诊断治疗试剂的制作方法
本发明主题总体上涉及一系列在近红外(nir)询问窗口(700-900nm)具有吸收的有机小分子化合物及其在光声成像(pai)和光热治疗(ptt)中的应用。
背景技术:
光诊断治疗试剂的出现为癌症的研究开启了一个新的方向。诊断治疗试剂可以有助于将实时诊断和原位光疗功能整合在一个平台中。在各种光触发的诊断/治疗技术中,光声成像(pai)联合光热治疗(ptt)在准确探测肿瘤位置和有效抑制肿瘤生长方面有很好的效果,并且对正常组织的副作用最小。pai是一种非常具有前景的非侵入式成像方法,它联合了超声成像中的深层组织穿透性和高分辨率以及光学成像中的高对比度。通常与pai伴随的治疗技术是ptt,因为pai主要用于检测光热产生的超声信号。pai/ptt应用的最重要的先决条件是采用在近红外(nir)询问窗口(700-900nm)具有强吸收的高效造影剂,因为已知nir光能够渗透更深的组织并且对活体的光损伤更小。
目前,各种纳米材料如金属纳米材料(例如,金和银纳米结构)、碳纳米材料(例如,碳纳米管和石墨烯)、过渡金属二硫化物(例如,mos2、ws2和ag2s)以及基于有机材料的纳米粒子被广泛用作pai/ptt试剂。与无机纳米试剂不同,有机材料(如聚合物和小分子)具有非常好的生物相容性、潜在的生物可降解性和易加工性等优点。因此,最近将半导体聚合物纳米粒子(spn)用作pai的造影剂以及具有优异性能的ptt应用。然而,尽管有机小分子具有化学结构明确、高纯度、良好的重复性、易于改性和加工等优点,但是适用于pai/ptt的有机小分子的开发还比较少。一些有机小分子面临的一个典型的挑战是小分子在pai/ptt中的不稳定性,目前这也限制了在该领域的发展。
临床上,一些常规的花菁染料被研究用作光介导的生物医学应用的中间体。例如,吲哚菁绿(icg)是一种在700-850nm的nir光谱区域具有强吸收的离子化合物,其已经被美国食品和药物管理局(fda)批准用于临床,这也体现了有机小分子在临床转化和实际应用中的前景。然而,这些花菁染料存在改良困难和稳定性差的问题,有可能导致安全问题和不可靠的诊疗结果。例如,许多花菁染料易于被活性氧/氮物质(rons)分解。因此,许多花菁染料可以用作检测活体中rons的敏感探针。花菁染料中交替排列的单键和双键很容易被高反应性rons氧化,这会导致特征的近红外吸收和荧光信号减弱或者消失。虽然利用花菁染料的反应特征进行rons的比率检测是合理的,但是花菁染料的不稳定性会在pai/ptt应用中产生严重问题,例如误导性pai信号、ptt的功效受损以及由体内分解引发的有害副作用。
许多目前可用的nir吸收的有机小分子面临各种挑战,包括光热不稳定性、光漂白和对rons分解的敏感性。
因此,需要开发用于有效的pai/ptt应用的高度稳定的nir有机小分子试剂。
技术实现要素:
本发明涉及可用于光声成像(pai)和光热治疗(ptt)应用中的诊断治疗试剂。该诊断治疗试剂包括在近红外(nir)询问窗口(700-900nm)具有吸收的有机小分子化合物。该化合物可以是生物相容性有机纳米粒子。可以将诊断治疗试剂施用于患者,从而采用光声成像确定患者体内的肿瘤部位。确定肿瘤部位以后,就可以用近红外光照射该肿瘤部位,当近红外光与本发明的化合物联合时,可以停止或抑制肿瘤的生长。
在一个实施方案中,该化合物具有:
选自由以下结构组成的组中的给体单元:
选自由以下结构组成的组中的受体单元(a):
其中d和d'代表给体单元;
其中所述化合物具有选自由d-a、d-a-d、a-d-a、d-d-a-d-d、a-a-d-a-a、d-a-d-a-d、a-d-a-d-a组成的组中的形式的结构排列,
其中a代表受体单元;
其中x和x'各自选自由o、s、se和te组成的组;
其中r、r′、r″、r″′或r″″各自是未取代的或取代的,并且选自由f、h、烷基、不饱和烷基、杂烷基、环烷基、杂环烷基、芳基、杂芳基、羧基、氨基、磺酸基、烷硫基和烷氧基组成的组;并且
其中r、r′、r″、r″′或r′′′′中的至少一者是具有取代基的末端官能团,该取代基独立地选自由n3、ncs、sh、nh2、cooh、炔烃、n-羟基琥珀酰亚胺酯、马来酰亚胺、酰肼、硝酮基、-cho、-oh、卤化物和带电离子基团组成的组;并且
其中r、r′、r″,r″′或r″″中的至少一者不是h。
在进一步的实施方案中,该化合物具有以下结构式之一:
其中x和x'各自选自由o、s、se和te组成的组;
其中d和d'各自选自由以下结构组成的组:
其中r、r′、r″和r″′各自为未取代的或取代的,并且选自由f、h、烷基、不饱和烷基、杂烷基、环烷基、杂环烷基、芳基、杂芳基、羧基、氨基、磺酸基、烷硫基和烷氧基组成的组;
其中r、r′、r″和r′′′中的至少一者为具有取代基的末端官能团,该取代基独立地选自由n3、ncs、sh、nh2、cooh、炔烃、n-羟基琥珀酰亚胺酯、马来酰亚胺、酰肼、硝酮基、-cho、-oh、卤化物和带电离子基团组成的组;并且
其中r、r′、r″和r′′′中的至少一者不是h。
在一个实施方案中,该化合物具有以下结构式:
其中x选自由o、s、se和te组成的组;
其中r、r′、r″、r′′′、r′′′′、r′′′′′、r″″″和r′′′′′′′各自为未取代的或取代的,并且选自由f、h、烷基、不饱和烷基、杂烷基、环烷基、杂环烷基、芳基、杂芳基、羧基、氨基、磺酸基、烷硫基和烷氧基组成的组;
其中r、r′、r″、r′′′、r′′′′、r′′′′′、r″″″和r″″″′中的至少一者为具有取代基的末端官能团,该取代基独立地选自由n3、ncs、sh、nh2、cooh、炔烃、n-羟基琥珀酰亚胺酯、马来酰亚胺、酰肼、硝酮基、-cho、-oh、卤化物和带电离子基团组成的组;并且
其中r、r′、r″、r″′、r″″r″″′、r″″″和r″″″′中的至少一者不是h。
在一个实施方案中,该化合物具有以下结构式:
其中r、r′、r″、r″′、r″″、r″″′、r″″″和r″″″′各自为未取代的或取代的,并且选自由f、h、烷基、不饱和烷基、杂烷基、环烷基、杂环烷基、芳基、杂芳基、羧基、氨基、磺酸基、烷硫基和烷氧基组成的组;
其中r、r′、r″、r″′、r″″、r″″′、r″″″和r″″″′中的至少一者是具有取代基的末端官能团,该取代基独立地选自由n3、ncs、sh、nh2、cooh、炔烃、n-羟基琥珀酰亚胺酯、马来酰亚胺、酰肼、硝酮基、-cho、-oh、卤化物和带电离子组;并且
其中r、r′、r″、r″′、r″″、r″″′、r″″″和r″″″′中的至少一者不是h。
在一个实施方案中,示例性化合物是:
附图说明
现将参考附图详细描述各种实施方案。
图1描绘了cdcl3中的化合物10的1hnmr谱。
图2描绘了cdcl3中的化合物10的13cnmr谱。
图3描绘了化合物10的hrms。
图4描绘了cdcl3中的化合物tpa-t-tq的1hnmr谱。
图5描绘了cdcl3中的化合物tpa-t-tq的13cnmr谱。
图6描绘了化合物tpa-t-tq的hrms。
图7描绘了tpa-t-tq的热重分析(tga)曲线。
图8a描绘了通过纳米沉淀法制备有机小分子纳米粒子的示意图。
图8b描绘了thf溶液中的tpa-t-tq的uv-vis-nir吸收光谱和水中的封装的onp的uv-vis-nir吸收光谱。
图8c描绘了thf溶液中的tpa-t-tq的光致发光(pl)光谱和水中的封装的onp的光致发光(pl)光谱。
图8d描绘了tpa-t-tqonp的tem图像。
图8e描绘了tpa-t-tqonp的dls结果。
图9a描绘了在用808nm激光(0.8w/cm2)照射不同时间时,pbs溶液(100μm)中onp和icg的ir热图像。
图9b描绘了在功率强度为0.8w/cm2的808nm光照射下,tpa-t-tqonp在不同浓度(5μm-100μm)下的光热转化行为。
图9c描绘了比较在相同浓度(100μm)的pbs溶液中tpa-t-tqonp和icg的光热转化行为的图(0.8w/cm2的808nm光照射5分钟)。
图9d描绘了在808nm光照射不同时间后pbs溶液中的onp和icg的照片。
图9e描绘了i/i0相对于不同照射时间的图(i和i0分别是激光照射之前和之后pbs溶液中onp/icg的最大nir吸收强度)。
图9f描绘了在五个加热-冷却循环过程期间,onp和icg(100μm)的抗光漂白性质(用于照射的激光是808nm光,功率密度为0.8w/cm2)。
图9g描绘了在于1分钟内加入400μm的onoo-和·oh之前和之后,pbs溶液中tpa-t-tqonp的吸收光谱(插图显示了在加入rons之前和之后onp溶液的照片)。
图9h描绘了在于1分钟内加入400μm的onoo-和·oh之前和之后,pbs溶液中icg的吸收光谱(插图显示了在加入rons之前和之后icg溶液的照片)。
图9i描绘了i/i0相对于rons(onoo-和·oh)的图。i和i0分别是存在和不存在rons的pbs溶液中onp/icg的最大nir吸收强度。
图10描绘了在于1分钟内加入0.4mmonoo-和·oh之前和之后,icg在水中的光致发光(pl)光谱。
图11a描绘了pbs溶液中的tpa-t-tqonp的pa光谱(基于tpa-t-tq的110μg/ml)。
图11b描绘了770nm处tpa-t-tqonp的pa振幅(amplitude)与tpa-t-tq浓度的关系。
图11c描绘了肿瘤部位的pa强度与注射后时间的关系。
图11d描绘了以指定的时间间隔全身施用tpa-t-tqonp后肿瘤部位的pa图像。
图12a描绘了在808nm激光照射(0.5w/cm2)下,对于不同时间点的4t1荷瘤小鼠的ir热成像图。
图12b描绘了显示肿瘤的平均温度与808nm激光(0.5w/cm2)照射时间的关系的图(对于(a)和(b),在静脉施用tpa-t-tqonp或盐水6小时后进行激光照射)。
图12c描绘了显示肿瘤生长曲线的图(不同治疗组的小鼠(**代表p<0.01,“onp+激光”组和其他治疗组之间的比较)。
图12d描绘了显示不同小鼠治疗组的体重变化图。
图13a显示在分别用onp和激光治疗、仅用onp治疗、用盐水和激光治疗、以及仅用盐水治疗后第16天,肿瘤切片的组织学h&e染色、荧光tunel染色和pcna染色。
图13b显示在分别用onp和激光治疗、仅用onp治疗、用盐水和激光治疗、以及仅用盐水治疗后第16天,肝脏和脾脏的组织学h&e染色。
图14a描绘了用tpa-t-tqonp治疗7天后的健康balb/c小鼠的血液生化数据图。
图14b描绘了用tpa-t-tqonp治疗7天后的健康balb/c小鼠的血液学数据图(未治疗的小鼠用作对照)。
具体实施方式
以下定义是为了理解本发明的主题和构建所附权利要求。
定义
应该理解,以上或以下描述的附图仅用于说明目的。附图不一定按比例绘制,重点通常在于说明本发明教导的原理。附图不旨在以任何方式限制本发明的范围。
在整个申请中,其中组合物被描述为具有、包括或包含特定组分时,或者其中方法被描述为具有、包括或包含特定的方法步骤时,预期本发明教导的组合物也可以基本上由所述组分组成、或者由所述部分组成,并且本发明教导的方法也可以基本上由所述方法步骤组成、或由所述方法步骤组成。
在本申请中,其中元素或组分被称为包括在所列举的元素或组分的列表中和/或从所列举的元素或组分的列表中选择时,应当理解,所述元素或组分可以是所列举的元素或组分中的任何一个,或者所述元素或组分可选自由两种或更多种所列举的元素或组分组成的组。此外,应当理解,本发明描述的组合物、装置或方法的要素和/或特征可以以各种方式组合而不脱离本发明的精神和范围,无论是明确的还是隐含的。
除非另外特别说明,否则术语“包括(include)”、“包含(includes)”、“含有(including)”、“具有(have)”、“有(has)”或“拥有(having)”的使用通常应理解为开放式和非限制性的。
除非额外特别说明,否则本发明中单数的使用包括复数(反之亦然)。另外,在术语“约”的使用在定量值之前的情况下,除非另外特别说明,否则本发明教导还包括具体的定量值本身。除非另有说明或推断,否则本发明所用的术语“约”是指与标称值间存在±10%的变化。
应当理解,只要本发明仍然可操作,则步骤的顺序或执行某些动作的顺序是不重要的。此外,可以同时进行两个或更多个步骤或动作。
本发明所用的“杂芳基”是指含有至少一个选自氧(o)、氮(n)、硫(s)、硅(si)和硒(se)的环杂原子的芳族单环体系,或者多环体系,其中在该多环体系中,环体系中存在的至少一个环是芳族环并含有至少一个环杂原子。多环杂芳基包括两个或更多个稠合在一起的杂芳基环和与一个或多个芳族碳环、非芳族碳环和/或非芳族环杂烷基环稠合的单环杂芳基环。杂芳基作为整体可具有例如5至22个环原子并含有1-5个环杂原子(即5-20元杂芳基)。杂芳基可以在任何杂原子或碳原子上与所定义的化学结构连接,从而产生稳定的结构。通常,杂芳基环不含o-o、s-s或s-o键。然而,杂芳基中的一个或多个n或s原子可被氧化(例如,吡啶n-氧化物,噻吩s-氧化物,噻吩s,s-二氧化物)。杂芳基的实例包括(例如)如下所示的5元单环体系或6元单环体系和5-6双环体系:
其中t为o、s、nh、n-烷基、n-芳基、n-(芳基烷基)(例如,n-苄基)、sih2、sih(烷基)、si(烷基)2、sih(芳基烷基)、si(芳基烷基)2、或si(烷基)(芳基烷基)。这种杂芳基环的实例包括吡咯基、呋喃基、噻吩基、吡啶基、嘧啶基、哒嗪基、吡嗪基、三唑基、四唑基、吡唑基、咪唑基、异噻唑基、噻唑基、噻二唑基、异噁唑基、噁唑基、噁二唑基、吲哚基、异吲哚基、苯并呋喃基、苯并噻吩基、喹啉基、2-甲基喹啉基、异喹啉基、喹喔啉基、喹唑啉基、苯并三唑基、苯并咪唑基、苯并噻唑基、苯并异噻唑基、苯并异噁唑基、苯并噁二唑基、苯并噁唑基、噌嗪基、1h-吲唑基、2h-吲唑基、吲嗪基、异苯并呋喃基、萘啶基、酞嗪基、蝶啶基、嘌呤基、噁唑并吡啶基、噻唑并吡啶基、咪唑并吡啶基、呋喃并吡啶基、噻吩并吡啶基、吡啶并嘧啶基、吡啶并吡嗪基、吡啶并哒嗪基、噻吩并噻唑基、噻吩并噁唑基、噻吩并咪唑基等。杂芳基的其他实例包括4,5,6,7-四氢吲哚基、四氢喹啉基、苯并噻吩并吡啶基、苯并呋喃并吡啶基等。在一些实施方案中,杂芳基可如本发明所述被取代。
本发明所用的“卤代”或“卤素”是指氟、氯、溴和碘。
本发明所用的“烷基”是指直链或支链的饱和烃基。烷基的实例包括甲基(me)、乙基(et)、丙基(例如,正丙基和z'-丙基)、丁基(例如,正丁基,z'-丁基,仲丁基,叔丁基)、戊基(例如,正戊基,z'-戊基,-戊基)、己基等。在各种实施方案中,烷基可具有1-40个碳原子(即,c1-40烷基),例如1-30个碳原子(即,c1-30烷基)。在一些实施方案中,烷基可具有1至6个碳原子,并且可称为“低级烷基”。低级烷基的实例包括甲基、乙基、丙基(例如正丙基和z'-丙基)和丁基(例如正丁基、z'-丁基、仲丁基、叔丁基)。在一些实施方案中,烷基可如本发明所述被取代。烷基通常不被另一个烷基、烯基或炔基取代。
本发明所用的“链烯基”是指具有一个或多个碳-碳双键的直链或支链烷基。链烯基的实例包括乙烯基、丙烯基、丁烯基、戊烯基、己烯基、丁二烯基、戊二烯基、己二烯基等。一个或多个碳-碳双键可以是内双键(例如2-丁烯中的双键)或末端双键(例如1-丁烯中的双键)。在各种实施方案中,链烯基可具有2-40个碳原子(即c2-40烯基),例如,2-20个碳原子(即,c2-20链烯基)。在一些实施方案中,链烯基可如本发明所述被取代。链烯基通常不被另一个链烯基、烷基或炔基取代。
本发明所用的“稠环”或“稠环部分”是指具有至少两个环的多环体系,其中至少一个环是芳环并且这种芳环(碳环或杂环)具有与至少另一个环(该环可以是芳环或非芳环,并且为碳环或杂环)共用一个键。这些多环体系可以是高度p-共轭的并且如本发明所述任选被取代。
本发明所用的“杂原子”是指除碳或氢之外的任何元素的原子,并且包括(例如)氮、氧、硅、硫、磷和硒。
本发明所用的“芳基”是指芳族单环烃环体系或多环体系,其中在该多环体系中,两个或更多个芳族烃环稠合(即,具有共同的键)在一起或至少一个芳族单环烃环与一个或多个环烷基和/或环杂烷基环稠合。芳基在其环体系中可具有6-24个碳原子(例如,c6-24芳基),其可包括多个稠环。在一些实施方案中,多环芳基可具有8至24个碳原子。芳基的任何合适的环位置可以与限定的化学结构共价连接。仅具有芳族碳环的芳基的实例包括苯基、1-萘基(双环)、2-萘基(双环)、蒽基(三环)、菲基(三环)、稠五苯基(五环)等基团。其中至少一个芳族碳环与一个或多个环烷基环和/或环杂烷基环稠合的多环体系的实例包括环戊烷的苯并衍生物(即,茚满基,其为5,6-双环环烷基/芳环体系)、环己烷的苯并衍生物(即,四氢萘基,其为6,6-双环环烷基/芳环体系)、咪唑啉的苯并衍生物(即,苯并咪唑啉基,其为5,6-双环环杂烷基/芳环体系)和吡喃的苯并衍生物(即,苯并吡喃基,其为6,6-双环环杂烷基/芳环体系)。芳基的其他实例包括苯并二噁烷基、苯并二氧杂环戊烯基、苯并二氢吡喃基、二氢吲哚基等。在一些实施方案中,芳基可如本发明所述被取代。在一些实施方案中,芳基可具有一个或多个卤素取代基,并且可称为“卤代芳基”。全卤芳基,即所有氢原子均被卤素原子取代的芳基(例如-c6f5)包括在“卤代芳基”的定义内。在某些实施方案中,芳基被另一个芳基取代并且可以称为联芳基。联芳基中的每个芳基可以如本文所公开的那样被取代。
本发明所用的“给体”材料是指具有作为主要电流或电荷载体的空穴的有机材料,例如,有机纳米粒子材料。
本发明所用的“受体”材料是指具有作为主要电流或电荷载体的电子的有机材料,例如,有机纳米粒子材料。
本发明所用的“诊断治疗试剂”是指同时具有诊断和治疗能力的有机材料,例如,有机纳米粒子材料。
除非另有定义,否则本发明使用的所有技术和科学术语具有与当前描述的主题所属领域的普通技术人员通常理解的含义相同的含义。
在提供数值范围的情况下,例如,浓度范围、百分比范围或比率范围,应理解的是,除了上下文另有明确规定之外,否则在上限和下限之间的每个中间值、至下限单位的十分之一以及所声明范围内的任何其他所声明值或中间值均包含在所述主题内。这些较小范围的上限和下限可以独立地包括在较小范围内,并且这些实施方案也包括在所描述的主题内,受所声明范围内的任何特别排除的限制。在所声明范围包括一个或两个界限值的情况下,排除所包括的界限值中的一个或两个界限值的范围也包括在所描述的主题中。
在整个申请中,各种实施例的描述使用表述“包含”。然而,本领域技术人员将理解,在一些特定情况下,可替代地使用表述“基本上由......组成”或“由......组成”来描述实施方案。
为了更好地理解本发明的教导并且决不限制本发明教导的范围,除非另有说明,否则在说明书和权利要求中使用的表示数量、百分比或比例以及其他数值的所有数字在所有情况下均应理解为术语“约”修饰。因此,除非有相反的指示,否则在以下说明书和所附权利要求书中列出的数值参数是近似值,其可以根据试图获得的所需性质而变化。至少,每个数值参数至少应根据公布的有效数字的个数并通过应用普通的舍入技术来解释。
诊断治疗试剂
本发明主题涉及诊断治疗试剂,或用于诊断和治疗目的的试剂。如本发明所预期的,诊断治疗试剂可包括至少一种在近红外(nir)询问窗口(700-900nm)中具有吸收的小分子有机化合物。该化合物可以是有机纳米粒子(onp)。本发明所述的诊断治疗试剂可为光触发诊断/治疗技术(例如与光热治疗(ptt)相关的光声成像(pai))提供理想的造影剂。本发明所述的诊断治疗试剂表现出优异的热稳定性和光热稳定性,以及对光漂白和rons的显着抗性。当暴露于nir光时,本发明所述的诊断治疗试剂还表现出优异的光热转换性能。
在一个实施方案中,本发明的诊断治疗试剂是小分子有机化合物,其具有:
选自由以下结构组成的组中的给体单元:
选自由以下结构组成的组中的受体单元(a):
其中所述化合物以选自d-a、d-a-d、a-d-a、d-d-a-d-d、a-a-d-a-a、d-a-d-a-d、a-d-a-d-a的形式排列,
其中d和d'代表给体单元;
其中a代表受体单元;
其中x和x'各自选自由o、s、se和te构成的组;
其中r、r′、r″、r″′和r″″各自是未取代的或取代的,并且选自由f、h、烷基、不饱和烷基、杂烷基、环烷基、杂环烷基、芳基、杂芳基、羧基、氨基、磺酸基、烷硫基和烷氧基组成的组;并且
其中r、r′、r″、r″′和r″″中的至少一者是具有取代基的末端官能团、所述取代基独立地选自由n3、ncs、sh、nh2、cooh、炔烃、n-羟基琥珀酰亚胺酯、马来酰亚胺、酰肼、硝酮基、-cho、-oh、卤化物和带电离子基团组成的组;并且
其中r、r′、r″、r″′和r″″中的至少一者不是h。
在进一步的实施方案中,该化合物具有以下结构式之一:
其中x和x'各自选自由o、s、se和te组成的组;
其中d和d'各自选自由以下结构组成的组:
其中r、r′、r″和r″′各自是未取代的或取代的,并且选自由f、h、烷基、不饱和烷基、杂烷基、环烷基、杂环烷基、芳基、杂芳基、羧基基团、氨基、磺酸基、烷硫基和烷氧基组成的组;
其中r、r′、r″和r″′中的至少一者是具有取代基的末端官能团,该取代基独立地选自由n3、ncs、sh、nh2、cooh、炔烃、n-羟基琥珀酰亚胺酯、马来酰亚胺、酰肼、硝酮基、-cho、-oh、卤化物和带电离子基团组成的组;并且
其中r、r′、r″和r″′中的至少一者不是h。
在一个实施方案中,该化合物具有以下结构式:
其中x选自由o、s、se和te组成的组;
其中r、r′、r″、r″′、r″″、r″″′、r″″″和r″″″′是未取代的或取代的,并且选自由f、h、烷基、不饱和烷基、杂烷基、环烷基、杂环烷基、芳基、杂芳基、羧基、氨基、磺酸基、烷硫基和烷氧基组成的组;
其中r、r′、r″、r″′、r″″r″″′、r″″″和r″″″′中的至少一者是具有取代基的末端官能团,所述取代基独立地选自由n3、ncs、sh、nh2、cooh、炔烃、n-羟基琥珀酰亚胺酯、马来酰亚胺、酰肼、硝酮基、-cho、-oh、卤化物和带电离子基团组成的组;并且
其中r、r′、r″、r″′、r″″r″″′、r″″″和r″″″′中的至少一者不是h。
在一个实施方案中,该化合物具有以下结构式:
其中r、r′、r″、r″′、r″″r″″′、r″″″和r″″″′是未取代的或取代的,并且选自由f、h、烷基、不饱和烷基、杂烷基、环烷基、杂环烷基、芳基、杂芳基、羧基、氨基、磺酸基、烷硫基和烷氧基组成的组;
其中r、r′、r″、r″′、r″″r″″′、r″″″和r″″″′中的至少一者是具有取代基的末端官能团,所述取代基独立地选自由n3、ncs、sh、nh2、cooh、炔烃、n-羟基琥珀酰亚胺酯、马来酰亚胺、酰肼、硝酮基、-cho、-oh、卤化物和带电离子基团组成的组;并且
其中r、r′、r″、r″′、r″″r″″′、r″″″和r″″″′中的至少一者不是h。
在一个实施方案中,该化合物是:
以下提供用于制备tpa-t-tq化合物的示例性反应方案:
在一个实施方案中,化合物以纳米粒子的形式提供。如图8a中描绘的反应方案中所示,可以使用两亲性基质合成纳米粒子,并在本发明中详细描述。
识别肿瘤并阻止或抑制肿瘤生长
本发明的化合物可以作为造影剂用于患者,用于通过使用体内成像技术(例如光声成像)确定患者的肿瘤部位。例如,化合物可以通过静脉内注射来施用。如本发明详细阐述的,体内成像研究表明,所述化合物能够以高对比度方式用作pai的有效探针。确定肿瘤部位以后,就可以用近红外光照射肿瘤部位,当近红外光与本发明化合物联合使用时,可以停止或抑制肿瘤的生长。在一个实施方案中,可以在pa成像和肿瘤的ptt治疗之前6小时将化合物施用于患者。
用异种移植4t1荷瘤小鼠模型的体内肿瘤生长动力学揭示,使用本发明化合物的ptt可以有效抑制并阻止肿瘤生长。本发明化合物在nir光照射下在肿瘤部位显示出快速的温度升高,这导致热引发的肿瘤抑制。如本发明所述,通过肿瘤切片的组织学和免疫组织化学染色也证实了这种优异的抗肿瘤功效。可以将各种功能性和靶向基团引入化合物中,以促进所需生物物质的特异性靶向。根据一个实施方案,一种或多种肽可以与本发明化合物缀合。
由于本发明化合物是完全有机的,因此这些化合物显示出良好的生物相容性,并且基于组织学检查和血液测试,没有可检测的毒副作用。该化合物显示出超高稳定性和良好的光热/光声性能,使其非常有希望用于体内诊断和治疗应用。
通过以下实施例说明本发明
实施例
材料与设备
通过使用cdcl3或dmso-d6作为溶剂,在brukerav400光谱仪上记录1h(400mhz)和13c(100mhz)核磁共振(nmr)谱。在maldi-tof模式的gctpremiercab048质谱仪上测量高分辨率质谱(hrms)。在tatgaq5000上,在氮气氛下以10℃/min的加热速率进行热重分析(tga)测量。uv-vis-nir吸收光谱在perkinelmerlambda365分光光度计上进行。使用horibafluorolog-3分光荧光计测定光致发光(pl)光谱。动态光散射(dls)在90plus粒度分析仪上进行。透射电子显微镜(tem)图像在jem-2010f透射电子显微镜上获得,加速电压为200kv。
定量数据表示为平均值±标准偏差(sd)。通过anova分析和双样本studentt检验进行统计学比较。p值<0.05被认为具有统计学意义。
实施例1
tpa-t-tq的合成
4-(叔丁基)-n-(对甲苯基)苯胺(3)
将4-(叔丁基)苯胺(1.94g,13mmol)、1-溴-4-甲基苯(1.71g,10mmol)、叔丁醇钠(1.25g,13mmol)和乙酸钯(ii)(pd(oac)2)(45mg,0.2mmol)加入到100ml的双颈圆底烧瓶中。将烧瓶抽真空并用干燥氮气吹扫三次。然后加入三叔丁基膦(p(tbu)3,0.25mmol,1m甲苯溶液,0.25ml)和无水甲苯(50ml),将所得混合物加热至回流并在无光条件下搅拌24小时。冷却至室温后,加入水,混合物用二氯甲烷萃取。合并有机相,用mgso4干燥。减压除去溶剂后,残余物通过硅胶柱色谱纯化,用二氯甲烷/己烷(v/v1:4)作为洗脱剂,得到为无色固体的4-(叔丁基)-n-(对甲苯基)苯胺(产率75%)。1hnmr(400mhz,cdcl3,25℃)δ(ppm):7.29(d,2h),7.09(d,2h),7.03-6.97(m,4h),5.56(br,1h),2.32(s,3h),1.33(s,9h)。13cnmr(100mhz,cdcl3,25℃)δ(ppm):143.48,141.20,140.93,130.29,129.83,126.10,118.19,117.16,34.14,31.51,20.67。
4-溴-n-(4-(叔丁基)苯基)-n-(对甲苯基)苯胺(5)
将4-(叔丁基)-n-(对甲苯基)苯胺(1.68g,7mmol)、1-溴-4-碘苯(1.98g,7mmol)、1,10-菲咯啉(0.27g,1.5mmol)、氯化亚铜(i)(0.15g,1.5mmol)和氢氧化钾(1.68g,30mmol)加入到100ml的双颈圆底烧瓶中。将烧瓶抽真空并用干燥氮气吹扫三次。然后加入无水甲苯(50ml),将所得混合物加热至回流并搅拌24小时。冷却至室温后,加入水,混合物用二氯甲烷萃取。合并有机相,用mgso4干燥。在减压下除去溶剂后,通过硅胶柱色谱法纯化残余物,使用二氯甲烷/己烷(v/v1:6)作为洗脱剂,得到为白色固体的4-溴-n-(4-(叔丁基)苯基)-n-(对甲苯基)苯胺(产率73%)。1hnmr(400mhz,cdcl3,25℃)δ(ppm):7.22(d,2h),7.09-6.95(m,8h),6.93(s,2h),2.30(s,3h),1.30(s,9h)。13cnmr(100mhz,cdcl3,25℃)δ(ppm):145.52,144.75,142.74,131.91,130.01,129.76,126.14,125.88,124.88,124.07,123.77,122.74,34.21,31.46,20.80。
噻吩-2-基硼酸(6)
向100ml双颈圆底烧瓶中加入噻吩(1.68g,20mmol)和无水thf(40ml)。然后将烧瓶抽真空并用干燥氮气吹扫三次。然后将混合物用干冰-丙酮冷却至-78℃,并保持15分钟,然后加入正丁基锂(nbuli,2.5m己烷溶液,8.8ml,22mmol)。将反应混合物在-78℃下搅拌30分钟,然后缓慢升温至-20℃,再搅拌30分钟。然后,将混合物冷却至-78℃,加入硼酸三乙酯(2.5ml,22mmol)。将混合物在-78℃下继续搅拌1小时,然后缓慢升温至室温,搅拌过夜。然后将反应混合物用hcl水溶液(1m)处理,并用二氯甲烷萃取三次。合并有机相,用mgso4干燥。在减压下除去溶剂后,通过重结晶进一步纯化产物,得到为白色固体的噻吩-2-基硼酸(产率65%)。1hnmr(400mhz,dmso-d6,25℃)δ(ppm):8.20(d,2h),7.74(dd,1h),7.67(dd,1h),7.17(dd,1h)。13cnmr(100mhz,dmso-d6,25℃)δ(ppm):136.41,132.02,128.54。
4-(叔丁基)-n-(4-(噻吩-2-基)苯基)-n-(对甲苯基)苯胺(7)
将4-溴-n-(4-(叔丁基)苯基)-n-(对甲苯基)苯胺(1.97g,5mmol),噻吩-2-基硼酸(0.64g,5mmol)、pd(pph3)4(115mg,0.1mmol)和k2co3(2.76g,20mmol)加入100ml双颈圆底烧瓶中。将烧瓶抽真空并用干燥氮气吹扫三次。然后加入无水thf(40ml)和水(10ml),将混合物加热至回流并在无光的情况下搅拌24小时。冷却至室温后,加入水,混合物用二氯甲烷萃取。合并有机相,用mgso4干燥。减压除去溶剂后,残余物通过硅胶柱色谱纯化,用二氯甲烷/己烷(v/v1:5)作为洗脱剂,得到为淡黄色固体的4-(叔丁基)-n-(4-(噻吩-2-基)苯基)-n-(对甲苯基)苯胺(产率81%)。1hnmr(400mhz,cdcl3,25℃)δ(ppm):7.46-7.42(m,2h),7.25-7.23(m,2h),7.19(d,2h),7.10-7.00(m,9h),2.32(s,3h),1.31(s,9h)。13cnmr(100mhz,cdcl3,25℃)δ(ppm):147.65,145.80,145.03,144.85,144.54,132.84,129.96,127.95,127.71,126.63,126.09,124.96,123.84,123.76,122.75,121.98,34.31,31.46,20.87。
4-(叔丁基)-n-(对甲苯基)-n-(4-(5-(三丁基甲锡烷基)噻吩-2-基)苯基)苯胺(8)
将4-(叔丁基)-n-(4-(噻吩-2-基)苯基)-n-(对甲苯基)苯胺(1.2g,3mmol)加入到100ml双颈圆底烧瓶中。将烧瓶抽真空并用干燥氮气吹扫三次。然后加入无水thf(50ml),将得到的混合物用干冰-丙酮冷却至-78℃,并保持15分钟,然后加入正丁基锂(nbuli,2.5m己烷溶液,1.25ml,3.2mmol)。将混合物在-78℃下搅拌2小时。然后加入三正丁基氯化锡(0.9ml,3.3mmol),将混合物缓慢升温至室温,搅拌过夜。加入水以淬灭反应,并将混合物用二氯甲烷萃取三次。合并有机相,用mgso4干燥。在减压下除去溶剂后,得到为褐色油状物的4-(叔丁基)-n-(对甲苯基)-n-(4-(5-(三丁基甲锡烷基)噻吩-2-基)苯基)苯胺,无需进一步纯化即可直接使用。
4,7-二溴-5,6-二硝基苯并[c][1,2,5]噻二唑(9)
将4,7-二溴苯并[c][1,2,5]噻二唑(8.82g,30mmol)、硫酸(60ml)、发烟硫酸(20ml)、发烟硝酸(50ml)的混合物在0℃下搅拌4小时以完成硝化反应。然后将混合物缓慢倒入冰水(500ml)中,得到悬浮液,通过布氏漏斗过滤,用水洗涤数次,真空干燥,得到为浅黄色粉末的4,7-二溴-5,6-二硝基苯并[c][1,2,5]噻二唑(产率:88%)。13cnmr(100mhz,dmso-d6,25℃)δ(ppm):151.98,144.13,111.84。
4,4'-((5,6-二硝基苯并[c][1,2,5]噻二唑-4,7-二基)双(噻吩-5,2-二基))双(n-(4-(叔丁基)苯基)-n-(对甲苯基)苯胺)(10)
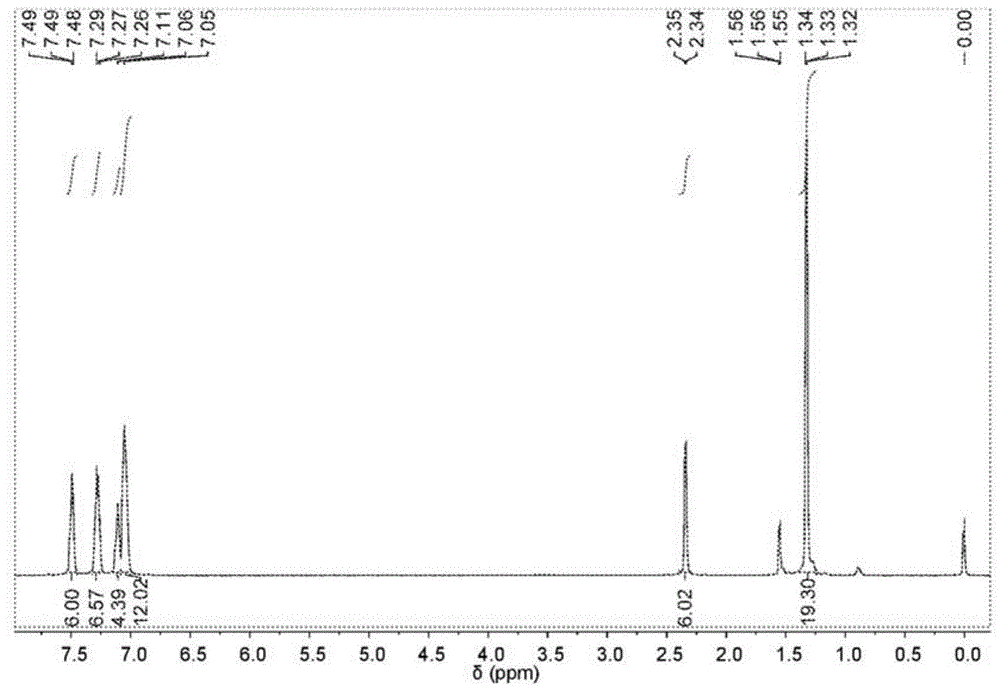
将4-(叔丁基)-n-(对甲苯基)-n-(4-(5-(三丁基甲锡烷基)噻吩-2-基)苯基)苯胺(2.06g,3mmol)、4,7-二溴-5,6-二硝基苯并[c][1,2,5]噻二唑(0.46g,1.2mmol)和pd(pph3)4(58mg,0.05mmol)加入到100ml双颈圆底烧瓶中。将烧瓶抽真空并用干燥氮气吹扫三次。然后加入无水thf(40ml),将混合物加热至回流并在无光的情况下搅拌24小时。冷却至室温后,加入水,混合物用二氯甲烷萃取。合并有机相,用mgso4干燥。在减压下除去溶剂后,将粗产物通过硅胶柱色谱纯化,用二氯甲烷/己烷(v/v1:3)作为洗脱液,得到为深蓝色固体的4,4'-((5,6-二硝基苯并[c][1,2,5]噻二唑-4,7-二基)双(噻吩-5,2-二基))双(n-(4-(叔丁基)苯基)-n-(对甲苯基)苯胺)(产率79%)。化合物10的1hnmr谱如图1所示。化合物10的13cnmr谱如图2所示。化合物10的hrms如图3所示。1hnmr(400mhz,cdcl3,25℃)δ(ppm):7.49(m,6h),7.28(m,6h),7.11(m,4h),7.05(m,12h),2.34(s,6h),1.33(m,18h)。13cnmr(100mhz,cdcl3,25℃)δ(ppm):152.02,151.40,148.86,146.44,144.61,144.43,141.20,133.44,132.18,130.06,127.53,126.88,126.21,125.73,125.38,124.38,122.80,121.78,120.28,34.37,31.44,20.91。hrms(maldi-tof,m/z):c60h52n6o4s3的[m]+计算值为1016.3212;试验值为1016.3249。
4,7-双(5-(4-((4-(叔丁基)苯基)(对甲苯基)氨基)苯基)噻吩-2-基)苯并[c][1,2,5]噻二唑-5,6-二胺(11)
将4,4'-((5,6-二硝基苯并[c][1,2,5]噻二唑-4,7-二基)双(噻吩-5,2-二基))双(n-(4-(叔丁基)苯基)-n-(对甲苯基)苯胺(0.92g,0.9mmol)、铁粉(1.1g,20mmol)和乙酸(50ml)悬浮在100ml圆底烧瓶中,将混合物加热至80℃,搅拌4小时。冷却至室温后,加入水,混合物用二氯甲烷萃取。用水和nahco3水溶液洗涤有机层。合并有机相,用mgso4干燥。在减压下除去溶剂后,获得为深红色固体的4,7-双(5-(4-((4-(叔丁基)苯基)(对甲苯基)氨基)苯基)噻吩-2-基)苯并[c][1,2,5]噻二唑-5,6-二胺,无需进一步纯化即可使用。
4,4'-((6,7-二苯基-[1,2,5]噻二唑并[3,4-g]喹喔啉-4,9-二基)双(噻吩-5,2-二基))双(n-(4-(叔丁基)苯基)-n-(对甲苯基)苯胺)(tpa-t-tq)
将4,7-双(5-(4-((4-(叔丁基)苯基)(对甲苯基)氨基)苯基)噻吩-2-基)苯并[c][1,2,5]噻二唑-5,6-二胺(0.86g,0.9mmol)溶于100ml圆底烧瓶中的氯仿(20ml)和乙酸(20ml)的混合物中,并加入苯偶酰(0.32g,1.5mmol)。然后将混合物加热至回流并搅拌12小时。冷却至室温后,加入水,混合物用二氯甲烷萃取。用水和nahco3水溶液洗涤有机层。合并有机相,用mgso4干燥。在减压下除去溶剂后,将粗产物通过硅胶柱色谱纯化,用二氯甲烷/己烷(v/v1:3)作为洗脱液,得到为黄绿色固体的4,4'-((6,7-)二苯基-[1,2,5]噻二唑并[3,4-g]喹喔啉-4,9-二基)双(噻吩-5,2-二基))双(n-(4-(叔丁基)苯基)-n-(对甲苯基)苯胺)(产率76%)。tpa-t-tq的1hnmr谱如图4所示。tpa-t-tq的13cnmr谱如图5所示。tpa-t-tq的hrms如图6所示。1hnmr(400mhz,cdcl3,25℃)δ(ppm):8.94(br,2h),7.77(m,4h),7.55(br,4h),7.45-7.32(m,8h),7.29(d,4h),7.15-7.01(m,16h),2.35(s,6h),1.34(s,18h)。13cnmr(100mhz,cdcl3,25℃)δ(ppm):152.39,151.66,147.86,145.95,144.97,144.80,138.08,134.90,134.47,132.98,130.88,130.00,129.50,128.94,128.46,128.07,126.44,126.14,125.14,124.04,122.56,122.19,122.80,120.50,34.35,31.48,20.91。hrms(maldi-tof,m/z):c74h62n6s3的[m]+计算值为1130.4198;试验值为1130.4208。
实施例2
tpa-t-tq有机纳米粒子(onp)的合成
将含有1mgtpa-t-tq化合物和2mg1,2-二硬酯酰-sn-甘油-3-磷酰乙醇胺-n-[甲氧基(聚乙二醇)-2000](dspe-peg2000)的1ml四氢呋喃(thf)溶液倒入10ml去离子水中。使用输出功率为12w的微探针超声波仪(xl2000,misonixincorporated,纽约)将混合物超声处理2分钟。通过在室温下在通风橱中剧烈搅拌悬浮液以蒸发残留的thf溶剂过夜,得到胶体溶液并直接使用。图8a描绘了使用纳米沉淀法制备有机小分子纳米粒子的示意图。图8b描绘了在thf溶液中的tpa-t-tq和在水中的封装的onp的uv-vis-nir吸收光谱。插图显示了(i)tpa-t-tqthf溶液和(ii)所制备的在水中的onp的照片。图8c描绘了在thf溶液中的tpa-t-tq的光致发光(pl)光谱和在水中的封装的onp的光致发光(pl)光谱。图8d描绘了tpa-t-tqonp的tem图像。图8e描绘了tpa-t-tqonp的dls结果。用乙酸双氧铀对纳米粒子进行负染色。
实施例3
光稳定性和rons稳定性研究
关于光稳定性研究,用808nm激光(0.8w/cm2)照射tpa-t-tqonp和icg的pbs溶液(ph7.4),并在不同时间点测定吸收光谱。对于抗光漂白研究,在五个加热和冷却循环期间记录样品溶液的温度。在一个加热-冷却循环中,首先用nir激光照射样品5分钟以达到稳定状态,然后移除激光并在6分钟内将样品自然冷却至环境温度。
关于rons稳定性研究,使用两种rons,即onoo-和·oh,根据文献进行制备。在加入0.4mmrons之前和之后记录吸收谱和光致发光光谱。造影剂在热稳定性和光热稳定性以及光漂白抗性和rons抗性方面的稳定性对于体内pai/ptt应用至关重要,这是因为如果造影剂的结构被破坏,则会导致不正确的信号或误导信号、治疗效果减弱和有害的副作用,特别是在活体系统中。首先进行热重分析(tga)以测定tpa-t-tq的热稳定性。tpa-t-tq的5%重量损失的分解温度高于400℃(图7),表明具有优异的热稳定性。然后在连续的808nm激光照射(0.8w/cm2)下,通过记录激光照射不同时间后的表观颜色和吸收光谱,从而分析tpa-t-tqonp和icg的光稳定性。图9a描绘了在808nm激光照射(0.8w/cm2)不同时间后,onp和icg在pbs溶液(100μm)中的ir热图像。图9b描绘了显示在808nm光照射下(功率强度为0.8w/cm2),tpa-t-tqonp在不同浓度(5μm-100μm)下的光热转化行为。图9c描绘了比较相同浓度(100μm)的tpa-t-tqonp和icg在pbs溶液中的光热转化行为(808nm光(0.8w/cm2)照射5分钟)。
如图9d和9e所示,tpa-t-tqonp的颜色和吸收光谱在15分钟的nir光照射持续时间内几乎不变,而icg溶液的蓝绿色逐渐消失,并且在暴露于nir光15分钟后,最大吸收强度几乎降至零。值得注意的是,即使在连续激光照射(0.8w/cm2)一小时后,tpa-t-tqonp的光学性质也与原始状态相同。然后,通过交替的加热和冷却过程评估了tpa-t-tqonp和icg的抗光漂白性能(首先用nir激光照射样品5分钟以加热样品,然后移除激光,然后在6分钟内使样品自然冷却至环境温度)。有趣的是,在五个加热和冷却过程的循环中,onp的光热转换能力几乎不变,而在两个加热-冷却过程的循环之后,icg的温度升高显著降至原始值(δt~44℃)的约20%(δt~10℃)(图9f)。这些结果证实了与现有的有机小分子相比,tpa-t-tqonp具有优异的抗光漂白性。
与体内应用相关的另一个重要问题是对高反应性分子(例如rons)的生理稳定性。rons是一种调节生理功能所必需的信号分子,但由于存在多种疾病(包括炎症、癌症和心血管疾病)而会过量产生rons。对于癌症的诊断和治疗,使用抗rons试剂以获得可靠的成像信号和治疗功效是非常关键的。因此,在生理条件下测定tpa-t-tqonp和icg在过氧亚硝基阴离子(onoo-)和羟基自由基(·oh)这两种rons的存在下的稳定性。在rons试剂处理之前和之后,tpa-t-tqonp和icg的吸收光谱和溶液照片分别示于图9g和9h。在添加onoo-和·oh之后,icg的最大吸收强度(在780nm处)相对于原始值分别降至约60%和4%。形成鲜明对比的是,在加入每种rons后,onp的吸收光谱和溶液外观几乎没有变化(图9i)。图10描绘了在加入0.4mmonoo-和·oh之前和加入之后1分钟时,icg在水中的光致发光(pl)光谱。
实施例4
光热性能
将不同浓度的tpa-t-tqonp和icg的pbs溶液(ph7.4)连续暴露于808nm的nir激光(功率强度为0.8w/cm2)5分钟。每20秒测量一次温度,并在温度接近平台期时停止。还记录了相应的ir热图像。为了评价光热转换性能,我们定量测量了不同浓度的tpa-t-tqonp溶液的温度变化与808nm激光(0.8w/cm2)照射时间之间的关系。如图9b所示,开始温度升高非常快,并且在激光照射3分钟后达到平台期。值得注意的是,平台期的最终温度取决于onp浓度。比较暴露于808nm激光照射时onp溶液和icg溶液的温度升高。如图9c所示,tpa-t-tqonp(5分钟内δt~53℃)表现出比icg(5分钟内δt~43℃)高得多的平台温度和更快的升温速率,因此揭示了onp的优异光热转换性能。可以从红外(ir)热图像直观地看到两个样品在不同照射时间的温度升高差异(图9a)。
实施例5
动物模型
为了建立异种移植4t1荷瘤小鼠模型,将悬浮在50μl的rpmi-1640培养基中的小鼠4t1乳腺癌细胞(1×106)皮下注射到每只小鼠的右腋窝中。约7天后,使用肿瘤体积为约80mm3-120mm3的小鼠。
实施例6
体内光声成像
注射onp后在指定的时间间隔获取770nm处的pa图像。研究通过将tpa-t-tqonp静脉内注射到异种移植4t1荷瘤裸鼠的体内而获得的体内pai。注射onp之前(0小时),在770nm处存在弱的pa信号,这可能归因于nir光谱区内源性黑色素和血红蛋白的吸收。图11a描绘了pbs溶液中的tpa-t-tqonp的pa谱图(基于tpa-t-tq为110μg/ml)。图11b描绘了tpa-t-tqonp在770nm处的pa强度与tpa-t-tq浓度的关系。
与0小时时肿瘤的pa图像相比,静脉注射tpa-t-tqonp后肿瘤部位的pa亮度随时间显著增加,在注射后6小时达到最大值(图11c),表明注射后6小时是肿瘤的pa成像和ptt治疗的最佳时间点。图11d是肿瘤的时间依赖性pa图像。6小时的pa信号是肿瘤背景的pa信号的2.4倍,表明onp具有显著的高通透性和滞留(epr)效应,导致onp在肿瘤组织中的有效积累。
实施例7
体内光热治疗
将异种移植4t1荷瘤小鼠随机分成4组(每组n=6),分别命名为“仅生理盐水”、“生理盐水+激光”、“仅onp”和“onp+激光”。对于“仅生理盐水”组和“仅onp”组,分别通过尾静脉注射生理盐水和tpa-t-tqonp(基于tpa-t-tq为250μg/ml)到4t1荷瘤小鼠中,没有随后的激光照射。对于“生理盐水+激光”组和“onp+激光”组,在分别静脉注射生理盐水和tpa-t-tqonp(基于tpa-t-tq为250μg/ml)6小时后,用808nm激光(0.5mw/cm2)连续照射各组中小鼠的肿瘤5分钟。在各种治疗后,每隔一天测量肿瘤体积和小鼠体重,持续16天。通过下式计算肿瘤体积:体积=宽度2×长度/2。
用异种移植4t1肿瘤小鼠模型验证onp的ptt能力。将荷瘤小鼠随机分为4组,分别命名为“仅生理盐水”、“生理盐水+激光”、“仅onp”和“onp+激光”。对于“仅生理盐水”组和“仅onp”组,分别通过尾静脉注射生理盐水和tpa-t-tqonp(基于tpa-t-tq为250μg/ml)到4t1荷瘤小鼠中,没有随后的激光照射。对于“生理盐水+激光”组和“onp+激光”组,分别静脉注射生理盐水和tpa-t-tqonp(基于tpa-t-tq为250μg/ml)6小时后,用808nm激光(0.5mw/cm2)连续照射各组中小鼠的肿瘤5分钟。首先,为了验证tpa-t-tqonp能够在活小鼠中通过激光照射产生热量,通过红外热成像监测“onp+激光”治疗的小鼠和“生理盐水+激光”治疗的小鼠在不同激光照射时间刻度下的肿瘤温度。
如图12a和12b所示,“生理盐水+激光”组中的小鼠的肿瘤在nir光照射5分钟时表现出微弱的温度升高(δt~2.5℃),这意味着单独的激光照射对热引发的肿瘤抑制效果可以忽略。相比之下,“onp+激光”治疗的小鼠的肿瘤中观察到快速的温度升高,在光暴露3分钟时,肿瘤温度从36℃升高至约64℃的平台温度。这种体内温度升高速率和升高的温度与目前可用的有机光热剂所达到的最佳光热转换性能相当。这些结果表明tpa-t-tqonp可以在nir光照射下引发肿瘤部位的快速升温,这表明其是一种活体肿瘤ptt的有效试剂。
通过监测肿瘤体积16天从而研究单独的ptt的“onp+激光”的体内抗肿瘤功效。如图12c所示,与对照组(“仅生理盐水”)相比,“生理盐水+激光”组的治疗完全无法抑制肿瘤生长,表明单独的808nm激光照射不具有抗肿瘤作用。此外,“仅onp”组中小鼠的肿瘤生长动力学也与“仅生理盐水”组类似,表明onp自身具有可忽略不计的抗癌活性行为。令人惊讶的是,与其他三组快速增长的肿瘤体积相比,“onp+激光”组显示出惊人的抗肿瘤功效。“onp+激光”组第16天的平均肿瘤体积甚至略小于第0天,表明tpa-t-tqonp的ptt能够使肿瘤生长停止,这对于肿瘤抑制是非常有效的。在16天的研究期间,每隔一天对各治疗组中的小鼠进行称重。如图12d所示,与对照组相比,“仅onp”组、“生理盐水+激光”组和“onp+激光”组的小鼠中未观察到明显的体重减轻,表明“onp+激光”治疗的毒副作用比较低。
实施例8
组织学研究
光热治疗16天后,处死上述四组小鼠,切除肿瘤和重要的器官,切片并染色。根据常规免疫组织化学步骤进行荧光pcna染色。根据deadend荧光tunel系统试剂盒(promega,美国)的使用说明进行荧光tunel染色。对于苏木精和曙红(h&e)染色,将小鼠的组织固定在4%福尔马林中,加工至石蜡中,并以5μm厚度切片。用数字显微镜(leicaqwin)检查切片(图13a)。为了研究tpa-t-tqonp是否引起体内毒性,在结束时间点也将各治疗组中的小鼠的肝脏和脾脏切除并切片,并进行h&e染色,这是因为通常认为纳米材料倾向于富集在网状内皮系统(res)器官(包括肝脏和脾脏)中。在所有小鼠治疗组的肝脏和脾脏器官中,未发现明显的组织损伤和/或炎性病变(图13b)。
实施例9
血清生化检测和全血细胞计数
将健康的balb/c小鼠随机分成2组(每组n=3)。将150μl的tpa-t-tqonp(基于tpa-t-tq为250μg/ml)静脉内注射到一组小鼠中。对于另一组,未进行任何治疗。一周后,收集所有小鼠的血液,然后使用自动血液分析仪检测。为了进一步研究tpa-t-tqonp的潜在毒理学,静脉注射了tpa-t-tqonp(基于tpa-t-tq为250μg/ml)的健康balb/c小鼠以及未治疗的健康小鼠在注射后第7天接受了血清生化检测和全血细胞计数。包括丙氨酸氨基转移酶(alt)、天冬氨酸转氨酶(ast)、白蛋白(alb)、总胆红素(tbil)、碱性磷酸酶(alp)和γ-球蛋白转移酶(ggt)在内的肝功能指标的测量结果均为正常的(图14a),并且未发现“onp+激光”治疗小鼠的明显肝肾疾病。全血检查(panel)的检测(包括白细胞(wbc)、淋巴细胞(lym)、血细胞比容(hct)、血红蛋白(hgb)、红细胞(rbc)、红细胞分布宽度(rdw)、红细胞血红蛋白浓度(chc)、血小板(plt)以及平均血小板体积(mpv))表明,在onp组和未治疗组之间这些指标没有统计学差异(图14a-14b)。此外,考虑到tpa-t-tqonp对小鼠体重的影响可以忽略(图12d)和重要器官都很健康(图13b),可以合理地推断tpa-t-tqonp是具有高度生物相容性的光诊断治疗纳米试剂,其对活小鼠没有明显的副作用。
以上描述了本发明的主题,显而易见的是可以通过许多方式修改或改变本发明的主题。不应将这些修改和变化视为脱离本发明主题的精神和范围,并且所有这些修改和变化旨在包括在所附权利要求的范围内。
- 还没有人留言评论。精彩留言会获得点赞!