fab'-sh、f(ab')2;双抗体;线性抗体;单链抗体分子(例如scfv);和由抗体片段形成的多特异性抗体。与参照抗体“结合相同表位的抗体”是指在竞争测定法中阻断参照抗体与其抗原结合的50%或更多的抗体,相反,参照抗体在竞争测定法中阻断该抗体与其抗原结合的50%或更多。用于进行竞争测定法的各种方法在本领域中是公知的。用于本文目的的“受体人框架”是包含衍生自人免疫球蛋白框架或共有框架的轻链可变结构域(vl)框架或重链可变结构域(vh)框架的氨基酸序列的框架,如下定义。“衍生自”人免疫球蛋白框架或人共有框架的受体人框架可以包含其相同的氨基酸序列,或者可以包含氨基酸序列变化。在一些实施方案中,氨基酸变化的数量为10或更少、9或更少、8或更少、7或更少、6或更少、5或更少、4或更少、3或更少、或2或更少。在一些实施方案中,vl受体人框架在序列上与vl人免疫球蛋白框架序列或人共有框架序列相同。术语“嵌合”抗体是指其中重链和/或轻链的一部分衍生自特定来源或物种、而重链和/或轻链的其余部分衍生自不同来源或物种的抗体。抗体的“类别”是指其重链具有的恒定结构域或恒定区的类型。存在五种主要类型的抗体:iga、igd、ige、igg和igm,这些中的一些可以进一步分为亚类(同种型),例如igg1、igg2、igg3、igg4、iga1和iga2。对应于不同类别免疫球蛋白的重链恒定结构域分别称为α、δ、ε、γ和μ。“效应功能”是指可归因于抗体fc区的那些生物学活性,其随抗体同种型而变化。抗体效应功能的实例包括:c1q结合和补体依赖性细胞毒性(cdc);fc受体结合;抗体依赖性细胞介导的细胞毒性(adcc);吞噬作用;细胞表面受体(例如b细胞受体)的下调;和b细胞激活。试剂(例如药物制剂)的“有效量”是指在必要的时间段内实现所需治疗或预防结果的有效剂量。本文的术语“fc区”用于定义免疫球蛋白重链的c末端区,其含有至少一部分恒定区。该术语包括天然序列fc区和变体fc区。在一个实施方案中,人igg重链fc区从cys226或从pro230延伸至重链的羧基末端。然而,fc区的c-末端赖氨酸(lys447)可以存在或不存在。除非本文另有说明,否则fc区或恒定区中氨基酸残基的编号根据eu编号系统,也称为eu索引,如kabat等人,sequencesofproteinsofimmunologicalinterest,第5版publichealthservice,nationalinstitutesofhealth,bethesda,md,1991中所述。“框架”或“fr”是指除高变区(hvr)残基之外的可变结构域残基。可变结构域的fr通常由四个fr域组成:fr1、fr2、fr3和fr4。因此,hvr和fr序列通常以下列顺序出现在vh(或vl)中:fr1-h1(l1)-fr2-h2(l2)-fr3-h3(l3)-fr4。术语“全长抗体”、“完整抗体”和“整个抗体”在本文中可互换使用,是指与天然抗体结构具有基本相似的结构的抗体或具有包含fc区的重链的抗体,如本文所定义。术语“宿主细胞”、“宿主细胞系”和“宿主细胞培养物”可互换使用,指其中已引入外源核酸的细胞,包括此类细胞的后代。宿主细胞包括“转化体”和“转化细胞”,其包括原代转化细胞和由其衍生的后代,而不考虑传代次数。后代可能与亲本细胞的核酸内容不完全相同,但可能含有突变。本文包括与最初转化细胞中所筛选或选择的功能或生物学活性具有相同的功能或生物学活性的突变后代。“人抗体”是具有氨基酸序列的抗体,所述氨基酸序列对应于由人或人细胞产生的抗体的氨基酸序列、或衍生自利用人抗体库或其他人抗体编码的序列的非人源的抗体的氨基酸序列。人抗体的这种定义特异性地排除了包含非人抗原结合残基的人源化抗体。“人共有框架”是代表选择的人免疫球蛋白vl或vh框架序列中最常出现的氨基酸残基的框架。通常,人免疫球蛋白vl或vh序列的选择来自可变结构域序列的亚组。通常,序列亚组是如kabat等人,sequencesofproteinsofimmunologicalinterest,第5版,nihpublication91-3242,bethesdamd(1991),vols.1-3中所述的亚组。在一个实施方案中,对于vl,亚组是如kabat等人,同上中的亚组kappai。在一个实施方案中,对于vh,亚组是如kabat等人,同上中的亚组iii。“人源化”抗体是指嵌合抗体,其包含来自非人hvr的氨基酸残基和来自人fr的氨基酸残基。在某些实施方案中,人源化抗体将包含至少一个,通常两个可变结构域中的基本上全部,其中全部或基本上全部hvr(例如,cdr)对应于非人抗体的那些,并且全部或基本上全部fr对应于人抗体的那些。人源化抗体任选地可以包含衍生自人抗体的抗体恒定区的至少一部分。抗体(例如非人抗体)的“人源化形式”是指已经历人源化的抗体。术语“高变区”或“hvr”是指抗体可变结构域的每个区域,其在序列上是高变的和/或形成结构上限定的环(“高变环”)。通常,天然四链抗体包含六个hvr;vh中的三个(h1、h2、h3)和vl中的三个(l1、l2、l3)。hvr通常包含来自高变环和/或来自“互补决定区”(cdr)的氨基酸残基,后者通常具有最高的序列可变性和/或参与抗原识别。如本文所用的hvr区包含位于24-36位(对于hvrl1)、46-56位(对于hvrl2)、89-97位(对于hvrl3)、26-35b位(对于hvrh1)、47-65位(对于hvrh2)和93-102位(对于hvrh3)内的任何数目的残基。“个体”或“患者”或“受试者”是哺乳动物。哺乳动物包括但不限于驯养动物(例如,牛、绵羊、猫、狗和马)、灵长类动物(例如,人和非人灵长类动物,如猴子)、兔子和啮齿动物(例如,小鼠和大鼠)。在某些实施方案中,个体或患者或受试者是人。在一些实施方案中,本文的“个体”或“患者”或“受试者”是经历或已经历一种或多种体征、症状或哮喘或呼吸病症的其他指征的任何有资格接受治疗的单个人受试者。旨在作为受试者包括的是参与临床研究试验的未显示任何疾病临床体征的任何受试者、或参与流行病学研究的受试者、或曾经用作对照的受试者。受试者可能先前已用il-13拮抗剂或另一种药物治疗,或未经此治疗。当本文的治疗开始时,受试者可能未经il-13拮抗剂处理,即,受试者先前可能未用例如“基线”上(即,在本文的治疗方法中在施用第一剂量的il-13拮抗剂之前的设定时间点上,如在开始治疗之前筛选受试者的那天)的il-13拮抗剂治疗。这种“未经处理的”受试者通常被认为是用这种药物治疗的候选者。“儿科”个体或患者或受试者是从出生到18岁(或0至18岁)的人。在一些实施方案中,儿科个体或患者或受试者为2至6岁、2至17岁、6至11岁、6至18岁、6至17岁、8至17岁、12至17岁或12至18岁。“分离的”抗体是已经与其天然环境的组分分离的抗体。在一些实施方案中,如通过例如电泳(例如,sds-page、等电聚焦(ief)、毛细管电泳)或色谱法(例如,离子交换或反相hplc)所测定的,将抗体纯化至大于95%或99%的纯度。关于评估抗体纯度的方法的综述,参见,例如,flatman等人,j.chromatogr.b848:79-87(2007)。“分离的”核酸是指已经与其天然环境的组分分离的核酸分子。分离的核酸包括通常含有核酸分子的细胞中含有的核酸分子,但该核酸分子存在于染色体外或不同于其天然染色体位置的染色体位置。“编码抗靶抗体的分离的核酸”是指编码抗体重链和轻链(或其片段)的一种或多种核酸分子,包括单个载体或分开的载体中的一种或多种这样的核酸分子,存在于宿主细胞的一个或多个位置的一种或多种这样的核酸分子。术语“单克隆抗体”是指从基本上同质的抗体群获得的抗体,即,群包含的各抗体是相同的和/或结合相同的表位,除了可能的变体抗体,例如,含有天然发生突变的变体抗体或在单克隆抗体制剂的生产过程中产生的变体抗体,这些变体通常以少量存在。与通常包括针对不同决定簇(表位)的不同抗体的多克隆抗体制剂相反,单克隆抗体制剂的每种单克隆抗体针对抗原上的单个决定簇。因此,修饰语“单克隆”表示抗体的特征是从基本上同质的抗体群获得的抗体,而不应解释为需要通过任何特定方法产生抗体。例如,可以通过多种技术制备根据本文提供的方法使用的单克隆抗体,包括但不限于杂交瘤方法、重组dna方法、噬菌体展示方法和利用含有全部或部分人免疫球蛋白基因座的转基因动物的方法,此类方法和用于制备单克隆抗体的其他示例性方法在本文中描述。“裸抗体”是指未与异源部分(例如,细胞毒性部分)或放射性标记缀合的抗体。裸抗体可以存在于药物制剂中。“天然抗体”是指具有不同结构的天然发生的免疫球蛋白分子。例如,天然igg抗体是约150,000道尔顿的异四聚体糖蛋白,其由二硫键连接的两条相同的轻链和两条相同的重链组成。从n-末端到c-末端,每条重链具有可变区(vh),也称为可变重链结构域或重链可变结构域,接着是三个恒定结构域(ch1、ch2和ch3)。类似地,从n-末端到c-末端,每条轻链具有可变区(vl),也称为可变轻链结构域或轻链可变结构域,接着是恒定轻链(cl)结构域。基于其恒定结构域的氨基酸序列,抗体的轻链可以被划分为称为kappa(κ)和lambda(λ)的两种类型中的一种。术语“包装说明书”用于指通常包括在治疗产品的商业包装中的说明书,其包含关于适应症、用法、剂量、施用、组合治疗、禁忌症和/或关于使用此类治疗产品的警告的信息。术语“包装说明书”还用于指通常包含在诊断产品的商业包装中的说明书,其包含关于预期用途、测试原理、试剂的制备和处理、标本收集和制备、测定法的校准和测定步骤、性能和精确度数据如测定法的灵敏度和特异性的信息。相对于参照多肽序列的“百分比(%)氨基酸序列同一性”定义为在比对序列(如果需要,引入缺口以实现最大的百分比序列同一性)后,候选序列中与参照多肽序列中的氨基酸残基相同的氨基酸残基的百分比,不考虑任何保守取代作为序列同一性的一部分。可以以本领域技术范围内的各种方式实现用于确定百分比氨基酸序列同一性的目的的比对,例如,使用公众可获得的计算机软件,如blast、blast-2、align或megalign(dnastar)软件。本领域技术人员可确定用于比对序列的适当参数,包括在所比较序列的全长上实现最大比对所需的任何算法。然而,为了本文的目的,使用序列比较计算机程序align-2产生%氨基酸序列同一性值。align-2序列比较计算机程序由genentech,inc.编写,源代码与用户文档已经在华盛顿特区20559的美国版权局提交,其以美国版权登记号txu510087注册。align-2程序可从加利福尼亚州南旧金山的genentech,inc.公开获得,或者可以从源代码编译。应编译align-2程序以在unix操作系统上使用,包括数字unixv4.0d。所有序列比较参数均由align-2程序设置,没有改变。在使用align-2进行氨基酸序列比较的情况下,给定氨基酸序列a相对于、与、针对给定氨基酸序列b的氨基酸序列同一性%(可以替代地表达为给定氨基酸序列a具有或包含某种相对于、与、或针对给定氨基酸序列b的氨基酸序列同一性%),如下计算:100乘以x/y的分数,其中x是在该程序的a和b的比对中通过序列比对程序align-2评分为相同匹配的氨基酸残基的数目,其中y是b中氨基酸残基的总数。应理解,当氨基酸序列a的长度不等于氨基酸序列b的长度时,a相对于b的%氨基酸序列同一性将不等于b相对于a的%氨基酸序列同一性。除非另有说明,本文使用的所有%氨基酸序列同一性的值均使用align-2计算机程序如前一段中所述获得。术语“药物制剂”或“药物组合物”是指这样的形式的制剂,其允许其中包含的活性成分的生物学活性有效,并且不含有对施用该制剂的受试者具有不可接受的毒性的其它成分。“药学上可接受的载体”是指药物制剂中除活性成分以外的成分,其对受试者无毒。药学上可接受的载体包括但不限于缓冲液、赋形剂、稳定剂或防腐剂。除非另有说明,术语“靶标”是指来自任何脊椎动物来源的任何天然分子,包括哺乳动物如灵长类动物(例如人)和啮齿动物(例如小鼠和大鼠)。该术语包括“全长”、未加工的靶标以及在细胞中加工产生的任何形式的靶标。该术语还包括天然发生的靶标变体,例如剪接变体或等位基因变体。如本文所用,术语“治疗”(及其语法变体,如“治疗”或“治疗”)是指试图改变所治疗个体的自然病程的临床干预,其可以用于预防或在临床病理学过程中进行。理想的治疗效果包括但不限于预防疾病的发生或复发、缓解症状、减轻疾病的任何直接或间接病理结果、预防转移、降低疾病进展的速率、改善或缓解疾病状态、缓解或改善预后。在一些实施方案中,抗体用于延迟疾病的发展或减缓疾病的进展。术语“可变区”或“可变结构域”是指抗体重链或轻链的结构域,其参与抗体与抗原的结合。天然抗体的重链和轻链的可变结构域(分别为vh和vl)通常具有相似的结构,每个结构域包含四个保守构架区(fr)和三个高变区(hvr)。(参见,例如kindt等人kubyimmunology,第6版,w.h.freemanandco.,第91页(2007))。单个vh或vl结构域可足以赋予抗原结合特异性。此外,可以使用来自结合抗原的抗体的vh或vl结构域分离结合特定抗原的抗体,以分别筛选互补vl或vh结构域的文库。参见,例如,portolano等人,j.immunol.150:880-887(1993);clarkson等人,nature352:624-628(1991)。术语“载体”是指能够繁殖与其连接的另一核酸的核酸分子。该术语包括作为自我复制核酸结构的载体以及掺入其所导入的宿主细胞基因组中的载体。某些载体能够指导与其可操作地连接的核酸的表达。此类载体在本文中称为“表达载体”。在诊断患有特应性皮炎的患者中术语“加剧”表示在持续治疗下至少3天期间病灶的程度或严重性的可测量增加,并且相对应地疾病严重性临床显著增加(如治疗医师和/或患者所评估的),需要治疗升级(参见,例如,darsow等人,jeuracaddermatolvenereol2010;24:317-28)。可以使用“hanifin/rajka标准”诊断特应性皮炎。hanifin/rajka诊断标准描述于actadermvenereol(stockh)1980;suppl92:44-7。为了建立特应性皮炎的诊断,患者需要存在至少3个“基本特征”和下面列出的3个或更多个次要特征。基本特征包括瘙痒、典型的形态学和分布(如弯曲苔藓化或线纹(linearity)、慢性或慢性复发性皮炎)、以及特应性反应的个人或家族史,如哮喘、过敏性鼻炎、特应性皮炎。次要特征包括干燥、鱼鳞藓、手掌线纹过多或毛发角化、立即(1型)皮肤试验反应性、血清ige升高、发病年龄早、皮肤感染倾向(特别是金黄色葡萄球菌(staph.aureus)和单纯疱疹(herpessimplex))、细胞介导的免疫力受损、非特异性手足皮炎倾向、乳头湿疹、唇炎、复发性结膜炎、dennie-morgan眶下皱褶、圆锥角膜、前囊下白内障、眼眶变黑、面部苍白/面部红斑、白糠疹、前颈褶、出汗时发痒。其他次要标准包括对羊毛和脂质溶剂的不耐受、毛囊周围加重、食物不耐受、受环境或情感因素影响的病程、以及白色皮肤划痕现象/变白延迟。可通过如rajkag和langelandt,actadermvenereol(stockh)1989;144(suppl):13-4所述的“rajka/langeland标准”确定特应性皮炎的严重性。三个疾病严重性评估分类评分为1至3:i)所涉及身体面积的程度,ii)病程,例如,在一年中多于或少于3个月,或持续病程,以及iii)强度,从轻度发痒到严重发痒,通常会扰乱夜晚的睡眠。评分为1.5或2.5是允许的。整体疾病严重性由三个疾病评估分类的各评分的总和确定,严重性由这些评分的总和确定,其中轻度定义为总评分3-4、中等的评分为4.5-7.5、严重的总评分为8-9。术语“特应性皮炎疾病严重性结果测量”或“addsom”意指确定与特应性皮炎相关并且可以定量或定性评估的某些体征、症状、特征或参数。示例性addsom包括但不限于“湿疹面积和严重性指数”(easi)、“特应性皮炎的严重性评分”(scorad)、“研究者全面评估”(iga)、“体征的研究者全面评估”(igsa)、rajka/langeland特应性皮炎严重性评分,患者报告的结果包括但不限于瘙痒直观模拟标度(作为scorad的一部分评估的疾病严重性的一个方面)、睡眠丧失直观模拟标度(作为scorad的一部分评估的疾病严重性的一个方面)、特应性皮炎症状日记(adsd)、特应性皮炎影响问卷(adiq)、皮肤病生活质量指数(dlqi)(finlay和khan,clinexperdermatol1994;19:210)和5-d瘙痒标度(elman等人,brjdermatol2010;162(3):587-593)。“湿疹面积和严重性指数”或“easi”是在临床环境中用于评估ad的严重性和程度的经验证的量度,hanifin等人,expdermatol2001;10:11-18。由临床医生或其他医疗专业人员评估四个个体身体区域:头颈部(h&n)、上肢(ul;包括外腋和手)、躯干(包括内腋和腹股沟)和下肢(ll;包括臀部和脚)。对于每个身体区域,评估受影响的体表面积(bsa)并为受影响的bsa的百分比(0%-100%)指定0至6(或任选0-7,其中0等于无出疹)的评分;每个区域单独评定严重性的平均程度(0-3:无、轻度、中度、重度),其中四个临床体征中的每一个允许半步骤:红斑、硬结丘疹、表皮脱落和苔藓化。每个身体区域指定0到12的总评分;基于以下指定总身体区域评分:个体临床体征评分的总和(最大=12)×受影响面积评分(最大=6)×身体区域指数(h&n-0.1,ul-0.2,躯干-0.3,ll-0.4)。基于四个身体区域评分中的每一个的总评分的总和指定总评分(0-72)。“研究者全面评估”或“iga”是在临床环境中使用的评估测量,其基于五点标度确定ad的严重性和对治疗的临床反应。评分为0(干净)意味着没有特应性皮炎的炎症体征,评分为1(几乎干净)意味着只有可察觉的红斑和只有可察觉的丘疹硬结。评分为2、3或4(轻度、中度、重度)是基于红斑、丘疹硬结、渗出和结痂的严重性。“体征的研究者全面评估”或“igsa”基于对红斑、水肿、苔藓斑块或丘疹以及表皮脱落的评估,使用范围从干净到严重的受损评估等级。此外,评估的受损等级可以根据皮肤病灶的程度和位置进行升级或降级。“特应性皮炎的严重性评分”或“scorad”是由欧洲特应性皮炎特别小组开发的ad严重性的临床评估(共识报告,dermatology1993;186:23-31)。对疾病严重性的三个方面评分:(i)评估为0-100评分的受炎症影响的身体面积程度,在整体总评分中指定为“a”,(ii)六个临床体征(红斑、水肿/群、渗出/结痂、表皮脱落、苔藓化和干燥)的强度,每个根据严重性(无、轻度、中度、重度)分别指定为0-3的评分,总评分范围为0-18,在整体总评分中指定为“b”,以及(iii)使用患者报告的结果的两个主观量度:瘙痒直观模拟评分(范围从0[无瘙痒]到10[可想象最糟糕的瘙痒])和睡眠丧失直观模拟评分(范围从0[无睡眠丧失]到10[可想象最糟糕的睡眠丧失]),最后三天或晚上的各平均值,在整体总评分中指定为“c”。根据公式a/5+(7b/2)+c确定整体总评分(0-103)。术语“患者报告的结果”或“pro”表示在临床实践或临床试验中使用的经验证的问卷或工具,其从患者的角度评估患者特应性皮炎疾病症状相关的生活质量,可包括该疾病的情感和功能影响。pro是直接来自患者的患者健康状况的报告,没有临床医生或任何其他人对患者反应的解释。可以用绝对术语(例如,疾病的症状、体征或状态的严重性)或作为与先前测量的变化来测量该结果。在临床试验中,pro仪器可用于测量医学干预对一个或多个概念(即,被测量的事物,如症状或症状组,对特定功能或功能组的影响,或显示测量健康状况严重性的症状或功能组)的影响。用于评估ad的示例性pro工具包括但不限于瘙痒直观模拟标度(作为scorad的一部分评估的疾病严重性的一个方面)、睡眠丧失直观模拟标度(作为scorad的一部分评估的疾病严重性的一个方面)、特应性皮炎症状日记(adsd)、特应性皮炎影响问卷(adiq)、皮肤病生活质量指数(dlqi)(finlay和khan,clinexperdermatol1994;19:210)和5-d瘙痒标度(elman等人,brjdermatol2010;162(3):587-593)。组合物和方法本发明至少部分基于惊人和意想不到的发现,即,如通过若干功效结果测量所评估的,当使用本文提供的给药方案施用于特应性皮炎患者(包括同时使用局部皮质类固醇的患者)时,抗il-13拮抗剂单克隆抗体lebrikizumab提供治疗益处。因此,本文提供用il-13拮抗剂(包括抗il-13抗体,如lebrikizumab)治疗特应性皮炎的方法。lebrikizumab是人源化的单克隆igg4抗体,其以高亲和力结合il-13并通过活化的il-4rα/il-13rα1异二聚体阻断信号传导。已在哮喘中临床测试了不同剂量的lebrikizumab,结果已在文献中报道,如下所述。在milly研究中,向尽管吸入皮质类固醇治疗但哮喘控制不良的成年人每四周施用一次250mglebrikizumab或安慰剂,持续共六个月。corren等人,nengljmed2011;365:1088-98。如通过fev1测量的,lebrikizumab治疗的患者,特别是生物标记物高(血清骨膜蛋白高)的亚组的患者,显示比安慰剂组更大的肺功能改善。corren等人,同上。molly研究是剂量范围研究,其中未接受吸入皮质类固醇的哮喘患者每四周接受一次125mglebrikizumab或250mglebrikizumab或500mglebrikizumab或安慰剂,持续共12周。noonan等人,jallergyclinimmunol2013;132:567-74。虽然与安慰剂组相比,全部lebrikizumab剂量组的fev1的平均相对变化在数值上更高,但这些差异在统计学上和临床上都不显著。noonan等人,同上。此外,molly研究未满足证明剂量反应的目的。noonan等人,同上。这些结果的一个结论是,在molly研究中如通过fev1测量的,与现有治疗相比阻断哮喘患者群中的单个细胞因子il-13不足以改善肺功能。noonan等人,同上。在lute和verse研究中,在吸入皮质类固醇未控制的中度至重度哮喘患者中测试了每四周施用一次三个不同剂量的lebrikizumab,37.5mg、125mg和250mg。hanania等人,thorax2015;70:748–756。lebrikizumab治疗降低了哮喘发作率,这在生物标记物高(血清骨膜蛋白高)的患者中(所有剂量:减少60%)比生物标记物低(血清骨膜蛋白低)的患者中(所有剂量:减少5%)更明显;然而,没有明显的剂量反应。hanania等人,2015,同上。lebrikizumab治疗后肺功能也有所改善,与骨膜蛋白低的患者相比骨膜蛋白高的患者中fev1增加最多。hanania等人,2015,同上。如本文所述,ad和哮喘患者均具有升高水平的与il-13生物学和th-2介导的超敏反应相关的生物标记物。因此,认为在哮喘中产生临床益处的lebrikizumab的剂量水平也可以在ad患者中产生生物学活性。因此,如实施例中所述,在临床研究i和临床研究ii中所述用于在ad患者中测试的建议初始剂量基于如上所述的公布的哮喘临床经验。在启动本文所述的临床研究i和临床研究ii之后,分析了哮喘中lebrikizumab的3期临床研究结果(lavoltai和lavoltaii)并于2016年9月5日在线发表在hanania等人,lancetrespirmed2016,可从dx(dot)doi(dot)org(slash)s2213-2600(16)30265-x获得。lavoltai和ii是重复的3期研究,以评估lebrikizumab在尽管吸入皮质类固醇和至少一个第二控制物但未控制的哮喘患者中的疗效和安全性。设计这些3期研究的目的是寻求全球卫生当局的上市批准,前提是在两项研究中都证明了统计学上显著的疗效和可接受的安全性特征。在lavoltai和ii中,用37.5mglebrikizumab或125mglebrikizumab或安慰剂治疗患者,每4周施用一次,持续52周。主要疗效端点是恶化率降低。根据生物标记物高(血清骨膜蛋白高或血液嗜酸性粒细胞计数高)或生物标记物低(血清骨膜蛋白低或血液嗜酸性粒细胞计数低)对患者进行分层。如hanania等人,2016,同上,所报道,两项研究的疗效结果不一致。lebrikizumab显著降低了lavoltai中生物标记物高的患者的恶化率,但在lavoltaii则没有。hanania等人,2016,同上。虽然药代动力学和药效学结果与上述ii期研究结果一致,但两项研究均未显示明显的剂量反应,表明药物暴露相似且il-13通路受到抑制。hanania等人,2016,同上。因此,尽管在本文所述的特应性皮炎临床研究i和临床研究ii中测试的lebrikizumab给药方案基于哮喘的临床经验,但不确定在针对临床研究i和临床研究ii描述的给药方案中lebrikizumab是否将为特应性皮炎患者提供临床上有意义的益处。哮喘患者中lebrikizumab的结果不一致,以及上文总结的多项研究中缺乏明确的剂量反应,包括针对3期lavoltai和ii研究报告的结果,导致lebrikizumab在特应性皮炎患者中治疗效益方面的这种不确定性。在本文所述的发明之前,不能预期lebrikizumab是否可以在本文提供的给药方案中为特应性皮炎患者提供临床上有意义的益处。示例性抗体抗il-13抗体在一个方面,本发明提供了与人il-13结合的分离的抗体。示例性抗il-13抗体是已知的并且包括,例如但不限于,lebrikizumab、ima-026、ima-638(也称为anrukinzumab,innno.910649-32-0;qax-576)、tralokinumab(也称为cat-354,casno.1044515-88-9);aer-001、abt-308(也称为人源化13c5.5抗体。此类抗il-13抗体和il-13的其他抑制剂的实例公开于例如wo2005/062967、wo2008/086395、wo2006/085938、us7,615,213、us7,501,121、wo2007/036745、wo2010/073119、wo2007/045477、wo2014/165771。在一个实施方案中,抗il-13抗体是人源化igg4抗体。在一个实施方案中,抗il-13抗体是lebrikizumab。在一个实施方案中,抗il-13抗体包含三个重链hvr,hvr-h1(seqidno:5)、hvr-h2(seqidno:6)和hvr-h3(seqidno.:7)。在一个实施方案中,抗il-13抗体包含三个轻链hvr,hvr-l1(seqidno:8)、hvr-l2(seqidno:9)和hvr-l3(seqidno.:10)。在一个实施方案中,抗il-13抗体包含三个重链hvr和三个轻链hvr,hvr-h1(seqidno:5),hvr-h2(seqidno.:6),hvr-h3(seqidno:7),hvr-l1(seqidno.:8),hvr-l2(seqidno.:9)和hvr-l3(seqidno.:10)。在一个实施方案中,抗il-13抗体包含可变重链区vh,其具有选自seqidno.1、3和24的氨基酸序列。在一个实施方案中,抗il-13抗体包含可变轻链区vl,其具有选自seqidno:2、4和25的氨基酸序列。在一个实施方案中,抗il-13抗体包含可变重链区vh和可变轻链区vl,所述可变重链区vh具有选自seqidno.1、3和24的氨基酸序列,所述可变轻链区vl具有选自seqidno:2、4和25的氨基酸序列。在另一个实施方案中,抗体包含可变区序列seqidno:1和seqidno:2。在另一个实施方案中,抗体包含可变区序列seqidno:1和seqidno:4。在另一个实施方案中,抗体包含可变区序列seqidno:1和seqidno:25。在另一个实施方案中,抗体包含可变区序列seqidno:3和seqidno:2。在另一个实施方案中,抗体包含可变区序列seqidno:3和seqidno:4。在另一个实施方案中,抗体包含可变区序列seqidno:3和seqidno:25。在另一个实施方案中,抗体包含可变区序列seqidno:24和seqidno:2。在另一个实施方案中,抗体包含可变区序列seqidno:24和seqidno:4。在另一个实施方案中,抗体包含可变区序列seqidno:24和seqidno:25。在任何上述实施方案中,抗il-13抗体可以是人源化的。在一个实施方案中,抗il-13抗体包含如任何上述实施方案中的hvr,并且还包含受体人框架,例如人免疫球蛋白框架或人共有框架。在另一个方面,抗il-13抗体包含重链可变结构域(vh)序列,其与seqidno:1的氨基酸序列具有至少90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%的序列同一性。在某些实施方案中,具有至少90%、91%、92%、93%、94%、95%、96%、97%、98%或99%同一性的vh序列相对于参照序列含有取代(例如,保守取代)、插入或缺失,但包含该序列的抗il-13抗体保留了与人il-13结合的能力。在某些实施方案中,在seqidno:1中已经取代、改变插入和/或缺失总共1至10个氨基酸。在某些实施方案中,取代、插入或缺失发生在hvr以外的区域(即,在fr中)。任选地,抗il-13抗体包含seqidno:1中的vh序列,包括该序列的翻译后修饰。任选地,抗il-13抗体包含seqidno:3中的vh序列,包括该序列的翻译后修饰。任选地,抗il-13抗体包含seqidno:24中的vh序列,包括该序列的翻译后修饰。在另一个方面,提供了抗il-13抗体,其中所述抗体包含轻链可变结构域(vl),其与seqidno:2的氨基酸序列具有至少90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%的序列同一性。在某些实施方案中,具有至少90%、91%、92%、93%、94%、95%、96%、97%、98%或99%同一性的vl序列相对于参照序列含有取代(例如,保守取代)、插入或缺失,但包含该序列的抗il-13抗体保留了与il-13结合的能力。在某些实施方案中,在seqidno:2中已经取代、插入和/或缺失总共1至10个氨基酸。在某些实施方案中,取代、插入或缺失发生在hvr以外的区域(即fr中)。任选地,抗il-13抗体包含seqidno:2中的vl序列,包括该序列的翻译后修饰。任选地,抗il-13抗体包含seqidno:4中的vl序列,包括该序列的翻译后修饰。任选地,抗il-13抗体包含seqidno:25中的vl序列,包括该序列的翻译后修饰。在另一个方面,提供了抗il-13抗体,其中所述抗体包含如上文提供的任何实施方案中的vh,和如上文提供的任何实施方案中的vl。在另一方面,本发明提供了与本文提供的抗il-13抗体结合相同表位的抗体。例如,在某些实施方案中,提供了与抗il-13抗体结合相同的表位或竞争性抑制抗il-13抗体的抗体,所述抗il-13抗体包含seqidno:1的vh序列和seqidno:2的vl序列。在本发明的另一方面,根据任何上述实施方案的抗il-13抗体可以是单克隆抗体,包括嵌合抗体、人源化抗体或人抗体。在一个实施方案中,抗il-13抗体是抗体片段,例如fv、fab、fab'、scfv、双抗体或f(ab')2片段。在另一个实施方案中,抗体是全长抗体,例如完整的igg1或igg4抗体或本文定义的其他抗体类别或同种型。根据另一个实施方案,抗体是双特异性抗体。在一个实施方案中,双特异性抗体包含hvr或包含上述vh和vl区。在一个实施方案中,抗il-13抗体包含三个重链hvr,hvr-h1(seqidno:13)、hvr-h2(seqidno:14)和hvr-h3(seqidno:15)。在一个实施方案中,抗il-13抗体包含三个轻链hvr,hvr-l1(seqidno:16)、hvr-l2(seqidno:17)和hvr-l3(seqidno:18)。在一个实施方案中,抗il-13抗体包含三个重链hvr和三个轻链hvr,hvr-h1(seqidno:13)、hvr-h2(seqidno:14)、hvr-h3(seqidno:15)、hvr-l1(seqidno:16)、hvr-l2(seqidno.:17)和hvr-l3(seqidno.:18)。在一个实施方案中,抗il-13抗体包含可变重链区vh,其具有seqidno:12的氨基酸序列。在一个实施方案中,抗il-13抗体包含可变轻链区vl,其具有seqidno:11的氨基酸序列。在一个实施方案中,抗il-13抗体包含具有seqidno:12的氨基酸序列的可变重链区vh和具有seqidno:11的氨基酸序列的可变轻链区vl。在任何上述实施方案中,抗il-13抗体可以是人源化的。在一个实施方案中,抗il-13抗体包含如任何上述实施方案中的hvr,并且还包含受体人框架,例如人免疫球蛋白框架或人共有框架。在另一个方面,抗il-13抗体包含重链可变结构域(vh)序列,其与seqidno:12的氨基酸序列具有至少90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%的序列同一性。在某些实施方案中,具有至少90%、91%、92%、93%、94%、95%、96%、97%、98%或99%同一性的vh序列相对于参照序列含有取代(例如,保守取代)、插入或缺失,但包含该序列的抗il-13抗体保留了与人il-13结合的能力。在某些实施方案中,在seqidno:12中已经取代、改变插入和/或缺失总共1至10个氨基酸。在某些实施方案中,取代、插入或缺失发生在hvr以外的区域中(即,在fr中)。任选地,抗il-13抗体包含seqidno:12中的vh序列,包括该序列的翻译后修饰。在另一个方面,提供了抗il-13抗体,其中所述抗体包含轻链可变结构域(vl),其与seqidno:11的氨基酸序列具有至少90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%的序列同一性。在某些实施方案中,具有至少90%、91%、92%、93%、94%、95%、96%、97%、98%或99%同一性的vl序列相对于参照序列含有取代(例如,保守取代)、插入或缺失,但包含该序列的抗il-13抗体保留了与il-13结合的能力。在某些实施方案中,在seqidno:11中已经取代、插入和/或缺失总共1至10个氨基酸。在某些实施方案中,取代、插入或缺失发生在hvr以外的区域(即fr中)。任选地,抗il-13抗体包含seqidno:11中的vl序列,包括该序列的翻译后修饰。在另一个方面,提供了抗il-13抗体,其中所述抗体包含如上文提供的任何实施方案中的vh,和如上文提供的任何实施方案中的vl。在另一方面,本发明提供了与本文提供的抗il-13抗体结合相同表位的抗体。例如,在某些实施方案中,提供了与抗il-13抗体结合相同表位或被抗il-13抗体竞争性抑制的抗体,所述抗il-13抗体包含seqidno:12的vh序列和seqidno:11的vl序列。在本发明的另一方面,根据任何上述实施方案的抗il-13抗体可以是单克隆抗体,包括嵌合抗体、人源化抗体或人抗体。在一个实施方案中,抗il-13抗体是抗体片段,例如fv、fab、fab'、scfv、双抗体或f(ab')2片段。在另一个实施方案中,抗体是全长抗体,例如完整的igg1或igg4抗体或本文定义的其他抗体类别或同种型。根据另一个实施方案,抗体是双特异性抗体。在一个实施方案中,双特异性抗体包含hvr或包含上述vh和vl区。在另一方面,根据任何上述实施方案的抗il-13抗体可以单独或组合掺入任何特征,如以下第1-7节中所述:i.抗体亲和力在某些实施方案中,本文提供的抗体具有≤1μm、≤100nm、≤10nm、≤1nm、≤0.1nm、≤0.01nm或≤0.001nm的解离常数(kd)(例如10-8m或更小,例如10-8m至10-13m,例如10-9m至10-13m)。在一个实施方案中,通过放射性标记的抗原结合测定法(ria)测量kd,如以下测定法所述使用目标抗体的fab形式及其抗原进行。通过在滴定系列的未标记抗原存在下用最小浓度的(125i)标记的抗原平衡fab、然后用抗fab抗体包被的平板捕获结合的抗原来测量fab对抗原的溶液结合亲和力(参见,例如,chen等人,j.mol.biol.293:865-881(1999))。为了建立测定条件,用5μg/ml捕获抗fab抗体(cappellabs)的50mm碳酸钠(ph9.6)溶液过夜包被多孔平板(thermoscientific),随后在室温下(约23℃)用2%(w/v)牛血清白蛋白的pbs溶液封闭2至5小时。在非吸附平板(nunc#269620)中,将100pm或26pm[125i]-抗原与目的fab的系列稀释液混合(例如,与presta等人,cancerres.57:4593-4599(1997)中的抗vegf抗体fab-12的评估一致)。然后将目的fab过夜孵育;然而,孵育可以持续更长的时间(例如,约65小时)以确保达到平衡。此后,将混合物转移到捕获平板中,在室温下孵育(例如,1小时)。然后除去溶液,用0.1%聚山梨醇酯20的pbs溶液洗涤平板8次。当平板干燥后,加入150μl/孔的闪烁剂(microscint-20tm;packard),并将平板在topcounttm伽马计数器(packard)上计数10分钟。选择产生小于或等于20%最大结合的各fab浓度用于竞争性结合测定法。根据另一个实施方案,使用或(biacore,inc.,piscataway,nj)的表面等离子体共振测定法在25℃下测量kd,其中以~10反应单位(ru)固定抗原cm5芯片。简言之,根据供应商的说明,用n-乙基-n'-(3-二甲基氨基丙基)-碳二亚胺盐酸盐(edc)和n-羟基琥珀酰亚胺(nhs)活化羧甲基化葡聚糖生物传感器芯片(cm5,biacore,inc.)。将抗原用10mm乙酸钠(ph4.8)稀释至5μg/ml(~0.2μm),然后以5μl/分钟的流速注射以获得约10反应单位(ru)的偶联蛋白。注射抗原后,注入1m乙醇胺以封闭未反应的基团。对于动力学测量,在25℃下以约25μl/分钟的流速注射含有0.05%聚山梨醇酯20(tween-20tm)表面活性剂的pbs溶液(pbst)中的fab两倍系列稀释液(0.78nm至500nm)。通过同时拟合结合和解离传感图,使用简单一对一langmuir结合模型(评估软件版本3.2)计算结合速率(kon)和解离速率(koff)。平衡解离常数(kd)计算为koff/kon比。参见,例如,chen等人,j.mol.biol.293:865-881(1999)。如果通过上述表面等离子体共振测定法获得的结合速率超过106m-1s-1,则可以通过使用荧光猝灭技术测定结合速率,该技术在25℃、20nm抗原抗体(fab形式)的pbs(ph7.2)溶液中、在递增浓度的抗原存在下测量荧光发射强度的增加或减少(激发=295nm;发射=340nm,16nm带通),如在光谱仪中测量,如停流配备的分光光度计(avivinstruments)或8000系列slm-amincotm分光光度计(thermospectronic)含有搅拌比色皿。2.抗体片段在某些实施方案中,本文提供的抗体是抗体片段。抗体片段包括但不限于fab、fab'、fab'-sh、f(ab')2、fv和scfv片段、以及下文描述的其他片段。有关某些抗体片段的综述,参见hudson等人nat.med.9:129-134(2003)。关于scfv片段的综述,参见例如pluckthün,thepharmacologyofmonoclonalantibodies,卷113,rosenburg和moore编辑,(springer-verlag,newyork),第269-315页(1994);还参见wo93/16185;以及美国专利号5,571,894和5,587,458。关于包含补救受体结合表位残基并具有增加的体内半衰期的fab和f(ab')2片段的讨论,参见美国专利号5,869,046。双抗体是具有两个抗原结合位点的抗体片段,其可以是二价或双特异性的。参见,例如,ep404,097;wo1993/01161;hudson等人,nat.med.9:129-134(2003);和hollinger等人,proc.natl.acad.sci.usa90:6444-6448(1993)。三抗体和四抗体也描述于hudson等人,nat.med.9:129-134(2003)。单结构域抗体是包含全部或部分抗体的重链可变结构域或全部或部分抗体的轻链可变结构域的抗体片段。在某些实施方案中,单结构域抗体是人单结构域抗体(domantis,inc.,waltham,ma;参见例如美国专利号6,248,516b1)。可以通过各种技术制备抗体片段,包括但不限于完整抗体的蛋白水解消化以及由重组宿主细胞(例如大肠杆菌或噬菌体)产生,如本文所述。3.嵌合和人源化抗体在某些实施方案中,本文提供的抗体是嵌合抗体。某些嵌合抗体描述于例如美国专利号4,816,567;和morrison等人,proc.natl.acad.sci.usa,81:6851-6855(1984))。在一个实施例中,嵌合抗体包含非人可变区(例如,衍生自小鼠、大鼠、仓鼠、兔或非人灵长类动物如猴的可变区)和人恒定区。在另一个实施例中,嵌合抗体是“类别转换的”抗体,其中类或亚类已经从亲本抗体的类或亚类改变。嵌合抗体包括其抗原结合片段。在某些实施方案中,嵌合抗体是人源化抗体。通常,将非人抗体人源化以降低对人的免疫原性,同时保留亲本非人抗体的特异性和亲和力。通常,人源化抗体包含一个或多个可变结构域,其中hvr,例如cdr(或其部分)衍生自非人抗体,fr(或其部分)衍生自人抗体序列。人源化抗体任选还包含人恒定区的至少一部分。在一些实施方案中,人源化抗体中的一些fr残基被来自非人抗体(例如,衍生hvr残基的抗体)的相应残基取代,例如,以恢复或改善抗体特异性或亲和力。人源化抗体及其制备方法在例如almagro和fransson,front.biosci.13:1619-1633(2008)中进行了综述,并且进一步描述在,例如,riechmann等人,nature332:323-329(1988);queen等人,proc.nat’lacad.sci.usa86:10029-10033(1989);美国专利号5,821,337,7,527,791,6,982,321和7,087,409;kashmiri等人,methods36:25-34(2005)(描述了sdr(a-cdr)接枝);padlan,mol.immunol.28:489-498(1991)(描述了“再表面化”);dall’acqua等人,methods36:43-60(2005)(描述了“fr改组”);和osbourn等人methods36:61-68(2005)和klimka等人,br.j.cancer,83:252-260(2000)(描述了fr改组的“引导选择”方法)。可用于人源化的人框架区包括但不限于:使用“最佳拟合”方法选择的框架区(参见,例如,sims等人j.immunol.151:2296(1993));衍生自轻链或重链可变区的特定亚组的人抗体共有序列的框架区(参见,例如,carter等人proc.natl.acad.sci.usa,89:4285(1992);和presta等人j.immunol.,151:2623(1993));人成熟(体细胞突变)框架区或人种系框架区(参见例如,almagro和fransson,front.biosci.13:1619-1633(2008));来自筛选fr文库的框架区(参见,例如,baca等人,j.biol.chem.272:10678-10684(1997)和rosok等人,j.biol.chem.271:22611-22618(1996))。4.人抗体在某些实施方案中,本文提供的抗体是人抗体。可以使用本领域已知的各种技术产生人抗体。人抗体一般描述于vandijk和vandewinkel,curr.opin.pharmacol.5:368-74(2001)和lonberg,curr.opin.immunol.20:450-459(2008)。可以通过向转基因动物施用免疫原来制备人抗体,所述转基因动物已被修饰以响应抗原攻击产生完整人抗体或具有人可变区的完整抗体。这些动物通常含有全部或部分人免疫球蛋白基因座,这些人免疫球蛋白基因座取代内源性免疫球蛋白基因座,或者存在于动物染色体外或随机整合到动物染色体中。在这种转基因小鼠中,内源性免疫球蛋白基因座通常被灭活。关于从转基因动物获得人抗体的方法的综述,参见lonberg,nat.biotech.23:1117-1125(2005)。还参见例如描述xenomousetm技术的美国专利号6,075,181和6,150,584;描述技术的美国专利号5,770,429;描述k-m技术的美国专利号7,041,870和描述技术的美国专利申请公开号us2007/0061900。可以进一步修饰由这些动物产生的完整抗体的人可变区,例如通过与不同的人恒定区组合。也可以通过基于杂交瘤的方法制备人抗体。已经描述了用于产生人单克隆抗体的人骨髓瘤和小鼠-人异源骨髓瘤细胞系(参见,例如,kozborj.immunol.,133:3001(1984);brodeur等人,monoclonalantibodyproductiontechniquesandapplications,第51-63页(marceldekker,inc.,newyork,1987);和boerner等人,j.immunol.,147:86(1991))。通过人b细胞杂交瘤技术产生的人抗体也描述于li等人,proc.natl.acad.sci.usa,103:3557-3562(2006)。其他方法包括例如美国专利号7,189,826(描述从杂交瘤细胞系产生单克隆人igm抗体)和ni,xiandaimianyixue,26(4):265-268(2006)(描述人-人杂交瘤)中描述的那些方法。人杂交瘤技术(trioma技术)也描述于vollmers和brandlein,histologyandhistopathology,20(3):927-937(2005),以及vollmers和brandlein,methodsandfindingsinexperimentalandclinicalpharmacology,27(3):185-91(2005)中。还可以通过分离选自人源噬菌体展示文库的fv克隆可变结构域序列来产生人抗体。然后可以将这种可变结构域序列与期望的人恒定结构域组合。从抗体文库中选择人抗体的技术如下描述。5.文库来源抗体可以通过筛选组合文库获得具有一种或多种所需活性的抗体来分离本发明的抗体。例如,本领域已知多种方法用于产生噬菌体展示文库并筛选这些文库以获得具有所需结合特征的抗体。例如,在hoogenboom等人的methodsinmolecularbiology178:1-37(o'brienetal.,ed.,humanpress,totowa,nj,2001)中对这些方法进行了综述,并进一步描述在例如mccafferty等人,nature348:552-554;clackson等人,nature352:624-628(1991);marks等人,j.mol.biol.222:581-597(1992);marks和bradbury,methodsinmolecularbiology248:161-175(lo,ed.,humanpress,totowa,nj,2003);sidhu等人j.mol.biol.338(2):299-310(2004);lee等人,j.mol.biol.340(5):1073-1093(2004);fellouse,proc.natl.acad.sci.usa101(34):12467-12472(2004);和lee等人,j.immunol.methods284(1-2):119-132(2004)中。在某些噬菌体展示方法中,通过聚合酶链反应(pcr)分别克隆vh和vl基因的库,并在噬菌体文库中随机重组,然后如winter等人,ann.rev.immunol.,12:433-455(1994)所述从中筛选抗原结合噬菌体。噬菌体通常展示抗体片段,即单链fv(scfv)片段或fab片段。来自免疫来源的文库提供针对免疫原的高亲和力抗体,而不需要构建杂交瘤。或者,可以(例如,从人)克隆天然库以提供针对多种非自身和自身抗原的单一抗体来源,而无需任何免疫,如griffiths等人,emboj,12:725-734(1993)所述。最后,如hoogenboom和winter,j.mol.biol.,227:381-388(1992)所述,也可以通过克隆来自干细胞的未重排v基因区段、并使用含有随机序列的pcr引物以编码高度可变的cdr3区并完成体外重排,合成地制备天然文库。描述人抗体噬菌体文库的专利出版物包括,例如:美国专利号5,750,373和美国专利公开号2005/0079574、2005/0119455、2005/0266000、2007/0117126、2007/0160598、2007/0237764、2007/0292936和2009/0002360。从人抗体文库分离的抗体或抗体片段在本文中被认为是人抗体或人抗体片段。6.多特异性抗体在某些实施方案中,本文提供的抗体是多特异性抗体,例如,双特异性抗体。多特异性抗体是对至少两个不同位点具有结合特异性的单克隆抗体。在某些实施方案中,结合特异性之一针对il-13,而另一个结合特异性针对任何其他抗原。在某些实施方案中,双特异性抗体可以结合il-13的两个不同表位。双特异性抗体也可用于将细胞毒性剂定位于细胞。双特异性抗体可以制备成全长抗体或抗体片段。制备多特异性抗体的技术包括但不限于具有不同特异性的两种免疫球蛋白重链-轻链对的重组共表达(参见milstein和cuello,nature305:537(1983))、wo93/08829和traunecker等人,emboj.10:3655(1991))和“旋钮入洞”工程化(参见例如美国专利号5,731,168)。还可以通过工程化用于制备抗体fc-异二聚体分子的静电转向效应来制备多特异性抗体(wo2009/089004a1);交联两种或更多种抗体或片段(参见,例如,美国专利号4,676,980,和brennan等人,science,229:81(1985));使用亮氨酸拉链产生双特异性抗体(参见,例如,kostelny等人,j.immunol.,148(5):1547-1553(1992));使用“双抗体”技术制备双特异性抗体片段(参见,例如,hollinger等人,proc.natl.acad.sci.usa,90:6444-6448(1993));以及使用单链fv(sfv)二聚体(参见,例如gruber等人,j.immunol.,152:5368(1994));以及例如,如tutt等人j.immunol.147:60(1991)所述制备三特异性抗体。本文还包括具有三个或更多个功能性抗原结合位点的工程化抗体,包括“章鱼抗体”(参见例如us2006/0025576a1)。本文的抗体或片段还包括“双作用fab”或“daf”,其包含结合il-13和另一种不同抗原的抗原结合位点(例如,参见us2008/0069820)。7.抗体变体在某些实施方案中,涵盖本文提供的抗体的氨基酸序列变体。例如,可能期望改善抗体的结合亲和力和/或其他生物学特性。可以通过将适当的修饰引入编码抗体的核苷酸序列中、或通过肽合成来制备抗体的氨基酸序列变体。此类修饰包括例如抗体氨基酸序列内残基的缺失、和/或插入和/或取代。可以制备缺失、插入和取代的任何组合以获得最终构建体,只要最终构建体具有期望的特征,例如抗原结合。取代、插入和缺失变体在某些实施方案中,提供了具有一个或多个氨基酸取代的抗体变体。用于取代诱变的目标位点包括hvr和fr。表1中在“保守取代”的标题下显示了保守取代。表1中在“示例性取代”的标题下提供了更多实质性变化,并且下文中参照氨基酸侧链类别进行了进一步的描述。可以将氨基酸取代引入目标抗体中,并筛选产物的期望活性,例如保留/改善的抗原结合、降低的免疫原性,或改善的adcc或cdc。表1原始残基示例性取代保守取代ala(a)val;leu;ilevalarg(r)lys;gln;asnlysasn(n)gln;his;asp,lys;argglnasp(d)glu;asnglucys(c)ser;alasergln(q)asn;gluasnglu(e)asp;glnaspgly(g)alaalahis(h)asn;gln;lys;argargile(i)leu;val;met;ala;phe;正亮氨酸leuleu(l)正亮氨酸;ile;val;met;ala;pheilelys(k)arg;gln;asnargmet(m)leu;phe;ileleuphe(f)trp;leu;val;ile;ala;tyrtyrpro(p)alaalaser(s)thrthrthr(t)val;sersertrp(w)tyr;phetyrtyr(y)trp;phe;thr;serpheval(v)ile;leu;met;phe;ala;正亮氨酸leu可根据常见的侧链性质对氨基酸进行分组:(1)疏水性:正亮氨酸、met、ala、val、leu、ile;(2)中性亲水性:cys、ser、thr、asn、gln;(3)酸性:asp、glu;(4)碱性:his、lys、arg;(5)影响链方向的残基:gly、pro;(6)芳香性:trp、tyr、phe。非保守取代需要将这些类别之一的成员交换为另一类别。一种类型的取代变体包括取代亲本抗体(例如人源化或人抗体)的一个或多个高变区残基。通常,选择用于进一步研究的所得变体相对于亲本抗体具有某些生物学特性的修饰(例如,改善)(例如,增加的亲和力、降低的免疫原性)和/或基本上保留了亲本抗体的某些生物学特性。示例性的取代变体是亲和力成熟的抗体,例如,其可以使用如本文所述的那些基于噬菌体展示的亲和力成熟技术方便地产生。简言之,突变一个或多个hvr残基并在噬菌体上展示变体抗体并筛选特定的生物学活性(例如结合亲和力)。可以在hvr中进行改变(例如,取代),例如,以改善抗体亲和力。这种改变可以在hvr“热点”中进行,该“热点”即在体细胞成熟过程中经历高频突变的密码子编码的残基(参见,例如,chowdhury,methodsmol.biol.207:179-196(2008)),和/或sdr(a-cdr),测试所得变体vh或vl的结合亲和力。通过构建和从二级文库中重新选择的亲和力成熟已经描述在例如hoogenboom等人methodsinmolecularbiology178:1-37(o’brien等人编辑,humanpress,totowa,nj,(2001))中。在亲和力成熟的一些实施方案中,通过多种方法中的任何一种(例如,易错pcr、链改组或寡核苷酸定向诱变)将多样性引入选择用于成熟的可变基因中。然后创建二级文库。然后筛选文库以鉴定具有期望亲和力的任何抗体变体。引入多样性的另一种方法包括hvr定向的方法,其中几个hvr残基(例如,每次4-6个残基)被随机分组。例如可以使用丙氨酸扫描诱变或建模特异性地鉴定参与抗原结合的hvr残基。具体地通常靶向cdr-h3和cdr-l3。在某些实施方案中,取代、插入或缺失可以在一个或多个hvr内发生,只要这种改变不实质上降低抗体结合抗原的能力。例如,可以在hvr中进行基本上不降低结合亲和力的保守改变(例如,如本文提供的保守取代)。这种改变可以在hvr“热点”或sdr以外。在上文提供的变体vh和vl序列的某些实施方案中,每个hvr未改变、或含有不超过一个、两个或三个氨基酸取代。用于鉴定可以靶向诱变的抗体残基或区域的有用的方法称为“丙氨酸扫描诱变”,如cunningham和wells(1989)science,244:1081-1085所述。在该方法中,鉴定残基或一组靶残基(例如带电荷的残基,如arg、asp、his、lys和glu)并用中性或带负电荷的氨基酸(例如丙氨酸或聚丙氨酸)代替以确定抗体与抗原的相互作用是否受到影响。可以在证明对初始取代是功能敏感的氨基酸位置上引入进一步的取代。或者,或另外地,抗原-抗体复合物的晶体结构用于鉴定抗体和抗原之间的接触点。可以作为取代候选物靶向或消除这种接触残基和邻近残基。可以筛选变体以确定它们是否含有期望的特性。氨基酸序列插入包括氨基-和/或羧基-末端融合(长度范围从一个残基到含有一百个或更多个残基的多肽),以及单个或多个氨基酸残基的序列内插入。末端插入的实例包括具有n-末端甲硫氨酰残基的抗体。抗体分子的其他插入变体包括抗体的n-或c-末端与酶(例如对于adept)或多肽的融合,所述酶或多肽增加抗体的血清半衰期。糖基化变体在某些实施方案中,改变本文提供的抗体以增加或降低抗体糖基化的程度。可以通过改变氨基酸序列以产生或除去一个或多个糖基化位点而方便地完成抗体糖基化位点的添加或缺失。当抗体包含fc区时,可以改变与其连接的碳水化合物。由哺乳动物细胞产生的天然抗体通常包含支链的双触角寡糖,其通常通过n-键连接至fc区的ch2结构域的asn297。参见,例如,wright等人tibtech15:26-32(1997)。寡糖可包括各种碳水化合物,例如甘露糖、n-乙酰葡糖胺(glcnac)、半乳糖和唾液酸、以及与双触角寡糖结构的"茎"中的glcnac连接的岩藻糖。在一些实施方案中,可以对本发明抗体中的寡糖进行修饰以产生具有某些改善的性质的抗体变体。在一个实施方案中,提供了具有碳水化合物结构的抗体变体,所述碳水化合物结构缺乏(直接或间接)与fc区连接的岩藻糖。例如,这种抗体中岩藻糖的量可以是1%至80%、1%至65%、5%至65%或20%至40%。通过计算相对于与asn297连接的所有糖结构(例如复合、杂合和高甘露糖结构)的总和,计算在asn297糖链内岩藻糖的平均量来确定岩藻糖的量,如通过maldi-tof质谱法测量,例如,如wo2008/077546中所述。asn297是指位于fc区约297位的天冬酰胺残基(fc区残基的eu编号);然而,由于抗体中的微小序列变异,asn297也可位于297位上游或下游的约±3个氨基酸,即294位和300位之间。此类岩藻糖基化变体可具有改善的adcc功能。参见,例如,美国专利公开号us2003/0157108(presta,l.);us2004/0093621(kyowahakkokogyoco.,ltd)。与“去岩藻糖基化”或“岩藻糖缺陷”抗体变体相关的出版物的实例包括:us2003/0157108;wo2000/61739;wo2001/29246;us2003/0115614;us2002/0164328;us2004/0093621;us2004/0132140;us2004/0110704;us2004/0110282;us2004/0109865;wo2003/085119;wo2003/084570;wo2005/035586;wo2005/035778;wo2005/053742;wo2002/031140;okazaki等人j.mol.biol.336:1239-1249(2004);yamane-ohnuki等人biotech.bioeng.87:614(2004)。能够产生去岩藻糖基化抗体的细胞系的实例包括蛋白岩藻糖基化缺陷的lec13cho细胞(ripka等人arch.biochem.biophys.249:533-545(1986);美国专利申请号us2003/0157108a1,presta,l;和wo2004/056312a1,adams等人,特别是实施例11)、和基因敲除细胞系,如α-1,6-岩藻糖基转移酶基因,fut8敲除的cho细胞(参见,例如,yamane-ohnuki等人biotech.bioeng.87:614(2004);kanda,y.等人,biotechnol.bioeng.,94(4):680-688(2006);和wo2003/085107)。进一步提供具有二等分寡糖的抗体变体,例如,其中与抗体fc区连接的双触角寡糖被glcnac二等分。此类抗体变体可具有降低的岩藻糖基化和/或改善的adcc功能。此类抗体变体的实例描述于例如wo2003/011878(jean-mairet等人);美国专利号6,602,684(umana等人);和us2005/0123546(umana等人)。还提供了与fc区连接的寡糖中具有至少一个半乳糖残基的抗体变体。此类抗体变体可具有改善的cdc功能。此类抗体变体描述于例如wo1997/30087(patel等人);wo1998/58964(raju,s.);和wo1999/22764(raju,s.)。fc区变体在某些实施方案中,可以将一个或多个氨基酸修饰引入本文提供的抗体的fc区,从而产生fc区变体。fc区变体可包含人fc区序列(例如,人igg1、igg2、igg3或igg4fc区),所述人fc区序列在一个或多个氨基酸位置处包含氨基酸修饰(例如取代)。在某些实施方案中,本发明考虑一种抗体变体,其具有一些但不是所有的效应功能,这使得其成为用于其中抗体体内半衰期是重要的但某些效应功能(如补体和adcc)是不必要的或是有害的应用中的理想候选者。可以进行体外和/或体内细胞毒性测定法以确认cdc和/或adcc活性的减少/消耗。例如,可以进行fc受体(fcr)结合测定法以确保抗体缺乏fcγr结合(因此可能缺乏adcc活性),但保留fcrn结合能力。介导adcc的主要细胞(nk细胞)仅表达fcγriii,而单核细胞表达fcγri、fcγrii和fcγriii。在ravetch和kinet,annu.rev.immunol.9:457-492(1991)的第464页的表3中总结了造血细胞上的fcr表达。用于评估目标分子adcc活性的体外测定法的非限制性实例描述于美国专利号5,500,362(参见,例如hellstrom,i.等人proc.nat’lacad.sci.usa83:7059-7063(1986))和hellstrom,i等人,proc.nat’lacad.sci.usa82:1499-1502(1985);5,821,337(参见bruggemann,m.等人,j.exp.med.166:1351-1361(1987))。或者,可以使用非放射性测定方法(参见,例如,用于流式细胞术的actitm非放射性细胞毒性测定法(celltechnology,inc.mountainview,ca;和cytotox非放射性细胞毒性测定法(promega,madison,wi)。用于此类测定法的有用效应细胞包括外周血单个核细胞(pbmc)和自然杀伤(nk)细胞。或者,或另外地,可在体内评估目的分子的adcc活性,例如在动物模型中,如clynes等人proc.nat’lacad.sci.usa95:652-656(1998)所公开的动物模型中评估。还可以进行c1q结合测定法以确认抗体不能结合c1q并因此缺乏cdc活性。参见例如wo2006/029879和wo2005/100402中的c1q和c3c结合elisa。为了评估补体激活,可以进行cdc测定法(参见,例如,gazzano-santoro等人,j.immunol.methods202:163(1996);cragg,m.s.等人,blood101:1045-1052(2003);以及cragg,m.s.和m.j.glennie,blood103:2738-2743(2004))。还可以使用本领域已知的方法进行fcrn结合和体内清除/半衰期测定(参见,例如,petkova,s.b.等人,int’l.immunol.18(12):1759-1769(2006))。具有降低的效应功能的抗体包括具有fc区残基238、265、269、270、297、327和329中的一个或多个的取代的抗体(美国专利号6,737,056)。此类fc突变体包括在氨基酸位置265、269、270、297和327中的两处或更多处具有取代的fc突变体,包括所谓的“dana”fc突变体,其残基265和297取代为丙氨酸(美国专利号7,332,581)。描述了具有改善或减弱的与fcr结合的某些抗体变体。(参见,例如,美国专利号6,737,056;wo2004/056312,和shields等人,j.biol.chem.9(2):6591-6604(2001)。在某些实施方案中,抗体变体包含具有一个或多个改善adcc的氨基酸取代的fc区,例如fc区的298、333和/或334位的取代(eu残基编号)。在一些实施方案中,在fc区中进行改变,其导致改变(即,改善或减少)c1q结合和/或补体依赖性细胞毒性(cdc),例如,如美国专利号6,194,551、wo99/51642和idusogie等人j.immunol.164:4178-4184(2000)所述。具有增加的半衰期和改善的与新生儿fc受体(fcrn)(其负责将母体igg转移至胎儿(guyer等人,j.immunol.117:587(1976)和kim等人j.immunol.24:249(1994)))结合的抗体描述于us2005/0014934a1(hinton等人)中。那些抗体包含其中具有一个或多个取代的fc区,这些取代改善fc区与fcrn的结合。此类fc变体包括在以下一个或多个fc区残基中具有取代的那些变体:238、256、265、272、286、303、305、307、311、312、317、340、356、360、362、376、378、380、382、413、424或434,例如,fc区残基434的取代(美国专利号7,371,826)。关于fc区变体的其他实例,还参见duncan&winter,nature322:738-40(1988);美国专利号5,648,260;美国专利号5,624,821;和wo94/29351。半胱氨酸工程化的抗体变体在某些实施方案中,可能期望产生半胱氨酸工程化的抗体,例如“thiomabs”,其中抗体的一个或多个残基被半胱氨酸残基取代。在特定的实施方案中,取代的残基发生在抗体的可及位点。通过用半胱氨酸取代那些残基,由此将反应性硫醇基团定位在抗体的可及位点,并且可以用于将抗体与其他部分(如药物部分或接头-药物部分)缀合,以产生免疫缀合物,如本文进一步描述的。在某些实施方案中,以下残基中的任何一个或多个可以被半胱氨酸取代:轻链的v205(kabat编号);重链的a118(eu编号);和重链fc区的s400(eu编号)。可以如例如美国专利号7,521,541中所述产生半胱氨酸工程化的抗体。抗体衍生物在某些实施方案中,可以进一步修饰本文提供的抗体以包含本领域已知且易于获得的其他非蛋白部分。适于抗体衍生化的部分包括但不限于水溶性聚合物。水溶性聚合物的非限制性实例包括但不限于聚乙二醇(peg)、乙二醇/丙二醇的共聚物、羧甲基纤维素、葡聚糖、聚乙烯醇、聚乙烯吡咯烷酮、聚-1,3-二氧戊环、聚-1,3,6-三噁烷、乙烯/马来酸酐共聚物、聚氨基酸(均聚物或无规共聚物)、和葡聚糖或聚(n-乙烯基吡咯烷酮)聚乙二醇、聚丙二醇均聚物、聚环氧丙烷/环氧乙烷共聚物、聚氧乙烯化多元醇(例如甘油)、聚乙烯醇及其混合物。聚乙二醇丙醛由于其在水中的稳定性而在制造中具有优势。聚合物可以是任何分子量,并且可以是支链或非支链的。与抗体连接的聚合物的数量可以变化,并且如果连接了多于一种聚合物,这些聚合物可以是相同或不同的分子。通常,用于衍生化的聚合物的数量和/或类型可以基于以下考虑因素来确定,包括但不限于待改善抗体的特定性质或功能、抗体衍生物是否将用于明确条件下的治疗中,等等。在另一个实施方案中,提供了抗体与非蛋白部分的缀合物,所述非蛋白部分可以通过暴露于辐射而选择性加热。在一个实施方案中,非蛋白部分是碳纳米管(kam等人,proc.natl.acad.sci.usa102:11600-11605(2005))。辐射可以是任何波长,包括但不限于这样的波长,其不损害正常细胞但是将非蛋白部分加热到抗体-非蛋白部分附近的细胞被杀死的温度。重组方法和组合物可以使用重组方法和组合物产生抗体,例如,如美国专利号4,816,567中所述。在一个实施方案中,提供了编码本文所述抗体的分离的核酸。此类核酸可编码包含vl的氨基酸序列和/或包含抗体vh的氨基酸序列(例如抗体的轻链和/或重链)。在进一步的实施方案中,提供了包含此类核酸的一种或多种载体(例如,表达载体)。在进一步的实施方案中,提供了包含此类核酸的宿主细胞。在一个这样的实施方案中,宿主细胞包含(例如,已转化有):(1)包含核酸的载体,所述核酸编码包含抗体vl的氨基酸序列和包含抗体vh的氨基酸序列,或(2)第一载体和第二载体,所述第一载体包含编码包含抗体vl的氨基酸序列的核酸,所述第二载体包含编码包含抗体vh的氨基酸序列的核酸。在一个实施方案中,宿主细胞是真核细胞,例如,中国仓鼠卵巢(cho)细胞或淋巴样细胞(例如,y0、ns0、sp20细胞)。在一个实施方案中,提供了制备抗体的方法,其中所述方法包括在适于表达抗体的条件下培养包含编码所述抗体的核酸的宿主细胞,和任选地从宿主细胞(或宿主细胞培养基)中回收抗体。为了重组产生抗体,分离例如如上所述的编码抗体的核酸,并将其插入一个或多个载体中,用于在宿主细胞中进一步克隆和/或表达。可以使用常规步骤容易地分离和测序这种核酸(例如,通过使用能够与编码抗体重链和轻链的基因特异性结合的寡核苷酸探针)。用于克隆或表达编码抗体的载体的合适宿主细胞包括本文所述的原核或真核细胞。例如,可以在细菌中产生抗体,特别是当不需要糖基化和fc效应功能时。对于在细菌中表达抗体片段和多肽,参见例如美国专利号5,648,237、5,789,199和5,840,523。(还参见charlton,methodsinmolecularbiology,第248卷(b.k.c.lo,编辑,humanapress,totowa,nj,2003),第245-254页,描述了抗体片段在大肠杆菌中的表达)。表达后,可以从细菌细胞糊中以可溶性级分分离抗体,并且可以进一步纯化。除原核生物外,真核微生物如丝状真菌或酵母是编码抗体的载体的合适的克隆或表达宿主,包括其糖基化通路已被“人源化”的真菌和酵母菌株,导致产生具有部分或完全人糖基化模式的抗体。参见gerngross,nat.biotech.22:1409-1414(2004),和li等人,nat.biotech.24:210-215(2006)。用于表达糖基化抗体的合适宿主细胞也来自多细胞生物(无脊椎动物和脊椎动物)。无脊椎动物细胞的实例包括植物和昆虫细胞。已经鉴定了许多杆状病毒株,其可以与昆虫细胞结合使用,特别是用于转染草地夜蛾(spodopterafrugiperda)细胞。植物细胞培养物也可用作宿主。参见,例如,美国专利号5,959,177、6,040,498、6,420,548、7,125,978和6,417,429(描述了用于在转基因植物中产生抗体的plantibodiestm技术)。脊椎动物细胞也可用作宿主。例如,适于在悬浮液中生长的哺乳动物细胞系可能是有用的。有用的哺乳动物宿主细胞系的其他实例是由sv40转化的猴肾cv1细胞系(cos-7);人胚肾细胞系(293或293细胞,如,例如graham等人,j.genvirol.36:59(1977)所述);小仓鼠肾细胞(bhk);小鼠睾丸支持细胞(tm4细胞,如,例如mather,biol.reprod.23:243-251(1980)所述);猴肾细胞(cv1);非洲绿猴肾细胞(vero-76);人宫颈癌细胞(hela);犬肾细胞(mdck;大鼠(buffalorat)肝细胞(brl3a);人肺细胞(w138);人肝细胞(hepg2);小鼠乳腺肿瘤(mmt060562);tri细胞,如例如mather等人,annalsn.y.acad.sci.383:44-68(1982)所述;mrc5细胞;和fs4细胞。其他有用的哺乳动物宿主细胞系包括中国仓鼠卵巢(cho)细胞,包括dhfr-cho细胞(urlaub等人,proc.natl.acad.sci.usa77:4216(1980));和骨髓瘤细胞系如y0、ns0和sp2/0。对于适合抗体产生的某些哺乳动物宿主细胞系的综述,参见,例如,yazaki和wu,methodsinmolecularbiology,vol.248(b.k.c.lo,ed.,humanapress,totowa,nj),第255-268页(2003)。药物制剂通过将具有所需纯度的抗体或分子与一种或多种任选的药学上可接受的载体混合以冻干制剂或水溶液的形式来制备如本文所述的抗il-3抗体的药物制剂,本文也称为抗il-3抗体的药物组合物(remington'spharmaceuticalsciences第16版,osol,a.编辑(1980))。药学上可接受的载体通常在所用剂量和浓度下对接受者无毒,其包括但不限于:缓冲液,如磷酸盐、柠檬酸盐和其他有机酸;抗氧化剂,包括抗坏血酸和蛋氨酸;防腐剂(如十八烷基二甲基苄基氯化铵;六甲基氯化铵;苯扎氯铵;苄索氯铵;苯酚、丁醇或苄醇;对羟基苯甲酸烷基酯,如对羟基苯甲酸甲酯或对羟基苯甲酸丙酯;儿茶酚;间苯二酚;环己醇;3-戊醇;和间甲酚);低分子量(少于约10个残基)的多肽;蛋白,如血清白蛋白、明胶或免疫球蛋白;亲水性聚合物,如聚乙烯吡咯烷酮;氨基酸,如甘氨酸、谷氨酰胺、天冬酰胺、组氨酸、精氨酸或赖氨酸;单糖、二糖和其他碳水化合物,包括葡萄糖、甘露糖或糊精;螯合剂如edta;糖类,如蔗糖、甘露醇、海藻糖或山梨糖醇;成盐抗衡离子如钠;金属配合物(例如锌-蛋白配合物);和/或非离子表面活性剂,如聚乙二醇(peg)。本文的示例性药学上可接受的载体还包括间隙(insterstitial)药物分散剂,如可溶性中性活性透明质酸酶糖蛋白(shasegp),例如人可溶性ph-20透明质酸酶糖蛋白,如rhuph20(baxterinternational,inc.)。某些示例性shasegp和使用方法,包括rhuph20,描述于美国专利公开号2005/0260186和2006/0104968中。在一个方面,shasegp与一种或多种另外的糖胺聚糖酶如软骨素酶组合。示例性的冻干抗体制剂描述于美国专利号no.6,267,958中。水性抗体制剂包括美国专利号6,171,586和wo2006/044908中描述的那些,后者的制剂包括组氨酸-乙酸盐缓冲液。本文的制剂还可含有一种以上所治疗的具体适应症所必需的活性成分,优选那些具有互补活性但不相互产生不利影响的活性成分。例如,可以期望进一步用th2通路抑制剂提供控制物。这样的活性成分适合以对预期目的有效的量组合存在。可以将活性成分包埋在微胶囊中,例如通过凝聚技术或通过界面聚合制备的例如羟甲基纤维素或明胶-微胶囊和聚-(甲基丙烯酸甲酯)微胶囊,在胶体药物递送系统中(例如,脂质体、白蛋白微球、微乳液、纳米颗粒和纳米胶囊)或在粗乳液中。这些技术公开于remington'spharmaceuticalsciences第16版,osol,a.编辑(1980)。可以制备持续释放制剂。持续释放制剂的合适实例包括含有抗体的固体疏水聚合物的半透性基质,该基质是成型制品的形式,例如,薄膜或微胶囊。用于体内施用的制剂通常是无菌的。例如,通过无菌过滤膜过滤可以容易地实现无菌。治疗方法和组合物在某些实施方案中,提供了治疗特应性皮炎的方法,其包括向特应性皮炎患者施用治疗有效量的lebrikizumab,其中如通过特应性皮炎疾病严重性结果测量(addsom)所测量或评估的,所述治疗导致疾病严重性降低。在一些实施方案中,如rajka/langeland标准评分所确定的,特应性皮炎是中度至重度的。4.5至9的rajka/langeland标准评分通常被认为是中度至重度特应性皮炎。在一些实施方案中,该方法还包括施用一种或多种局部皮质类固醇。局部皮质类固醇可以在施用il-13拮抗剂之前(即,前)施用、与施用il-13拮抗剂同时施用、或在施用il-13拮抗剂之后施用。示例性局部皮质类固醇包括但不限于曲安奈德、氢化可的松、以及曲安奈德和氢化可的松的组合。曲安奈德通常在乳膏中以0.1%的浓度配制,氢化可的松通常在乳膏中以1%或2.5%的浓度配制。某些局部皮质类固醇被认为具有非常高的疗效,例如,二丙酸倍他米松、丙酸氯倍他索、二乙酸二氟拉松、醋酸氟轻松和丙酸乌倍他索。某些局部皮质类固醇被认为是高疗效的,例如,安西奈德、去羟米松、哈西奈德和曲安奈德。某些局部皮质类固醇被认为是中等疗效,例如,戊酸倍他米松、氯可托龙戊酸酯、氟轻松、氟氢缩松、醋酸氟轻松、丙酸氟替卡松、丁酸氢化可的松、戊酸氢化可的松、糠酸莫米松和泼尼卡酯。某些局部皮质类固醇被认为是低疗效,例如,二丙酸阿氯米松、地奈德和氢化可的松。tcs可以每天一次、每天两次、每天三次或根据需要应用于受损害的区域。在一些实施方案中,局部皮质类固醇对患者的控制不充分。除了新的特应性皮炎影响问卷(adiq)之外,各种addsom是本领域已知的,并且描述于本文中。示例性addsom包括但不限于“湿疹面积和严重性指数”(easi)、“特应性皮炎的严重性评分”(scorad)、“研究者全面评估”(iga)、“体征的研究者全面评估”(igsa)、rajka/langeland特应性皮炎严重性评分和患者报告的结果,所述患者报告的结果包括但不限于瘙痒直观模拟标度(作为scorad的一部分评估的疾病严重性的一个方面)、睡眠丧失直观模拟标度(作为scorad的一部分评估的疾病严重性的一个方面)、特应性皮炎症状日记(adsd)、特应性皮炎影响问卷(adiq)、皮肤病生活质量指数(dlqi)(finlay和khan,clinexperdermatol1994;19:210)和5-d瘙痒标度(elman等人,brjdermatol2010;162(3):587-593)。本文提供的治疗剂可以通过任何合适的方式施用,包括肠胃外、皮下、腹膜内、肺内和鼻内。肠胃外输注包括肌内、静脉内、动脉内、腹膜内或皮下施用。在某些实施方案中,通过注射进行给药,例如静脉内或皮下注射。在另一个实施方案中,使用注射器(例如,预填充或非预填充)或自动注射器施用治疗剂。在某些实施方案中,局部应用治疗剂。在某些实施方案中,使用例如自注射装置、自动注射装置或设计用于自我施用的其他装置施用本发明的抗il13抗体。在某些实施方案中,使用皮下施用装置施用本发明的抗il13抗体。各种自注射装置和皮下施用装置,包括自动注射装置,是本领域已知的并且是可商购的。示例性装置包括但不限于预填充注射器(如来自bectondickinson的bdhypakreadyfilltm和sterifillscftm;来自baxter的clearshottm共聚物预填充注射器;和可获得自westpharmaceuticalservices的daikyoseikocrystal预填充注射器);一次性笔注射装置,如来自bectondickinson的bd笔;超锐微针装置(如来自bectondickinson的inject-easetm和微输注装置;和可获自valeritas的h-patchtm)以及无针注射装置(如可获自的bioject和和可获自medtronic的和贴剂装置(patchdevice)。在某些实施方案中,使用包含各种组件的自注射装置施用本发明的抗il-13抗体,例如,如美国专利公开号2014/0114247、2014/0148763和2013/0131590;美国专利号7,597,685、7,896,850和8,617,109中所述,所有这些都通过引用并入本文。预期使用这种自注射装置或皮下施用装置的抗il13抗体与至少第二治疗化合物共同制剂或共同施用。为了预防或治疗疾病,本发明抗体的适当剂量(当单独使用或与一种或多种其他治疗剂组合使用时)将取决于待治疗疾病的类型、抗体的类型、疾病的严重性和病程、抗体是用于预防还是治疗目的施用、先前的治疗、患者的临床病史和对抗体的反应、以及主治医师的判断。抗体适合一次或在一系列治疗中施用至患者。在某些实施方案中,本发明的抗体以125mg或250mg或约125mg或约250mg的均一剂量(不是基于重量的剂量)或110mg至140mg的均一剂量或120mg至130mg的均一剂量或225mg至275mg的均一剂量或240mg至260mg的均一剂量施用。在某些实施方案中,通过每四周一次或每八周一次皮下注射施用剂量一段时间。在某些实施方案中,该一段时间是12周、或20周、或24周、或9个月、或1年、2年、5年、10年、15年、20年或患者终生。通过常规技术和测定法可以容易地监测该治疗的进展。在某些实施方案中,患者患有中度至重度特应性皮炎,并且患者在用本发明的抗体治疗时根据需要施用tcs。在某些实施方案中,每天一次、或每天两次,或每天三次或更多次施用tcs。在某些实施方案中,患者能够在施用本发明抗体时减少tcs随时间的使用。这种tcs使用的减少被称为“逐渐减少(taping)”或“tcs节约(sparing)”。可能发生减少使用的一段时间是一周、两周、三周或四周、或者两个、三个、四个、五个或六个月。根据某些实施方案,本发明的il-13拮抗剂包括与一种或多种另外的治疗剂组合施用。术语“与......组合”是指在包含il-13拮抗剂的药物组合物之前、之后或与其同时施用另外的治疗剂。术语“与......组合”还包括依次或伴随施用il-13拮抗剂和第二治疗剂。例如,当在包含il-13拮抗剂的药物组合物“之前”施用时,另外的治疗剂可以在施用包含il-13拮抗剂的药物组合物约72小时、约60小时、约48小时、约36小时、约24小时、约12小时、约10小时、约8小时、约6小时、约4小时、约2小时、约1小时、约30分钟、约15分钟或约10分钟之前施用。当在包含il-13拮抗剂的药物组合物“之后”施用时,另外的治疗剂可以在施用包含il-13拮抗剂的药物组合物约10分钟、约15分钟、约30分钟、约1小时、约2小时、约4小时、约6小时、约8小时、约10小时、约12小时、约24小时、约36小时、约48小时、约60小时或约72小时之后施用。与包含il-13拮抗剂的药物组合物“同时”或一起施用意味着另外的治疗剂在施用包含il-13拮抗剂的药物组合物(之前、之后或同时)少于5分钟内以分开的剂型施用至受试者,或作为单一组合剂量制剂(包含另外的治疗剂和il-13拮抗剂)施用至受试者。另外的治疗剂可以是例如另一种il-13拮抗剂、il-4r拮抗剂、il-1拮抗剂、il-6拮抗剂、il-6r拮抗剂、tnf拮抗剂、il-8拮抗剂、il-9拮抗剂、il-17拮抗剂、il-5拮抗剂、ige拮抗剂、cd48拮抗剂、il-31拮抗剂、胸腺基质淋巴细胞生成素(tslp)拮抗剂、干扰素-γ(ifnγ)、抗生素、局部皮质类固醇、他克莫司、吡美莫司、霉酚酸酯、环孢菌素、硫唑嘌呤、甲氨蝶呤、色甘酸钠、蛋白酶抑制剂或其组合。在某些实施方案中,将包含抗il13拮抗剂的药物组合物与口服免疫抑制剂、局部钙依赖磷酸酶抑制剂(例如吡美莫司或他克莫司)或口服皮质类固醇(例如,泼尼松龙)联合施用至受试者。在某些实施方案中,将包含抗il13拮抗剂的药物组合物与非药物疗法(如紫外(uv)光疗法)联合施用至受试者。在某些实施方案中,本发明的抗体作为均一(即,非体重依赖)的负荷剂量施用一次或多次,接着作为均一的维持剂量施用一次或多次。在某些实施方案中,负荷剂量为250mg或500mg,维持剂量为125mg。在某些实施方案中,间隔15天或间隔29天施用两个250mg的负荷剂量。在某些实施方案中,维持剂量在最后负荷剂量两周后施用、或在最后负荷剂量四周后施用,然后每四周施用一次。在某些实施方案中,负荷剂量为500mg,维持剂量为250mg。在某些实施方案中,250mg的维持剂量在负荷剂量4周后施用,然后在其后的每4周或每8周施用。制品在本发明的另一个方面,提供了含有可用于治疗、预防和/或诊断上述疾患的物质的制品。制品包括容器和在容器上或与容器相关的标签或包装说明书。合适的容器包括例如瓶子、小瓶、注射器、iv溶液袋、自动注射器等。容器可以由多种材料(如玻璃或塑料)制成。容器容纳组合物(该组合物自身或与另一种有效治疗、预防和/或诊断病症的组合物组合)并且可具有无菌进入口(例如,容器可以是静脉内溶液袋或具有通过皮下注射针头可刺穿的塞子的小瓶)。组合物中的至少一种活性剂是本发明的抗体。标签或包装说明书表明该组合物用于治疗所选择的病症。此外,制品可包括(a)其中含有组合物的第一容器,其中所述组合物包含本发明的抗体;和(b)其中含有组合物的第二容器,其中所述组合物包含另外的治疗剂。本发明该实施方案中的制品还可包含包装说明书,其表明该组合物可用于治疗特定病症。或者或另外地,制品还可包含第二(或第三)容器,其包含药学上可接受的缓冲液,如抑菌性注射用水(bwfi)、磷酸盐缓冲盐水、林格氏溶液和右旋糖溶液。其还可以包括从商业和用户角度所需的其他物质,包括其他缓冲液、稀释液、过滤器、针头、注射器和自动注射器。实施例实施例1-抗il-13抗体先前已经描述了抗il-13抗体,包括单克隆人源化抗il-13抗体(参见,例如,wo2005062967)。其中描述的一种单克隆人源化抗il-13抗体是lebrikizumab,一种igg4抗体,也参见序列表。lebrikizumab用于下述研究中,以125mg/ml配制在药学上可接受的组合物中,并在预填充注射器中提供。实施例2-临床研究i临床研究i是ii期、随机、双盲、安慰剂对照研究,以评估lebrikizumab在患有持续中度至重度特应性皮炎的患者(18-75岁)中的安全性和疗效,所述特应性皮炎由局部皮质类固醇(tcs)控制不充分。在该研究中,将lebrikizumab与tcs组合进行测试。研究模式如图1提供。筛选。有资格参加研究的患者必须患有中度至重度ad至少1年,并且必须符合所有资格标准。如下评估患者:在筛选时评估进入磨合(run-in)期的资格并且在磨合期结束时再次评估进入治疗期和随机分组研究药物的资格。纳入标准包括以下:年龄18至75岁;通过hanifin/rajka标准诊断为ad并且在筛选时存在至少1年;根据rajka/langeland标准在筛选时分级为中度至重度ad;对至少每天tcs和常规润肤剂治疗ad的治疗方案反应不足的病史≥1个月(在筛选就诊前3个月内);在筛选和磨合期结束时(就诊3)easi评分≥14,在筛选和磨合期结束时(就诊3)iga评分≥3(5-分制);在筛选时ad涉及≥10%体表面积(bsa);和在筛选时瘙痒vas评分≥3(作为scorad的一部分测量)。排除标准包括以下:过去和/或当前使用任何抗il-13或抗il-4/il-13治疗,包括lebrikizumab;在筛选前4周内或在研究试剂的5个半衰期内使用研究试剂,以较长者为准;严重过敏反应或对生物制剂的过敏反应或已知对lebrikizumab注射的任何成分过敏的病史;对tcs或研究中使用的tcs产品所含任何其他成分的超敏反应;在磨合期前7天内使用任何补充、替代或顺势疗法药物,包括但不限于植物疗法、传统或非传统草药、必需脂肪酸或针灸,或在研究期间需要此类药物;体重<40kg或身体质量指数>38kg/m2;其他皮肤病症的证据;包括但不限于t细胞淋巴瘤或过敏性接触性皮炎;筛选时(第-15天)活动性皮肤感染的证据或持续治疗(包括局部抗生素);某些感染;近期(<1年)寄生虫感染史,特别是线虫(例如蛔虫属(ascaris)、钩虫属(ancylostoma))、扁形动物(例如血吸虫属(schistosoma))或李斯特菌感染史;活动性结核病需要在第1次就诊前12个月内接受治疗;急性或慢性肝炎或已知肝硬化的证据;已知的免疫缺陷,包括hiv感染;在筛选时使用tci(局部钙依赖磷酸酶抑制剂);在筛选就诊前4周内使用晒黑展台/店;筛选的3个月内过敏原免疫治疗;在基线就诊(第1天)前4周内接受减毒活疫苗;研究期间计划的手术;筛选ecg或实验室检查(血液学、血清化学和尿液分析)中显著的临床异常;筛选期间ast、alt或总胆红素升高≥2.0×正常上限(uln);当前已知的恶性肿瘤或当前评估潜在的恶性肿瘤,包括皮肤基底或鳞状细胞癌或原位癌;筛选前5年内恶性肿瘤病史,除外适当治疗的宫颈原位癌、非黑色素皮肤癌;i期子宫癌;以及尽管治疗但不受控制的其他临床上显著的医学疾病。磨合期。在筛选就诊后,合格的患者进入2周的磨合期(第-14天至第-1天),在此期间开始指定方案的局部治疗方案。在磨合期的第一天(第-14天),患者接受tcs,用于整个磨合期。患者需要每天应用以下局部治疗方案:至少每天一次对所有干燥皮肤表面施用润肤剂;每天两次仅对所有活动性皮肤病灶施用由中效tcs(曲安奈德0.1%)组成的tcs乳膏。对于影响面部或擦烂区域的病灶,可以由研究者决定使用低效tcs(氢化可的松2.5%乳膏)代替曲安奈德0.1%。在磨合期结束时,进行第二次随机分组为研究药物(lebrikizumab或安慰剂)资格的评估,如通过easi评分≥14和iga评分≥3所评估的,患者必须继续显示中度至重度ad。治疗期。在磨合期结束时,1)证明符合指定方案的tcs方案、和2)继续满足上述资格标准的患者被随机分组。计划将大约200名患者(1:1:1:1)随机指定到以下四个治疗组之一(见图1):(1)组1:皮下(sc)施用lebrikizumab250mg单剂量(第1天),接着2次安慰剂剂量(第4周和第8周),总共3个剂量+tcs乳膏;(2)组2:lebrikizumab125mgsc单剂量(第1天),接着2个安慰剂剂量(第4周和第8周),总共3个剂量+tcs乳膏;(3)组3:每4周lebrikizumab125mgsc(q4w),总共3个剂量+tcs乳膏;(4)组4:安慰剂scq4w,总共3个剂量+tcs乳膏。包括三个活性剂量以表征暴露-反应关系和给药频率要求。将患者随机指定到这四个接受研究药物(lebrikizumab或安慰剂sc)与tcs组合的研究治疗组。在整个12周的治疗期间,这些患者继续每天至少一次应用润肤剂和曲安奈德0.1%乳膏,如磨合期(每天两次向身体所有活动性皮肤病灶施用)所述。对于影响面部或擦烂区域的病灶,可以由研究者决定使用低效tcs(氢化可的松2.5%乳膏)代替曲安奈德0.1%。患者在整个治疗期间使用手持式ediary记录其使用局部乳膏和对pro相关问题的回答。在每次研究就诊时,患者需要将他们的tcs容器带到研究地点进行称重。安全性随访期。在安慰剂对照治疗期间(第1周至第12周)完成研究治疗的所有患者进行安全性随访另外8周(第13周至第20周)。在此安全性随访期间,患者不再需要应用磨合期和治疗期所指定的局部治疗方案。相反,如患者和研究人员所确定的,可以应用曲安奈德0.1%乳膏和/或氢化可的松2.5%乳膏。在每次研究就诊时,患者需要将他们的tcs容器带到研究地点进行称重。鼓励患者每天至少一次继续将润肤剂应用在干燥的皮肤上。在研究期间的任何时候如果在研究者看来出现了临床上指证,则允许升级ad治疗。在该研究期间的任何时候都不允许局部钙依赖磷酸酶抑制剂(tci)。如果患者同意在研究期间停止使用tci,并且如果在研究者看来停止使用tci并且使用指定方案的tcs乳膏代替是安全的,则允许在筛选时一直使用tci的患者参加研究。lebrikizumab剂量和时间表的基本原理。lebrikizumab对于ad的剂量是基于哮喘项目的临床经验和两个患者群体中il-13生物学的相似性以及lebrikizumab的预期pk特征。ad和哮喘患者均具有升高的与il-13生物学相关的生物标记物水平和th-2介导的超敏反应。特别是,il-13在哮喘患者的肺中升高并且在哮喘中具有致病作用,同时报道在ad皮肤中il-13的表达一致地增加。另外,鉴于lebrikizumab的主要清除机制是通过非特异性胞吞作用和分解代谢,而靶向介导的清除贡献最小,预期lebrikizumab的pk特征在哮喘和ad患者之间是相似的。尽管文献中存在的关于单克隆抗体向各种组织的分配的信息有限,但在动物模型中获得的数据表明在肺和皮肤组织中单克隆抗体的血浆-组织分配系数相似(shahdk等人,mabs2013;5(2):297-305)。因此,在哮喘中产生临床益处的lebrikizumab的剂量水平也可以在ad患者中产生生物学活性。如上所述,在该ii期研究中测试了三种不同的剂量方案:(1)包括125mgq4w剂量(组3)作为潜在的有效剂量。125mgq4w是在哮喘的lebrikizumab关键性iii期成年人研究(lavoltai和ii)中进行研究的最高剂量方案(即最高总暴露),在研究开始时持续进行。鉴于哮喘和ad之间il-13的作用和药代动力学的预期相似性,假设该剂量可能对ad患者有效;(2)建议将单一250mg剂量(组1)作为替代的潜在有效剂量。预期该剂量在前12周内产生的总暴露低于125mgq4w。在第12周的主要端点,测试单一250mg剂量以探索每季度给药的可能性;和(3)建议将单一125mg剂量(组2)作为潜在的部分有效剂量。鉴于预期该剂量的总暴露比125mgq4w和250mg单剂量方案低2-3倍,来自所有三个剂量组的数据可用于表征在ad患者中lebrikizumab的暴露-反应关系。然而,如上所述,lavoltai和ii研究的结果是不一致的。hanania等人,lancetrespirmed2016,可在2016年9月5日在线发表的dx(dot)doi(dot)org(slash)s2213-2600(16)30265-x获得。两项研究未能显示明确的剂量-反应,尽管药代动力学和药效学结果与先前描述的ii期研究结果一致,这表明药物暴露相似且il-13通路被抑制。hanania等人,2016,同上。因此,不确定lebrikizumab是否会在本文所述的给药方案中为特应性皮炎患者提供临床上有意义的益处。在第12周测量ii期研究的主要端点。预期第12周的平均血清lebrikizumab谷浓度超过接受125mgq4w的患者的稳态值的90%。此外,对于125mgq4w剂量,预期第12周的谷浓度将在lebrikizumab在ii期哮喘临床试验中证明生物学活性的范围内。对于250mg单剂量,预期经过12周的平均暴露高于37.5mgq4w方案的平均暴露,这也证明了ii期哮喘试验的生物学活性;hanania等人,thorax2015;70:748–756。对照组的基本原理。治疗第4组的患者接受安慰剂sc+tcs乳膏,因此用作活性比较组。该治疗组用作lebrikizumabsc的对照,以充分评估lebrikizumab作为tcs辅助治疗的疗效和安全性作用。包括活性tcs比较组是因为使用tcs是临床实践中的标准护理并且在启动升级治疗(例如,全身性试剂)的同时继续使用(欧洲皮肤病学和性病学研究院[eadv]2009指南;darsow等人,jeuracaddermatolvenereol2010;24:317-28)。此外,在患有持续中度至重度疾病的患者中停止tcs可能导致ad症状和疾病负担的显著恶化。疗效结果测量。主要疗效结果测量是在第12周实现easi-50(easi评分从基线降低50%)的患者百分比。该研究的次要疗效结果测量如下:(i)在第12周easi评分从基线的百分比和绝对变化;(ii)在第12周实现easi评分从基线降低75%(easi-75)的患者百分比;(iii)在第12周实现iga评分为0或1的患者百分比;(iv)在第12周iga从基线降低≥2点的患者百分比;(v)在第12周iga从基线的绝对变化;(vi)在第12周实现igsa评分为0或1的患者百分比;(vii)在第12周igsa从基线降低≥2点的患者百分比;(viii)在第12周igsa从基线的绝对变化;(ix)在第12周scorad从基线的百分比和绝对变化;(x)在第12周scorad-50/75从基线降低50%或75%的患者百分比;(xi)在第12周实现easi-50并在第16周和第20周维持easi-50的患者百分比;(xii)在第12周实现iga评分为0或1且在第16周和第20周维持iga评分为0或1的患者百分比;(xiii)在第12周实现igsa评分为0或1且在第16周和第20周维持igsa评分为0或1的患者百分比;(xiv)在第12周实现scorad-50且在第16周和第20周维持scorad50的患者百分比;(xv)在第12周受影响的总%体表面积(bsa)从基线的百分比变化;(xvi)在第12周由瘙痒vas(作为scorad的一部分评估)测量的瘙痒从基线的绝对和百分比变化;(xvii)在第12周由5-d瘙痒标度测量的瘙痒从基线的绝对和百分比变化;(xviii)从基线到第12周tcs的总使用量(克);(xvix)从第12周到研究结束或提前终止tcs的总使用量(克);(xx)从基线到第12周的疾病加剧的次数;(xxi)由adsd评估的ad症状从基线到第12周的变化;(xxii)由adiq评估的ad特异性健康相关qol从基线到第12周的变化;和(xxiii)由dlqi测量的健康相关qol从基线到第12周的变化。安全性结果测量。本研究的安全性结果测量如下:(i)治疗中出现的不良事件的频率和严重性;(ii)在基线和研究期间人抗治疗性抗体(ata)的发生率;(iii)整个研究中皮肤和其他器官系统感染的频率和严重性;皮肤感染的临床定义如下:感染的诊断基于研究者的临床评估,包括但不限于皮疹部位存在蜂蜜色结痂、浆液性分泌物、脓疱或疼痛,其可能与全身特征相关,包括发烧或ad疾病的加剧(如上所述);和(iv)研究人员评估中止研究药物后疾病反弹的发生率;疾病反弹的临床定义是:停止治疗后疾病严重性显著恶化,严重性水平大于开始治疗前的严重性(hijnen等人,jeuracaddermatolvenereol2007;21(1)85-9)。特应性皮炎影响问卷(adiq)。我们根据美国食品和药品管理局患者报告结果(pro)指南(可在www(dot)fda(dot)gov(slash)downloads(slash)drugs(slash)guidances(slash)ucm193282(dot)pdf.在线获得)开发了一种ad特异性健康相关生活质量工具,用于12岁及以上的患者。具体而言,进行文献综述以评估adpro情况。进行与中度或重度ad的18-75岁成年人(n=15)和青少年12-17(n=15)的概念引发(ce)访谈,以引出对患者重要的ad体征和症状。采用扎根理论方法对访谈记录进行定性分析。举办条款生成会议(igm)以回顾数据,并基于来自ce访谈的患者反馈开发初步的adiq。在与34名成年人和青少年患者的认知访谈中测试adiq草案,这些患者确认adiq中包括的概念的清晰度和相关性,如下表2所示。心理测量验证不包括在内。表2.特应性皮炎影响问卷(adiq)结果在该研究中,治疗了209名患者。与安慰剂相比,在第12周用lebrikizumab(每四周125mg)加tcs治疗的患者中的更大比例实现了easi-50(图2a和图3a)、easi-75(图3b)、easi-90(图3c)和scorad-50(图2b)。此外,与安慰剂(62.3%)相比,在第12周所有lebrikizumab组中实现easi-50(主要疗效结果)的患者百分比更高,但仅在多剂量组中达到统计学显著性(对于125mgq4w组为82.4%,[p=0.026]),如图11a所示。总体而言,与安慰剂(34.0%)相比,在第12周所有lebrikizumab剂量组中实现easi-75反应(次要疗效结果)的患者比例更高,但仅在125mgq4w组中显著更高(54.9%[p=0.036]),如图11b所示。与安慰剂相比,在第12周所有lebrikizumab组实现iga0/1的患者百分比更高,但没有达到统计学显著性。然而,与其他结果一样,数据表明iga的剂量-反应关系(图11c),并且125mgq4w组在治疗期的最后几周显示持续改善(图2c)。关于scorad-50,与安慰剂(26.4%;图11d)相比,在第12周lebrikizumab125mgq4w组(51.0%[p=0.018])和250mgsd组(47.2%[p=0.030])实现该端点的患者更多。所有lebrikizumab组均显示受影响的bsa的改善(表3)。在第12周,在lebrikizumab125mgq4w组中观察到最大降低了受影响的bsa(降低57.7%),尽管在安慰剂组也有实质性改善(47.4%)。对于bsa,安慰剂校正的疗效无统计学显著性(p=0.38)。lebrikizumab治疗导致瘙痒vas的改善(表3和表7)。在2周的tcs磨合期期间,瘙痒vas的改善在数量上大于严重性测量结果的改善。在所有治疗组中使用tcs的坚持服药率很高,其中从基线到第12周平均tcs使用的天数为86.8%(125mgsd)、86.7%(250mgsd)、91.9%(125mgq4w)和88.2%(安慰剂)。在第12周与安慰剂相比,用每4周125mglebrikizumab加tcs治疗的患者中睡眠丧失vas也有更大的改善,以及iga0/1、瘙痒vas和ad特异性健康相关生活质量(qol)测量有改善的趋势,特别是特应性皮炎影响问卷(adiq)(表3、表7、图12a、图12b和图12c)。dlqi评分也存在剂量依赖性改善,与安慰剂相比,所有lebrikizumab组在数值上都从基线降低更多(表3、表7和图12d)。还如下评估了药代动力学参数。针对所有给药方案,在第1天(给药前)和第1、4周(给药前)、第6、8周(给药前)、第12、16和20周获得用于分析lebrikizumab药代动力学的血清样品。使用接受lebrikizumab治疗方案之一的患者的描述性统计数据,根据治疗和就诊总结血清lebrikizumab浓度。报告的pk参数包括第1周的cmax、第4、8和12周的cmin以及清除半衰期。每种lebrikizumab给药方案的平均pk参数和各标准偏差显示在表8中。结果显示在ad患者中lebrikizumab的药代动力学与先前已发表文献中报道的针对成年人哮喘的lebrikizumab的药代动力学一致(参见例如,hanania等人,thorax70(8):748-756(2015)),表明线性和剂量比例特征,其中半衰期为19-22天。治疗组之间的不良事件率通常相似(所有lebrikizumab组为66.7%,安慰剂组为66.0%),并且大多数的严重性为轻度或中度(表9)。具体而言,lebrikizumab组(所有剂量组合)中3名(2%)患者和安慰剂组中1名(2%)患者经历了导致退出研究的ae(125mgsd组中的皮肤感染,125mgq4w组中的焦虑和肌病,以及安慰剂组中的特应性皮炎)。lebrikizumab组中的一名(1%)患者经历了导致剂量中断的ae(胃肠道病毒感染)。没有死亡、过敏反应事件、恶性肿瘤或方案限定的寄生虫或目标细胞内感染。注射部位反应很少发生(所有lebrikizumab组为1.3%,安慰剂组为1.9%);所有事件均为非严重,持续时间的中值为1-3天,并未导致治疗中断。疱疹感染很少发生且仅在lebrikizumab治疗的患者中发生(n=6[3.8%];单纯疱疹n=4[2.6%]和带状疱疹n=2[1.3%]);所有事件都不严重,没有导致停止治疗的事件。此外,没有疱疹性湿疹的事件。在lebrikizumab治疗组(所有剂量组合)中有14名(9%)患者皮肤感染,在安慰剂组中有9名(17%)患者。嗜酸性粒细胞相关的ae也很少报道,仅发生在5名lebrikizumab治疗的患者中(3.2%;3名“嗜酸性粒细胞增多”和2名“嗜酸性粒细胞计数增加”);所有事件均不严重、强度为轻度至中度,并未导致治疗中断。这5名患者中最大嗜酸性粒细胞计数范围为0.91至3.2×109/l,其中3名为2级嗜酸性粒细胞增多(1501-5000细胞/mm3)。在这5名患者中,没有相关的临床症状与报道的事件有关。此外,观察到的增加与先前的lebrikizumab研究中所见的一致(hanania等人,thorax2015:70(8):748-756;hanania等人,lancetrespirmed2016;4(10):781-796)。我们评估了先前在该疾病区域的生物学试验中报告的失衡引起的结膜炎(thaci等人,lancet2016:387(10013):40-52)。集合的lebrikizumab组中共有15名(9.6%)患者、安慰剂组中有4名患者(7.5%)患有结膜炎ae;所有事件都不严重,没有导致停止治疗的事件。表3.第12周关键疗效结果adiq=特应性皮炎影响问卷;bsa=体表面积;ci=置信区间;dlqi=皮肤病生活质量指数;easi=湿疹面积严重性指数;iga=研究者全面评估;scorad=评分特应性皮炎;se=标准误差;vas=直观模拟标度。表7.从筛选到基线(磨合)easi、scorad、iga、瘙痒vas和受影响的bsa百分比的变化百分比。bsa=体表面积;easi=湿疹面积严重性指数;iga=研究者全面评估;q4w=每4周一次;sd=单剂量scorad=评分的特应性皮炎;sd=单剂量;se=标准误差;vas=直观模拟标度。表8.单剂量或多剂量皮下施用后的平均lebrikizumab药代动力学参数。cmax,wk1=第1周的最大lebrikizumab浓度,cmin,wk4=第4周观察到的最小浓度,cmin,wk8=第8周观察到的最小浓度,cmin,wk12=第12周观察到的最小浓度,t1/2=清除半衰期表9.第0周至第20周的关键安全性信息概述。ae=不良事件;q4w=每4周一次;sae=严重不良事件;sd=单剂量。*对盲法数据进行回顾,根据sampson的标准判定为过敏反应的病例。研究人员评估了与研究药物相关的感染。我们还观察到图2a中所示数据的明显剂量-反应关系。与125mg单剂量相比,250mg单剂量在早期时间点在数值上更好。此外,图2a中显示的数据表明,每四周一次125mg剂量似乎在第12周时没有达到疗效平台期。这些数据表明较高的给药(例如,250mgq4w)或负荷剂量的潜在益处。此外,与安慰剂相比,在第20周在改变的意向治疗群体中,更大比例的用每4周125mglebrikizumab加tcs治疗的患者实现easi-50(图3a)、easi-75(图3b)和easi-90(图3c)。也参见表9。还观察到与安慰剂相比,lebrikizumab治疗的患者经过12周的easi从基线降低的百分比更高(图4)。值得注意的是,每4周接受一次125mglebrikizumab、持续12周的患者表现为70.5%的easi变化(减少),而安慰剂组表现为53.1%的变化(减少)。lebrikizumab治疗组和安慰剂组之间17.4%的差异具有统计学显著性,p=0.025。表9.第20周关键疗效结果的总结。bsa=体表面积;easi=湿疹面积严重性指数;iga=研究者全面评估;scorad=评分特应性皮炎;sd=单剂量;se=标准误差;vas=直观模拟标度。我们还通过scorad瘙痒直观模拟标度(vas)、睡眠丧失vas和adiq测量了患者的症状和与健康相关的生活质量。在瘙痒症vas、睡眠丧失vas和adiq中证实了从基线的改善(见表3)。事后(posthoc)分析还检查了从筛选的变化。每四周施用一次125mglebrikizumab治疗的患者表现瘙痒vas在第12周从筛选降低55.2%(p=0.03,与安慰剂相比),而安慰剂导致从筛选降低39.3%。从筛选到第12周,睡眠丧失vas也有所改善,用lebrikizumab治疗降低61.5%(p=0.04,与安慰剂相比),而安慰剂降低39.3%。使用lebrikizumab治疗的adiq从筛选降低64.7%(p=0.02,与安慰剂相比),而安慰剂降低41.8%。仅作为单剂量(125mg和250mg)给予lebrikizumab的组相对于安慰剂在这些端点的差异在大多数情况下不具有统计学意义。总之,这些结果表明,靶向中度至重度ad中的il-13在许多严重性结果中提供临床上有意义的安慰剂校正的改善,通常以剂量依赖性方式。每月给药显著改善了实现easi-50、easi-75和scorad-50的患者比例,具有改善实现iga0/1的患者和改善瘙痒vas的倾向。这些改善是在与安慰剂组中的实质性反应相关的集中的tcs应用的基础上看到的。治疗组之间的不良事件发生率通常相似,并且大多数在严重性上为轻度或中度。此外,结果显示lebrikizumab在中度至重度ad患者中在严格应用tcs的基础上提供治疗益处,所述患者对标准护理tcs治疗的反应不充分且具有指示患者群向程度更严重的结局发展的基线特征。该研究符合其主要端点-第12周时easi-50患者的百分比-在lebrikizumab125mgq4w组中,统计学上显著数量的患者达到基线easi评分中该水平降低。此外,治疗期最后几周的向上倾斜的反应曲线表明,对于lebrikizumab125mgq4w组,到第12周可能尚未达到反应平台,并且较长的治疗持续时间可导致改善的疗效。似乎也存在剂量-反应关系,lebrikizumab125mgq4w剂量在大多数端点,特别是在easi中显示数值上最高的反应率,而lebrikizumab125mgsd在第12周显示最小的益处。值得注意的是,lebrikizumab250mgsd组在几个结果的较早时间点显示出数值上更高的反应,表明更高的给药或负荷剂量的潜在益处。在所有主要疗效端点中,在经过12周治疗期间观察到持续改善。与安慰剂组的患者相比,在第12周lebrikizumab125mgq4w组中的患者以未调整的5%显著性水平在许多次要疗效端点中具有统计学显著的差异。这些包括具有easi-75的患者的百分比、具有scorad-50的患者的百分比、easi评分从基线的百分比变化、以及scorad从基线的百分比变化。iga和瘙痒vas的改善在数值上高于安慰剂。瘙痒被认为是中度至重度ad患者qol降低的主要原因,并且使用lebrikizumab125mgq4w给药,qol(如通过dlqi和adiq测量的)和睡眠在数值上大幅改善,可能是由于瘙痒的改善。一般来说,在几个结果的8周随访期间也保持了lebrikizumab的作用,包括easi、iga(0/1)和scorad反应维持到第20周。考虑到在多个端点观察到的剂量-反应关系,以及随着剂量和持续时间增加而具有改善疗效的倾向,假设进一步增加剂量(以负荷剂量或更高剂量如250mgq4w的形式)和/或治疗持续时间可导致疗效改善是合理的。此外,在哮喘患者中的fev1(hanania等人,thorax2015;70(8):748-756)与ad患者中的easi/iga端点之间观察到的lebrikizumab剂量-反应关系差异表明ad实际上可能需要更高剂量的lebrikizumab以达到反应平台。这与相对于哮喘患者,特应性皮炎患者中较高的il-13负荷一致。该研究方案要求患者在2周的磨合期期间顺应每日两次tcs,才有资格进行随机分组。此外,如果患者在此磨合期后表现出足够的ad严重性,则才符合随机分组条件。尽管本研究中包括的患者未得到tcs的充分控制,但包含该tcs磨合期导致该疾病的实质性改善。这种改善通过抑制的基线ad严重性评分、特别是瘙痒测量值得到证实,可能导致超出基线的额外改善证实更困难。在治疗期间,该方案强制要求继续应用每日两次的tcs,并且通过电子日记向患者提供每日提醒;在所有治疗组中,本研究中的患者以88%的高坚持服药率应用tcs。虽然通常推荐针对急性病灶每日使用tcs,而不是连续和无限期使用,但这一验证概念的2期研究试图了解lebrikizumab加上极其严格的tcs应用的潜在功效(eichenfield等人,jamacaddermatol.2014;71(1):116-132),并不是评估tcs节约。考虑以较低频率应用tcs,因为该tcs方案可能不反映长期治疗或临床实践。然而,该方案与tcs标签一致,并且以低于产品标签规定的频率使用存在标签外使用的监管问题。同时,担心针对背景tcs的不被允许的替代设计,如lebrikizumab单一疗法,会导致大量患者中途退出和/或对照组内的估算的患者失败。实际上,单一疗法设置中生物治疗的研究导致对照组内中途退出/估算的患者失败率大约为50%(simpson等人,nengljmed.2016;375(24):2335-2348),相反,本研究中安慰剂组的中途退出率为13%。在12周治疗期间严格的tcs应用可能解释了在安慰剂组中观察到的实质性反应。已表明长期和频繁的tcs使用导致ad逐渐改善,但大多数指南建议限制每日使用以避免ae。参见,例如,brunner等人,jallergyclinimmunol.2016;138(1):169-178;schneider等人,jallergyclinimmunol.2013;131(2):295-9.e1-27;和hanifin等人,jamacaddermatol.2004;50(3):391-404。此外,安慰剂组中的这种实质性反应可能是安慰剂和lebrikizumab治疗组之间未观察到较大治疗差异的原因。尽管长期和频繁使用tcs的疗效相对较高,但通过加入lebrikizumab治疗仍有显著改善,特别是在ad体征(easi)和全面评分(scorad)中。dupilumab,一种抗il-4rα单克隆抗体,在患有中度至重度ad的患者中显示出疗效,并且最近已被批准用于在ad中。il-4rα被认为是il-4和il-13信号传导的重要受体亚基。在ad患者中对dupilumab的研究提供了il-13阻断治疗ad的可能性的见解,但需要注意的是尚未确定在ad中il-4比il-13相对重要。il-13和il-4二者共享重叠的生物学和效应功能;例如,两者均在t辅助细胞2细胞发育、ige产生、嗜酸性粒细胞募集、通过下调抗微生物肽的上皮屏障完整性破坏、纤维化和降低的丝聚蛋白表达中起作用。参见,例如,paternoster等人,natgenet.2011;44(2):187-192;kagami等人,clinexpimmunol.2005;141(3):459-466。由于生物学中如此高的重叠,单独阻断il-13可能潜在地在ad中提供相当的改善以阻断il-13和il-4的组合,具有更具体的靶向作用。另外,靶向可溶性细胞因子,如il-13,可以提供线性pk曲线的优势,从而改善给药频率。这部分解释了给药lebrikizumabq4w剂量的能力,并且可以允许在维持期间较低频率地给药。相反,受体靶向与靶标介导的药物清除相关,这可能导致药物中止或中断后浓度迅速下降,如以q2w给药的dupilumab的情况(kovalenko等人,cptpharmacometricssystpharmacol.(2016)nov;5(11):617-624)。该研究的结果表明il-13介导的信号传导通路在ad的发病机理中起重要作用,并且该细胞因子的阻断可以导致显著的临床益处。使用lebrikizumab治疗,即使单剂量和加强的tcs使用,中度至重度ad的患者表现出改善。未来对于不同(或不存在)背景方案的更长持续时间、更高频率或更高给药剂量以及在更大人群中的研究将有助于阐明单独靶向il-13在ad中的作用。实施例3-替代给药方案为了评估是否可以开发替代的lebrikizumab给药方案并且是否可以预期显示疗效,如下所述我们开发了新型、基于机理的纵向pk-pd模型。该模型用于模拟各种替代给药方案的预期疗效(例如easi-75评分),例如,所述替代给药方案包括但不限于各种负荷剂量,随后是各种维持剂量,例如,以定期时间间隔施用均一剂量的lebrikizumab,或各种较低的均一剂量或较高的均一剂量,或使用lebrikizumab增加治疗间隔。模型开发始于在进行上述ii期临床研究i期间收集的原始药代动力学和疗效数据。特应性皮炎患者中的lebrikizumab药代动力学数据与来自lebrikizumab群pk模型的预期一致,该模型是基于从哮喘患者和健康志愿者获得的药物浓度开发的(数据未显示)。如下所述,这允许使用lebrikizumab群pk模型来促进特应性皮炎pk-pd模型的开发。基于临床研究i的长达12周的easi评分和先前建立的lebrikizumab群pk模型,开发了lebrikizumab的特应性皮炎pk-pd模型。pd反应效应是easi评分的降低。安慰剂和/或tcs施用的作用也包含在模型中。协变量分析表明,基线easi评分是kout(组织修复率常数)和econ(安慰剂/tcs作用)的显著协变量。最终模型如图5所示,模型参数如表4所示。表4.lebrikizumab特应性皮炎模型参数。置信区间是针对1000个引导样本估计的参数的第2.5和第97.5百分位数;参见图5的用于解释符号的图示说明。该模型包括某些假设,如假设所有药物浓度间的暴露-反应关系相似,然而,我们发现其充分描述了研究1中观察到的原始easi评分的变化。此外,该模型能够很好地预期研究1中至第12周的easi-50、easi-75和easi-90反应,尽管不是为此目的而设计的。该结果为模型提供了额外的验证,增加了我们对其准确性和稳健性的信心。我们使用最终模型来模拟多种给药方案,然后我们预期在治疗期间不同时间的easi评分。模拟的给药方案在下表5中提供。所有模拟均在24周的时间内进行。使用每四周施用一次125mg的给药方案作为参照点以比较所有模拟结果。如表5所示,模拟的给药方案概括地分为四组:第1组方案为负荷剂量和维持剂量排列,第2组方案为基于每四周施用一次250mg的排列,第3组方案为基于每8周施用一次250mg的排列,第4组方案为基于每4周施用一次37.5mg的较低给药剂量的排列。表5.使用lebrikizumabad模型模拟的给药方案。对于各个第1组lebrikizumab给药方案随时间推移实现easi-75的患者模拟显示在图6中。pk-pd模型预期所有负荷剂量排列导致比单独的每4周一次125mg在easi-75反应中有小幅改善。这种改善在曲线的中间部分(例如第56-84天)最明显,并且大部分在更后的时间点(第140-168天)被稀释。对于各个第2组lebrikizumab给药方案随时间推移实现easi-75的患者模拟显示在图7中。pk-pd模型预期基于每四周一次250mg的方案在曲线的中间部分开始显示与每四周一次125mg的方案的分离(即,easi-75的改善),并且该分离在更后的时间点维持。对于各个第3组lebrikizumab给药方案随时间推移实现easi-75的患者模拟显示在图8中。pk-pd模型预期每8周施用一次250mg的方案与每四周施用一次125mg的方案具有相当的疗效,在休息周(例如,在第12周和第20周)略微改善,并且在给予下一剂量之前的一周(例如,在第8周和第16周)期间具有稍差的结果。对于各个第4组lebrikizumab给药方案随时间推移实现easi-75的患者模拟显示在图9中。pk-pd模型预期每四周施用一次37.5mg的方案不如每四周施用一次125mg的给药方案有效。总之,lebrikizumab特应性皮炎pk-pd模型预期所有“每四周一次”lebrikizumab给药方案提供与每四周一次施用125mg(在上述ii期临床研究i中临床测试的剂量)所获得的easi-75评分相当或更好的easi-75评分,除了包括每四周施用一次37.5mglebrikizumab的剂量的给药方案外。这些模拟显示,如通过easi-75评分所评估的,预期每四周施用一次37.5mglebrikizumab的效果低于任何其他模拟给药方案。lebrikizumab特应性皮炎pk-pd模型还预期,使用每四周施用一次250mglebrikizumab(有或没有负荷剂量)、持续16-24周的治疗持续时间获得最佳easi-75结果。该模型还预期,包含负荷剂量可在较早的时间点(例如,第8周或第12周)改善easi-75结果,但是与缺少负荷剂量的给药方案相比,这种改善的影响随着时间的推移而减少(例如,在第20或24周)。因此,包含负荷剂量可以在治疗过程早期为患者提供改善的治疗益处。最后,该模型预期每8周一次250mglebrikizumab的施用导致与每四周一次125mglebrikizumab的施用所预期的easi-75结果相似的easi-75改善。对于“250mgq8w方案”,预期easi-75的整体改善在未施用250mg剂量的周(例如,在第4周)稍高,而在250mg剂量施用之前略低于“125mgq4w方案”(例如,在第8周)。因此,这里描述的建模结果表明预期许多lebrikizumab给药方案为特应性皮炎患者提供治疗益处。实施例4-临床研究ii临床研究ii是ii期、随机、开放标记的研究,以评估lebrikizumab单一疗法在患有持续中度至重度ad的成年患者(18-75岁)中的安全性和疗效,所述ad患者的tcs控制不充分。研究ii方案在图10中提供。筛选。有资格参加研究的患者必须符合下述所有资格标准。纳入标准包括以下:年龄18至75岁;通过hanifin/rajka标准诊断为ad并且在筛选时存在至少1年;根据rajka/langeland标准在筛选时分级为中度至重度ad;对至少每天tcs和常规润肤剂治疗ad的治疗方案反应不足的病史≥1个月的病史(在筛选就诊前3个月内);在筛选和磨合期结束时(就诊3)easi评分≥14,在筛选和磨合期结束时(就诊3)iga评分≥3(5-分制);在筛选时ad涉及≥10%体表面积(bsa);和在筛选时瘙痒vas评分≥3(作为scorad的一部分测量);在进入治疗期(第1天)时遵守方案指定的2周磨合期tcs方案(14天中至少10天)。排除标准包括以下:过去和/或当前使用任何抗il-13或抗il-4/il-13治疗,包括lebrikizumab;在筛选前4周内或在研究试剂的5个半衰期内使用研究试剂,以较长者为准;严重过敏反应或对生物制剂的过敏反应或已知对lebrikizumab注射的任何成分超敏的病史;对tcs或研究中使用的tcs产品所含任何其他成分的超敏反应;在磨合期前7天内使用任何补充、替代或顺势疗法药物,包括但不限于植物疗法、传统或非传统草药、必需脂肪酸或针灸,或在研究期间需要此类药物;体重<40kg或身体质量指数>38kg/m2;其他皮肤病症的证据;包括但不限于t细胞淋巴瘤或过敏性接触性皮炎;筛选时(第-15天)活动性皮肤感染的证据或持续治疗(包括局部抗生素);某些感染;近期或活性(6个月内)寄生虫感染史,特别是线虫(例如蛔虫属、钩虫属)、扁形动物(例如血吸虫属)或李斯特菌感染史;活动性结核病需要在第1次就诊前12个月内接受治疗;急性或慢性肝炎或已知肝硬化的证据;已知的免疫缺陷,包括hiv感染;在筛选时使用tci(局部钙依赖磷酸酶抑制剂);在筛选就诊前4周内使用晒黑展台/店;筛选的3个月内过敏原免疫治疗;在基线就诊(第1天)前4周内接受减毒活疫苗;研究期间计划的手术;筛选ecg或实验室检查(血液学、血清化学和尿液分析)中显著的临床异常;筛选期间ast、alt或总胆红素升高≥2.0×正常上限(uln);当前已知的恶性肿瘤或当前评估潜在的恶性肿瘤,包括皮肤基底或鳞状细胞癌或原位癌;筛选前5年内恶性肿瘤病史,除外适当治疗的宫颈原位癌、非黑色素皮肤癌;i期子宫癌;以及尽管治疗但不受控制的其他临床上显著的医学疾病。磨合期。筛选后,符合条件的患者进入2周的磨合期(第-14天至-1天)。在磨合期后,患者停止使用他们研究前的tcs(和其他ad药物),并开始如下所述的方案指定的局部治疗方案。在磨合期的第一天(第-14天),患者接受tcs用于整个磨合期。具体而言,患者接受曲安奈德0.1%乳膏用于身体和氢化可的松2.5%乳膏用于面部和擦烂区域。指导患者每天至少一次向所有干燥的皮肤表面施用润肤剂,并且每天两次向活动性皮肤病灶施用乳膏,而不向未受影响的区域施用乳膏。在磨合期结束时,进行疾病严重性的评估,如通过easi和iga评估的(参见上文的纳入标准),持续显示中度至重度ad的患者有资格随机分组为研究药物(lebrikizumab)或仅tcs。治疗期(第1-12周)。在磨合期结束时,已经证实遵守方案指定的tcs方案并且继续满足资格标准(参见上面的纳入标准)的患者被随机分组。总共约50名患者被随机分组(1:1)至以下两个治疗组之一(参见图10):第1组:每4周(q4w)通过皮下(sc)注射施用125mglebrikizumab,总共3个剂量;第2组:单独使用tcs乳膏。随机分组至第1组的患者未接受方案指定的tcs方案,随机分组至第2组的患者继续应用tcs,使用与在磨合期应用的tcs方案相同的tcs方案(每天两次仅施用至受损皮肤)。安全性随访期(第13-20周)。随访在治疗期间(第1周至第12周)完成研究治疗的所有患者的安全性另外8周(第13周至第20周)。在此安全性随访期间,第2组(仅tcs)的患者不再需要按照磨合期和治疗期间的指定应用方案指定的tcs方案。相反,如患者和研究人员所确定的,曲安奈德0.1%乳膏和/或氢化可的松2.5%乳膏仅可应用到活动性皮肤病灶。同样,如患者和研究人员所确定的,治疗第1组(lebrikizumab单一疗法)的患者可以仅在活动性皮肤病灶中使用曲安奈德0.1%乳膏和/或氢化可的松2.5%乳膏。鼓励所有患者每天至少一次将润肤剂应用于干燥的皮肤。在该研究期间的任何时间都不允许局部钙依赖磷酸酶抑制剂(tci)。如果患者同意在研究期间停止使用tci,并且如果在研究者看来停止使用tci并且使用指定方案的tcs乳膏代替是安全的,则允许在筛选时一直使用tci的患者参加研究。lebrikizumab剂量和时间表的基本原理。对于研究ii的lebrikizumab剂量和时间表的基本原理类似于上文对研究i所述的基本原理。在该ii期研究中测试了一种lebrikizumab剂量方案(125mgq4w[第1组])。在哮喘中lebrikizumab的关键性iii期成年人研究(lavoltai和ii)中包括125mgq4w剂量方案作为潜在有效剂量,并且是被研究的最高剂量方案。鉴于预期的在哮喘和ad之间il-13的作用和药代动力学的相似性,假设该剂量可能对ad患者有效。然而,如上所述,lavoltai和ii研究果是不一致的。hanania等人,lancetrespirmed2016,可在dx(dot)doi(dot)org(slash)s2213-2600(16)30265-x获得,在线公开于2016年9月5日。两个研究都不能表明明确的剂量-反应,尽管药代动力学和药效学结果与前面描述的ii期研究的那些结果一致,表明药物暴露相似且il-13通路受到抑制。hanania等人,2016,同上。因此,不清楚lebrikizumab是否会在本文所述的给药方案中为特应性皮炎患者提供临床上有意义的益处。在第12周测量ii期研究的主要端点。对于接受125mgq4w的患者,预期第12周的平均血清lebrikizumab谷浓度超过稳态值的90%。此外,对于125mgq4w剂量,预期第12周的谷浓度将恰好在ii期哮喘临床试验中证明的lebrikizumab生物学活性的浓度范围内。对照组的基本原理。治疗第2组中的患者单独接受tcs乳膏,因此用作活性比较组。该治疗组用作lebrikizumabsc的对照,以充分评估lebrikizumab作为单一疗法与tcs相比的安全性和疗效。包括活性tcs比较组,是因为使用tcs是临床实践中的标准护理(欧洲皮肤病学和性病学研究院[eadv]2009指南;darsow等人,jeuracaddermatolvenereol2010;24:317-28;美国皮肤病学会2014年指南;eichenfield等人,jamacaddermatol2014:71(1):116-32)。疗效结果测量。本研究中与tcs单独相比,lebrikizumab单一疗法的探索疗效结果测量依次如下:(i)第12周时从基线easi降低50%或75%(easi50/75)的患者百分比(easi-50和easi-75分别定义为在第12周与基线相比easi评分降低50%和75%);(ii)在第12周easi评分从基线的百分比和绝对变化;(iii)在第12周实现iga评分为0或1的患者百分比;(iv)在第12周iga从基线降低≥2点的患者百分比;(v)在第12周iga从基线的绝对变化;(vi)在第12周实现igsa评分为0或1的患者百分比;(vii)在第12周igsa从基线降低≥2点患者百分比;(viii)在第12周igsa从基线的绝对变化;(ix)在第12周scorad从基线的百分比和绝对变化;(x)在第12周scorad评分从基线降低50%或75%(scorad50/75)的患者百分比;(xi)在第12周受影响的总体表面积(bsa)百分比从基线的百分比变化;(xii)在第12周通过瘙痒vas(作为scorad的一部分评估)测量的瘙痒从基线的绝对和百分比变化;(xiii)从基线到第12周的疾病加剧次数;和(xiv)在第12周之前接受非方案指定的tcs的患者百分比。安全性结果测量。本研究的安全性结果测量如下:(i)lebrikizumab作为单一疗法与tcs单独相比,在第12周治疗中出现的不良事件的发生率;(ii)在基线和研究期间人抗治疗性抗体(ata)的发生率;(iii)整个研究中皮肤和其他器官系统感染的频率和严重性;皮肤感染的临床定义如下:感染的诊断基于研究者的临床评估,包括但不限于皮疹部位存在蜂蜜色结痂、浆液性分泌物、脓疱或疼痛,其可能与全身特征相关,包括发烧或ad疾病的加剧(如上所述);(iv)研究人员评估中止研究药物后疾病反弹的发生率;疾病反弹的临床定义是:停止治疗后疾病严重性显著恶化,严重性水平大于开始治疗前的严重性(hijnen等人,jeuracaddermatolvenereol2007;21(1)85-9);(v)从基线到第12周的注射部位反应的发生率;和(vi)治疗中出现的不良事件的频率和严重性,包括从基线到第12周的特定目的的不良事件。结果。临床研究ii的关键疗效结果总结于下表6中。数据显示,经过12周研究期间,lebrikizumab单一疗法是有效的。lebrikizumab和tcs治疗组之间easi-75和easi-90的结果相似,而lebrikizumab组中的iga、scorad和瘙痒结果略低于tcs组。表6.第12周的关键疗效结果。*基线瘙痒vas>3的患者中。抗il13抗体(lebrikizumab)的氨基酸序列下表显示了lebrikizumab的cdr-h1、cdr-h2、cdr-h3、cdr-l1、cdr-l2和cdr-l3区的氨基酸序列,以及vh、vl、重链序列和轻链序列。如下面的序列表中所示并且如上所述,vh和重链可以包括n-末端谷氨酰胺,重链也可以包括c-末端赖氨酸。如本领域众所周知的,n-末端谷氨酰胺残基可以形成焦谷氨酸,并且c-末端赖氨酸残基可以在制造过程中被剪切。序列表当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1