用于预防和治疗神经退行性疾病和疼痛的化合物的制作方法
本发明涉及用于预防和/或治疗神经退行性疾病或疼痛或二者的化合物。
发明背景
美国专利公开号US20120295863公开了用于预防和治疗神经退行性疾病的、靶向腺苷A2A受体和腺苷转运体的双重作用化合物。一种命名为CGS21680(缩写CGS)的选择性A2A腺苷受体(A2AR)激动剂,已表明在转基因小鼠模型中能够减轻亨廷顿氏舞蹈病(HD)症状,并通过提高泛素-蛋白酶系统的活性来拯救HD疾病的尿素循环缺陷(Chiang等.,2009Hum Mol Genet.18:2929-2942;Chou等.,2005J Neurochem.93:310-320)。然而,已知CGS具有强的免疫抑制作用和其它副作用,因此不适合临床使用。
N6-(4-羟基苄基)腺苷,在US20120295863中命名为T1-11并也是一种A2AR激动剂,已表明在治疗神经退行性疾病上具有治疗潜力。然而由于其生物利用度差(F<5%),因此T1-11开发为一种可口服的药物仍然困难。口服生物利用度是药物开发中的一个重要性质,因为它代表物质被吸收和代谢后,到达系统循环的百分比。
发明概要
一方面,本发明涉及式(I)或(IA)化合物:
或其药学上可接受盐,其中X为卤素。
卤素(F,Cl,Br或I)在式(I)中可位于3-、4-或5-位,或在式(IA)中位于2-、4-或5-位。
在本发明的一个实施例中,化合物选自下组:N6-[(3-卤代噻吩-2-基)甲基]腺苷、N6-[(4-卤代噻吩-2-基)甲基]腺苷、和N6-[(5-卤代噻吩-2-基)甲基]腺苷。
在本发明的另一个实施例中,化合物选自下组:N6-[(2-卤代噻吩-3-基)甲基]腺苷、N6-[(4-卤代噻吩-3-基)甲基]腺苷、和N6-[(5-卤代噻吩-3-基)甲基]腺苷。
进一步在本发明另一个实施例中,化合物选自下组:N6-[(5-溴噻吩-2-基)甲基]腺苷、N6-[(4-溴噻吩-2-基)甲基]腺苷、N6-[(3-溴噻吩-2-基)甲基]腺苷、N6-[(5-氯噻吩-2-基)甲基]腺苷、N6-[(4-氯噻吩-2-基)甲基]腺苷、和N6-[(3-氯噻吩-2-基)甲基]腺苷。
在本发明的另一个实施例中,化合物选自下组:N6-[(2-溴噻吩-3-基)甲基]腺苷、N6-[(4-溴噻吩-3-基)甲基]腺苷、N6-[(5-溴噻吩-3-基)甲基]腺苷N6-[(2-氯噻吩-3-基)甲基]腺苷、N6-[(4-氯噻吩-3-基)甲基]腺苷,和N6-[(5-氯噻吩-3-基)甲基]腺苷。
在另一方面,本发明涉及一种制备如上所提到的式(I)或式(IA)化合物的方法,包括步骤(I)或(II):
(I):
(a)在碱存在下,将6-氯嘌呤呋喃核糖苷与式3或3A所示的氟、氯、溴或碘取代的(噻吩基)甲胺进行反应,
从而得到式(I)或(IA)化合物;或
(II):
(a1)在碱存在下,将(2',3'-O-亚异丙基)腺苷与羟基保护试剂进行反应,从而得到具有羟基保护基的(2',3'-O-亚异丙基)腺苷衍生物;
(b)将具有羟基保护基的(2',3'-O-亚异丙基)腺苷衍生物与胺基保护试剂进行反应,从而得到具有羟基保护基和胺基保护基的(2',3'-O-亚异丙基)腺苷衍生物;
(c)将具有羟基保护基和胺基保护基的(2',3'-O-亚异丙基)腺苷衍生物与式7或7A所示的含取代的(噻吩基)甲基的化合物进行偶联反应:
其中X为F、Cl、Br、或I,和Y为X、OH、甲磺酸基(OSO2CH3、OMs)、对甲苯磺酸基(OSO2C6H4-p-CH3,OTs)、或三氟甲磺酸基(OSO2CF3,OTf),
从而得到包含保护基和取代的(噻吩基)甲基基团的产物;和
(d)在酸性条件下从步骤(c)的产物中除去保护基,从而得到式(I)或(IA)化合物。
在本发明的一个实施例中,步骤(a)中的碱可以是二异丙基乙胺。羟基保护试剂可以是叔丁基二甲基氯硅烷。胺基保护试剂可以是二碳酸二叔丁酯。步骤(a1)中的碱可以是咪唑。
在本发明的另一个实施例中,步骤(I)(a)中,碱为二异丙基乙胺,而取代的(噻吩基)甲胺为:(i)(5-溴噻吩-2-基)甲胺,从而得到N6-[(5-溴噻吩-2-基)甲基]腺苷;或(ii)(5-氯噻吩-2-基)甲胺,从而得到N6-[(5-氯噻吩-2-基)甲基]腺苷。
进一步在本发明的另一个实施例中,偶联反应在三苯基膦和偶氮二甲酸二异丙酯存在下进行。含取代的(噻吩基)甲基基团的化合物可以是(5-溴噻吩-2-基)甲醇。
进一步在另一方面,本发明涉及一种组合物,它包括:
(a)治疗有效量的如前述的化合物,或其药学上可接受的盐;和
(b)药学上可接受的载体、赋形剂或运载体。
在另一方面,本发明涉及如前所述化合物在制备药物中的应用,所述药物用于治疗有此需要的对象中的神经退行性疾病和/或疼痛。或者,本发明涉及如前所述化合物在治疗有此需要的对象中的神经退行性疾病和/或疼痛的应用。
在本发明的一个实施例中,神经退行性疾病选自下组:阿尔茨海默病、帕金森病、肌萎缩侧索硬化、朊病毒病、亨廷顿氏舞蹈病和脊髓小脑性共济失调。脊髓小脑性共济失调可选自下组:脊髓小脑性共济失调2型、脊髓小脑性共济失调3型和脊髓小脑性共济失调7型。疼痛可以是酸诱发的疼痛。酸诱发的疼痛可以是酸诱发的肌肉疼痛。酸诱发的肌肉疼痛可以是酸诱发的慢性肌肉疼痛。
在本发明的一个实施例中,疼痛选自下组:炎症性疼痛、癌疼痛、胸痛、背痛、颈部疼痛、肩痛、偏头痛、头痛、肌筋膜疼痛、关节疼痛、肌肉疼痛综合症、神经病理性疼痛、末梢性神经疼痛、交感神经痛、术后疼痛、创伤后疼痛和多发性硬化症疼痛。
在本发明的另一个实施例中,疼痛可以是功能失调性疼痛。功能失调性疼痛可选自下组:纤维肌痛、肌筋膜疼痛、膀胱疼痛综合症,和由肠易激综合症所致的疼痛。
基于以下附图和优选实施例的以下描述的结合中,可以明显看出这些方面以及其他方面,尽管可以在不脱离本发明公开内容的新颖概念的精神和范围的情况下,进行各种变化和改动。
所附附图用于阐述本发明的一个或多个实施例,并与书面描述一起解释本发明的原理。只要有可能,相同的引用编号在整个附图中指实施例中相同或相似的元素。
附图的简要说明
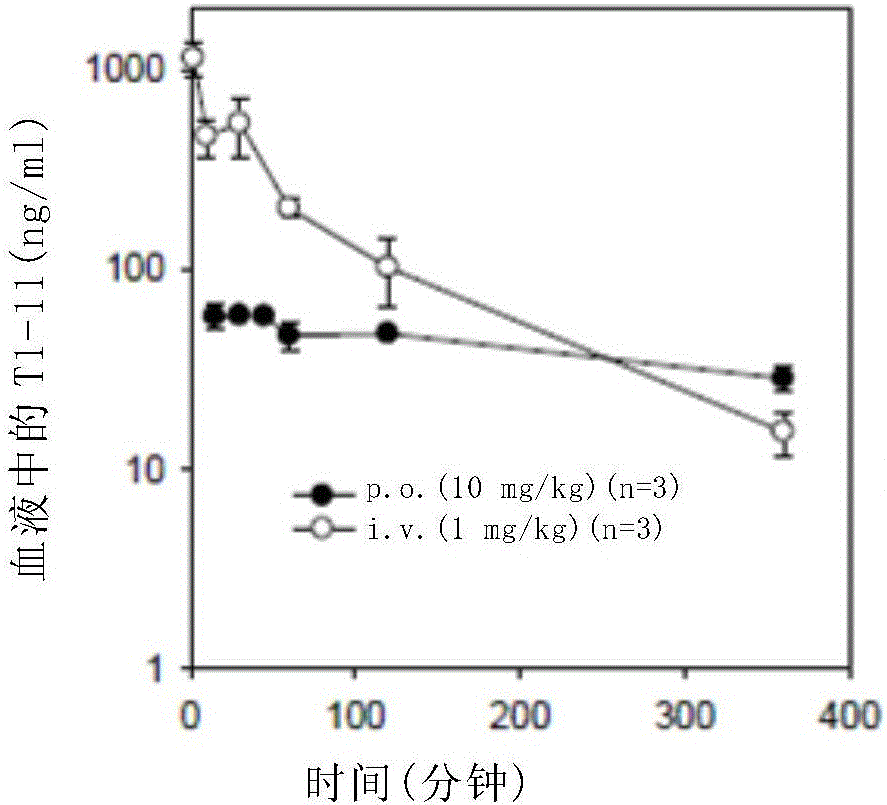
图1显示了接受T1-11或JMF3464静脉注射(1mg/kg)或口服(10mg/kg)的ICR小鼠中,T1-11(A)和JMF3464(B)在血液中的浓度。T1-11和JMF3464的口服生物利用度分别估计为2.8%和17.4%。
图2显示了A2AR激动剂(CGS21680、T1-11、JMF3464和JMF3818)对去血清诱导的细胞死亡的效果。去血清PC12细胞用或不用所指定试剂处理24小时。与含血清的对照组的平均值相比,以来自MTT试验结果的百分数来表示细胞活力。数据点表示为平均值±SEM(n=3~6)。
图3A-B显示了JMF3464对运动障碍和寿命的影响。使用ALZET渗透微泵对指定的7周龄小鼠皮下给药JMF3464(0.11μg/鼠/天),JMF1907(0.11μg/鼠/天)或运载体(对照,CON)6周。对这些小鼠的转动棒行为(A)和寿命(B)进行评估。*p<0.05;***p<0.005。
图4A-B显示了T1-11在预防突变型SCA2基因过表达诱导的蛋白集聚和SCA2转基因小鼠的行为表现方面的效果。(A)pSCA2-22Q-EGFP或pSCA2-104Q-EGFP-转染的细胞经10μM T1-11或10μM JMF1907(一种T1-11-衍生的A2A-R激动剂)处理24小时。收获细胞并进行过滤阻滞试验或图像采集。(B)野生型(WT)或转基因小鼠(SCA2)喝含有或不含有T1-11的水。用转动棒行为来测量小鼠的行为功能。
图5A-C显示了T1-11改善SCA3转基因小鼠的运动功能障碍和脑桥神经元死亡。(A)在4周龄开始给SCA3转基因小鼠饮用含运载体(0.2%DMSO)或T1-11(0.1mg/ml)的水。转动棒测试表明,相比于野生型(WT)小鼠,运载体(vehicle)处理的4月龄的共济失调蛋白(ataxin)-3-Q79转基因小鼠表现出明显更短的落下延迟和动作不协调性。T1-11处理的4月龄SCA3转基因小鼠(每天1mg)的转动棒行为明显优于运载体处理的相同年龄的共济失调蛋白-3-Q79小鼠。每个点显示了7-8只小鼠的平均值+S.E.。(B-C)神经元标记物NeuN的免疫组化染色表明,T1-11的每日口服给药(每天1mg)显著改善了4月龄的SCA3转基因小鼠的脑桥核中的神经元死亡(SCA3+T1-11)。比例尺为50μm。每条柱状显示7-8只小鼠的平均值+S.E.。与SAC3转基因小鼠相比*P<0.01。
图6A-C显示了JMF1907缓解SCA3转基因小鼠的共济失调症状和脑桥神经元死亡。(A)转动棒试验表明,相比于运载体处理的4月龄SCA3转基因小鼠,JMF1907处理的4月龄SCA3小鼠(每天1mg)的转动棒行为显著改善。每个点显示6只小鼠的平均值+S.E.。(B-C)NeuN免疫细胞化学染色显示,JMF1907口服给药(每天1mg)明显预防了4月龄SCA3小鼠脑桥核中的神经元死亡(SCA3+JMF1907)。比例尺为50μm。每条柱状显示6只小鼠的平均值+S.E.。与SAC3小鼠相比,*P<0.01。
图7A-C显示了JMF3464改善SCA3转基因小鼠的共济失调和脑桥神经元死亡。(A)SCA3转基因小鼠表现出受损的转动棒行为。每日给药JMF3464(每天0.3mg)大大改善了4月龄SCA3小鼠的转动棒行为。(B-C)神经元标记物NeuN的免疫细胞化学染色表明,每日口服JMF3464(每天0.3mg)治疗显著改善4月龄SCA3转基因小鼠的脑桥核中的神经元死亡。每条柱状显示6只小鼠的平均值+S.E.。相比SAC3转基因小鼠,#P<0.01。
图8A-B显示了经JMF1907处理,改善了TDP-43转基因小鼠的运动功能。测试了4种不同剂量的JMF1907(3.7,1.25,0.5,0.1mg/kg)。对照CTL:经DMSO处理的转基因小鼠。WT:经DMSO处理的野生型小鼠。采用两因素ANOVA进行统计。对于转动棒,1.5和0.5mg/kg的所有数据点,0.1mg/kg的12-21周和3.7mg/kg的10-12周的数据点,达到统计学显著性。对于握力,对于所有测试剂量的所有数据点均达到统计学显著性。N=18(CTL)、15(WT)、15(0.1)、15(0.5)、15(1.25)、5(3.7)。
图9显示了经JMF3464处理降低了NSC34细胞中TDP-43错误定位。细胞经JMF3464(30μM)预处理1小时,然后在JMF3464存在下用AICAR(1mM,Al)再处理24小时。通过免疫染色对TDP-43(红色)定位进行分析。细胞核位置由Hochest(蓝)标记。
图10A-C显示了JMF3464对小鼠纤维肌痛模型的镇痛效果。(A-1和A-2)T1-11口服给药显示对小鼠纤维肌痛模型没有镇痛效果,其中肌内酸注射和染料木黄酮处理后,小鼠发展为慢性肌肉疼痛。空心箭头表示小鼠接受酸注射的时间。黑色箭头表示小鼠接受Tl-11(口服)的时间。(B)JMF3464显示了对小鼠纤维肌痛模型的镇痛效果,其中在经间歇冷应激处理2天后,小鼠发展为慢性弥漫性疼痛。JMF3464的镇痛效果是剂量依赖性的。有效剂量起始于100μg/kg(i.p.)。(C)JMF3464口服给药(1mg/kg)显示出对间歇冷应激模型的镇痛效果。
发明详细说明
除非另有定义,本文使用的所有技术和科学术语具有相同的含义,该含义是本发明涉及领域的普通技术人员通常理解的。在冲突的情况下,本文(包括其定义)优先。
术语“治疗(treating)”或“治疗(treatment)”是指对有此需要的对象施用有效量的治疗试剂,目的是治愈、减轻、缓解、补救、改善,或预防所述疾病和/或疼痛,所述疾病和/或疼痛的症状,或所述疾病和/或疼痛的倾向,而该对象具有神经退行性疾病和/或疼痛,或有这种疾病和/或疼痛的症状或倾向。这样的对象可通过健康护理专业人士依据任何合适的诊断方法的结果加以确定。
“有效量”是指赋予治疗对象疗效所需要的活性化合物的量。如那些本领域技术人员所知晓,有效剂量可有所不同,这取决于施用途径、赋形剂的使用,和与其他治疗处理共同使用的可能性等。
美国食品与药品管理局健康与人类服务部出版的“评估成人健康志愿者治疗的临床试验的安全起始剂量的行业和评审人指南”公开,一种“治疗有效量”可由下面的公式计算而获得:
HED=动物剂量mg/kg×(动物体重kg/人体体重kg)0.33。
JMF 1907表示化合物N6-[2-(吲哚-3-基)乙基]腺苷,其作为化合物6披露在美国专利公开号No.20120295863A1中。
化学合成
在一种方法中,在碱存在下,将6-氯嘌呤呋喃核糖苷(2)用任选取代的噻吩甲胺(3)进行处理,得到所需的式(I)化合物。
例如,二异丙基乙胺被用作碱,在溶剂1-丙醇中,通过加热,进行6-氯嘌呤呋喃核糖苷与(5-溴噻吩-2-基)甲胺的取代反应,从而得到N6-[(5-溴噻吩-2-基)甲基]腺苷(JMF3464,结构式1)。
方案1
方案2
在另一种方法中,在碱咪唑存在下,(2',3'-O-异亚丙基)腺嘌呤(4)用叔丁基二甲基氯硅烷(TBDMSCl)处理,得到硅醚衍生物(5)。6-氨基基团被保护为带有叔丁氧基羰基(Boc)取代基的氨基甲酸酯6。引入任选取代的噻吩甲基,并在酸性条件下除去所有保护基后,得到所需式(I)化合物。
例如,使用三苯基膦和偶氮二甲酸二异丙酯(DIAD),来促进化合物6和(5-溴噻吩-2-基)甲醇(化合物7,其中X=5-溴,Y=OH)的偶联反应,从而得到化合物8。在酸性条件下对化合物8中的甲硅烷基、丙酮基和Boc基团全面去保护,从而得到化合物1。
使用相同化学方法,得到化合物(IA)。
方案3
口服生物利用度
为测量受试化合物的口服生物利用度,在口服(10mg/kg)或静脉内(1mg/kg)给予受试化合物后,从雄性ICR小鼠(6周龄;20-25g)采集血液样本。在指定时间采集的血液样本用含0.1%甲酸的甲醇提取,然后将每种提取样本10μL注入UPLC-MSMS进行定量。结果(图1A-B)表明,ICR小鼠中T1-11和JMF34964的口服生物利用度分别为2.8%和17.4%,这表明JMF3464口服吸收比T1-11高6倍多。
JMF3464结合于A2AR和ENT1以及保护神经元细胞凋亡
我们首先用放射性配体结合试验,对JMF3464药理性质进行表征。表1显示了T1-11、JMF1907和JMF3464的药理性质。使用标准结合方案,对指定化合物和A2A腺苷受体(A2AR)和腺苷转运体(ENT1)的结合进行表征。如表1所示,JMF3464结合于A2AR和腺苷转运体(平衡型核苷转运体1)。JMF3464对A2AR的亲和力与T1-11和JMF1907(T1-11类似物)的亲和力相似,然而它对ENT1的亲和力比T1-11和JMF1907的亲和力好很多。我们的研究还表明,N6-[(5-氯噻吩-2-基)甲基]腺苷(JMF3818)也抑制因血清去除所引起的PC12细胞凋亡。10μM的JMF3818分别抑制54%A2A受体和96%的腺苷转运体(ENT1)(图2)。
表1
JMF3464在HD转基因小鼠模型中对亨廷顿氏舞蹈病(HD)主要症状产生有益的效果
由于A2AR和ENT1位于纹状体中并与纹状体功能有关,因此我们假设用JMF3464进行长期治疗会调节HD的进展。我们测试了JMF3464在HD转基因小鼠模型(R6/2)中的效果,在该模型中A2AR激动剂具有有益效果。如转动棒行为(Rotarod performance)所评价的那样,对小鼠从7周大起添加JMF3464(0.11mg/kg/天)抵消了运动协调性中进行性恶化(图3A)。通过持续(长期)的JMF3464治疗,被缩短的R6/2小鼠的寿命也有提高。
JMF3464对脊髓小脑性共济失调2型(SCA2)的有益效果
由于活化的蛋白酶体活性可能是A2AR受体信号通路在预防突变型Htt集聚或R6/2转基因小鼠中行为表现的机制(Huang等人,2011PLoS One.6:e20934;Lee等人,2012PLoS One.7:e38865),因此A2AR激动剂可能在减轻其他多聚谷氨酰胺疾病(如SCA2)上也具有益处。事实上,我们的数据显示,T1-11在预防突变ATXN2集聚(图4A)和SCA2转基因小鼠(ATAXN2Q127)的行为表现(图4B)是有效的,这支持了上述假说。考虑到JMF3464的生物利用度明显优于T1-11,我们推断JMF3464也会对SCA2产生有益的效果。
T1-11及其类似物对脊髓小脑性共济失调3型(SCA3)的有益效果
与野生型小鼠相比,用运载体处理的表达多聚谷氨酰胺-扩展型(expanded)共济失调蛋白-3-Q79的SCA3转基因小鼠,表现出各种共济失调症状,包括受损的转动棒行为(rotarod performance)(图5A和6A),发病年龄约为3-4个月。如图5A和6A所示,4月龄SCA3转基因小鼠经每日口服给药T1-11(每天1mg)或JMF1907(每天1mg)治疗,表现出显著改善的转动棒行为。与SCA3患病动物相似,在SCA3转基因小鼠的脑桥核中发现明显的神经元死亡(图5B和6B)。与转动棒实验结果一致,每日T1-11(图5B和5C)或JMF1907(图6B和图6C)的口服治疗,明显缓解了SCA3转基因小鼠中脑桥神经元死亡(图5B,5C,6B和图6C)。这些结果提供了证据,即持续的T1-11或JMF1907口服治疗改善SCA3转基因小鼠的神经和神经病理学表型。
JMF3464具有更高的生物利用度。因此,JMF3464也被期望对SCA3转基因小鼠中突变型共济失调蛋白-3-Q79诱导的共济失调和神经退行性病变具有有益的效果。果然,每日使用JMF3464(每天0.3mg)缓解了SCA3转基因小鼠的共济失调(图7A)和脑桥神经元死亡(图7B和7C)。
JMF1907和JMF3464对肌萎缩侧索硬化(ALS)的有益效果
如图8A-B所示,被给予4个不同剂量(3.7,1.25,0.5和0.1mg/kg/天)JMF1907的转基因小鼠,明显比对照组小鼠在转动棒和握力测试中表现得更好。1.25mg/kg剂量表现出最大的益处。这些数据表现出明确的运动功能的改善。JMF3464(一种T1-11和JMF1907的类似物)具有更高的生物利用度。因此,JMF3464被期望在ALS小鼠中发挥有益的效果。用JMF3464对NSC34细胞进行处理,可使得由AMPK活化引起的TDP-43错误定位变得正常(图9),这将支持以下观点:JMF3464能够预防ALS发病的起始阶段。这些发现支持:JMF3464对ALS表现出有益效果。
JMF3464对疼痛的有益效果
尽管T1-11(腹腔内)对2种纤维肌痛小鼠模型(酸诱导的慢性弥漫性疼痛和间歇冷应激模型)表现出优异的镇痛效果,但其生物利用度非常低。T1-11口服给药(高达2mg/kg)对酸诱导慢性弥漫性疼痛模型没有表现出镇痛效果,在该模型中,在肌内酸注射和染料木黄酮处理后,小鼠发展为纤维肌痛样疼痛(图10A)。JMF3464(一种T1-11类似物)对纤维肌痛小鼠模型表现出镇痛效果,在该模型中,在经间歇冷应激处理2天后,小鼠发展为慢性弥漫性疼痛(图10B)。与其良好的生物利用度一致,JMF3464(1mg/kg)口服给药对间歇冷应激模型表现出镇痛效果(图10C)。
实施例1
所有试剂和溶剂为试剂级,并无需进一步纯化而使用,除非另外说明。四氢呋喃和乙醚从钠/二苯甲酮中蒸馏,CH2C12从CaH2中蒸馏。所有空气或湿气敏感的实验在氩气下操作。所有玻璃容器在烘箱干燥超过2小时并在干燥器中冷却至室温后使用。使用聚焦型单模微波装置(CEM Discover)进行微波反应。该机器由连续聚焦的微波功率输送系统组成,并且该系统具有操作者可选的功率输出。
熔点记录在Yanaco微型仪器中。旋光度在日本JASCO Co.DIP-1000数字旋光仪中测定。[α]D值以10–1度cm2g–1为单位。红外(IR)光谱用Nicolet Magna 550-11记录。NMR谱由Varian Unity Plus-400(400MHz)获得,化学位移(δ)以相对于CHCl3/CDCl3的δΗ7.24/δC77.0(t中心线),相对于(CH3)2CO/(CD3)2CO的δΗ2.05/δC 29.92,相对于CH3OH/CD3OD的δH3.31/δC49.0,和相对于(CH3)2SO/(CD3)2SO的δH 2.49(m)/δC 39.5(m)的每百万部分(ppm)(parts per million)记录。分裂模式被报告为s(单峰)、d(双峰)、t(三重峰)、q(四重峰)、m(多重峰)和br(宽峰)。耦合常数(J)以Hz给出。ESI-MS实验在Bruker Daltonics BioTOF III高分辨率质谱仪上进行。分析薄层色谱法(TLC)在E.Merck硅胶60F254板(0.25mm)上进行。化合物通过UV、茴香醛或茚三酮喷雾显示。在70-230目硅胶柱上进行柱层析。
通过HPLC(Agilent HP-1 100)在280nm波长检测,化合物的纯度为评估为≥95%。
2',3'-O-亚异丙基-5'-O-(叔丁基二甲基硅基)腺苷(5)
向在冰浴下冷却的2',3'-(O-亚异丙基)腺苷(4,614mg,2.00mmol)和咪唑(408mg,6mmol)的无水CH2Cl2溶液(12mL)中,在氮气氛下和0℃,添加叔丁基二甲基氯硅烷(TBDMSCl,452mg,3mmol)。移除冰浴,混合物在室温下搅拌12小时。加入甲醇(4mL),混合物再搅拌15分钟,然后旋转蒸发减压浓缩。固体残留物溶解在CH2Cl2,并依次用1M HCl,去离子水和盐水洗涤。收集有机层,MgSO4干燥并过滤。减压下浓缩滤液,得到化合物5(819mg,1.57mmol,78%收率),为白色固体。
6-N-叔丁氧羰基-2',3'-O-亚异丙基-5'-O-(叔丁基二甲基硅基)腺苷(6)
化合物5(819mg,1.57mmol)和4-(二甲基氨基)吡啶(DMAP,催化量)的无水THF溶液(10mL)在氮气下搅拌2分钟。将溶液在冰浴下冷却,滴加二碳酸二叔丁酯((Boc)2O,1.08mL,4.71mmol)。移除冰浴,混合物室温搅拌12小时。反应完成后,通过旋转蒸发减压浓缩混合物。粗品溶解在CH2Cl2中,依次用1M HCl、去离子水和盐水洗涤。收集有机层,MgSO4干燥,过滤。减压下浓缩滤液,得到淡黄色泡沫状的双-Boc化合物(859mg,1.38mmol,87%收率)。
向双-Boc化合物(400mg,0.64mmol)的甲醇溶液(8mL)中加入甲胺(0.25mL的40%的甲醇溶液,2.54mmol)。混合物室温搅拌20小时直到TLC分析表明起始原料完全消耗。通过旋转蒸发减压浓缩混合物。粗品在硅胶柱上并用EtOAc/正己烷(0:1到2:1梯度)洗脱液进行层析纯化,得到淡黄色油状的化合物6(300mg,0.57mmol,88%收率)。
6-N-(5-溴噻吩-2-基)甲基-6-N-叔丁氧羰基-2',3'-O-亚异丙基-5'-O-(叔丁基二甲基硅基)腺苷(8,X=5-溴)
化合物6(300mg,0.57mmol),(5-溴噻吩-2-基)甲醇(164mg,0.85mmol)和三苯基膦(226mg,0.85mmol)的无水THF溶液(9mL),在氮气氛下,于45℃搅拌2分钟。滴加偶氮二甲酸二异丙酯(DIAD,0.168mL,0.85mmol)。混合物再搅拌45分钟直到TLC分析显示化合物6消失。通过旋转蒸发减压浓缩混合物。粗品经快速柱层析法(EtOAc/正己烷=1:9)纯化,得到淡黄色油状的化合物8(X=5-溴)。
N6-[(5-溴噻吩-2-基)甲基]腺苷(JMF3464)
方法A:搅拌化合物8(X=5-溴,138mg,0.19mmol)在去离子水(5mL)和THF(1mL)中的悬浮液,并冰浴下冷却。0℃滴加三氟乙酸(5mL)。混合物搅拌10分钟,移除冰浴,混合物再搅拌30分钟。通过旋转蒸发减压浓缩混合物。粗品经快速柱层析法(MeOH/EtOAc=1:49)纯化,得到白色粉末JMF3464(化合物1)。
方法B:将(5-溴噻吩-2-基)甲胺(768mg,4mmol),6-氯嘌呤呋喃核糖苷(143mg,0.5mmol)和二异丙基乙胺(2mL,12mmol)在1-丙醇中的混合物(20mL),在70℃加热7小时。将包含所需的产物和未反应的(5-溴噻吩-2-基)甲胺的混合物,用含二碳酸二叔丁酯(0.92mL,4mmol)的THF(6mL)和NaHCO3(672mg,8mmol)在室温下处理2小时。混合物通过旋转蒸发浓缩,并经快速柱层析(硅胶,MeOH/EtOAc=1:9)纯化。所需产物JMF3464(59mg,27%收率)如果在MeOH中重结晶而获得。通过HC-C18柱(Agilent,4.6×250mm,5μm)并用50%含水MeOH梯度洗脱的HPLC显示,产物纯度为96%。
方法C:向密封管(15mL)中,加入(5-溴噻吩-2-基)甲胺(384mg,2mmol),6-氯嘌呤呋喃核糖苷(287mg,1mmol),和二异丙基乙胺(3mL,17mmol)(在乙醇(8mL)中)。密封管放置在聚焦型单模微波反应器(CEM Discover)的腔内并在80℃下100W照射20分钟。旋转蒸发移除溶剂。残留物经快速柱层析法纯化(硅胶,MeOH/EtOAc=1:9),并在MeOH中重结晶,得到所需产物JMF3464(159mg,36%收率)。
C15H16BrN5O4S;黄色粉末;mp 141.4–141.7℃;[α]25D=–43.0(DMSO,c=1.0);TLC(2-丙醇/己烷(2:3))Rf=0.38;1H NMR(DMSO-d6,400MHz)δ8.55(1H,br s),8.40(1H,s),8.29(1H,br s),7.03(1H,d,J=3.6Hz),6.78(1H,d,J=4.0Hz),5.90(1H,d,J=6.0Hz),5.45(1H,d,J=6.0Hz),5.34–5.32(1H,m),5.19(1H,d,J=4.4Hz),4.76(2H,s),4.63–4.59(1H,m),4.15–4.14(1H,m),3.96–3.95(1H,m),3.69–3.65(1H,m),3.57–3.52(1H,m),13C NMR(DMSO-d6,100MHz)δ153.9,152.2,148.5,145.0,140.2,129.6,126.3,120.0,109.7,87.9,85.8,73.5,70.6,61.6,42.9,C15H17BrN5O4S的ESI-MS计算值:442.0185,测得值m/z442.0189[M+H]+。
N6-(噻吩-3-基-甲基)腺苷(JMF3461)
将3-(氨基甲基)噻吩(0.25mL,2.5mmol)、6-氯嘌呤呋喃核糖苷(143mg,0.5mmol)和二异丙基乙胺(2mL,12mmol)在1-丙醇(25mL)中的混合物,在80℃加热6小时。旋转蒸发浓缩混合物,在MeOH中重结晶,得到所需产品JMF3461(136mg,75%收率)。通过HC-C18柱(Agilent,4.6×250mm,5μm)用50%含水MeOH梯度洗脱的HPLC显示,产物纯度为99%。C15H17N5O4S;黄色粉末;mp 134.3–135.1℃;[α]24D=–58.6(DMSO,c=1.0);TLC(2-丙醇/己烷,(2:3))Rf=0.33;1H NMR(DMSO-d6,400MHz)δ8.36(2H,br s),8.22(1H,s),7.43(1H,dd,J=3,5Hz),7.28(1H,d,J=1.6Hz),7.09(1H,d,J=4.8Hz),5.88(1H,d,J=6.4Hz),5.43(1H,d,J=6.0Hz),5.38(1H,q,J=4.6Hz),5.17(1H,d,J=4.4Hz),4.69(2H,s),4.63–4.16(1H,m),4.14–4.13(1H,m),3.97–3.95(1H,m),3.69–3.64(1H,m),3.57–3.52(1H,m),13C NMR(DMSO-d6,100MHz)δ154.4,152.3,148.5,140.8,139.9,127.9,125.1,120.0,119.8,87.9,85.9,73.5,70.7,61.7,42.9;ESI-MS计算值C15H18N5O4S:364.1080,测得值:m/z 364.1079[M+H]+。
N6-(噻吩-2-基-甲基)腺苷(JMF3462)
将2-(氨基甲基)噻吩(0.25mL,2.5mmol)、6-氯嘌呤呋喃核糖苷(143mg,0.5mmol)和二异丙基乙胺(2mL,12mmol)在1-丙醇(25mL)中的混合物,在80℃加热7小时。旋转蒸发浓缩混合物,在MeOH中重结晶,得到所需产品JMF3462(154mg,85%收率)。通过HC-C18柱(Agilent,4.6×250mm,5μm)用50%含水MeOH梯度洗脱的HPLC显示,产物纯度为99%。C15H17N5O4S;白色粉末;mp 149.2–149.7℃;[α]25D=–68.2(DMSO,c=1.0);TLC(2-丙醇/己烷,(2:3))Rf=0.35;1H NMR(DMSO-d6,400MHz)δ8.51(1H,br s),8.39(1H,s),8.27(1H,br s),7.32(1H,d,J=5.2Hz),7.28(1H,d,J=3.2Hz),6.93(1H,dd,J=1.8,2.6Hz),5.89(1H,d,J=6.0Hz),5.46–5.45(1H,m),5.36(1H,q,J=4.6Hz),5.20–5.18(1H,m),4.64(2H,s),4.16–4.13(1H,m),3.97–3.95(1H,m),3.70–3.65(1H,m),3.58–3.52(1H,m),3.57–3.52(1H,m),13C NMR(DMSO-d6,100MHz)δ154.1,152.2,148.5,142.9,140.0,126.5,125.3,124.7,120.0,87.9,85.9,73.5,70.6,61.6,42.9;ESI-MS计算值C15H18N5O4S:364.1080,测得值:m/z 364.1081[M+H]+。
N6-[(4-溴噻吩-2-基)甲基]腺苷
将(4-溴噻吩-2-基)甲胺(1152mg,6mmol)、6-氯嘌呤呋喃核糖苷(214mg,0.75mmol)和二异丙基乙胺(3mL,18mmol)在1-丙醇(30mL)中的混合物,在70℃下加热7小时。将包含所需产物和未反应的(3-溴噻吩-2-基)甲胺的混合物,用含二碳酸二叔丁酯(1.4mL,6mmol)的THF(8mL)和NaHCO3(1g,1.2mmol)在室温下处理2小时。混合物通过旋转蒸发浓缩并经快速柱层析(硅胶,MeOH/EtOAc=1:9)纯化,得到N6-[(4-溴噻吩-2-基)甲基]腺苷。
N6-[(3-溴噻吩-2-基)甲基]腺苷
向密封管(15mL)中,加入(3-溴噻吩-2-基)甲胺(576mg,3mmol)、6-氯嘌呤呋喃核糖苷(430mg,1.5mmol),和二异丙基乙胺(4.5mL,15.5mmol)在乙醇(10mL)中的混合物。密封管放置在聚焦型单模微波反应器(CEM Discover)的腔内,并在80℃下100W照射20分钟。旋转蒸发移除溶剂。残留物经快速柱层析法纯化(硅胶,MeOH/EtOAc=1:9),得到N6-[(3-溴噻吩-2-基)甲基]腺苷。
N6-[(2-溴噻吩-3-基)甲基]腺苷
向密封管(15mL)中,加入(2-溴噻吩-3-基)甲胺(384mg,2mmol)、6-氯嘌呤呋喃核糖苷(287mg,1mmol)和二异丙基乙胺(3mL,17mmol)在乙醇(8mL)中的混合物。密封管放置在聚焦型单模微波反应器(CEM Discover)的腔内,并在80℃下100W照射20分钟。旋转蒸发移除溶剂。残留物经快速柱层析法纯化(硅胶,MeOH/EtOAc=1:9),得到N6-[(2-溴噻吩-3-基)甲基]腺苷。
N6-[(4-溴噻吩-3-基)甲基]腺苷
将(4-溴噻吩-3-基)甲胺(768mg,4mmol)、6-氯嘌呤呋喃核糖苷(143mg,0.5mmol)和二异丙基乙胺(2mL,12mmol)在1-丙醇(20mL)中的混合物,在70℃下加热7小时。将包含所需产物和未反应的(4-溴噻吩-3-基)甲胺的混合物,用含二碳酸二叔丁酯(0.92mL,4mmol)的THF(6mL)和NaHCO3(672mg,8mmol)在室温下处理2小时。混合物通过旋转蒸发浓缩并经柱层析(硅胶,MeOH/EtOAc=1:9)纯化,得到N6-[(4-溴噻吩-3-基)甲基]腺苷。
6-N-(5-溴噻吩-3-基)甲基-6-N-叔丁氧羰基-2',3'-O-亚异丙基-5'-O-(叔丁基二甲基硅基)腺苷
化合物6(360mg,0.68mmol)、(5-溴噻吩-3-基)甲醇(197mg,1.02mmol)和三苯基膦(271mg,1.02mmol)的无水THF(10mL)溶液,在氮气氛下和45℃搅拌2分钟。滴加偶氮二甲酸二异丙酯(DIAD,0.20mL,1.02mmol)。搅拌混合物直到TLC分析显示化合物6消失。通过旋转蒸发减压浓缩混合物。粗品经快速柱层析法(EtOAc/正己烷=1:9)纯化,得到6-N-(5-溴噻吩-3-基)甲基-6-N-叔丁氧羰基-2',3'-O-亚异丙基-5'-O-(叔丁基二甲基硅基)腺苷。
N6-[(5-溴噻吩-3-基)甲基]腺苷
搅拌6-N-(5-溴噻吩-3-基)甲基-6-N-叔丁氧羰基-2',3'-O-亚异丙基-5'-O-(叔丁基二甲基硅基)腺苷(138mg,0.19mmol)在去离子水(5mL)和THF(1mL)中的悬浮液,并冰浴下冷却。0℃滴加三氟乙酸(5mL)。混合物搅拌10分钟,移除冰浴,混合物再搅拌30分钟。通过旋转蒸发减压浓缩混合物。粗品经快速柱层析法(MeOH/EtOAc=1:49)纯化,得到N6-[(5-溴噻吩-3-基)甲基]腺苷。
(5-氯噻吩-2-基)甲胺
将2-(氨基甲基)噻吩(1mL,10mmol)、二碳酸二叔丁酯(2.5mL,11mmol),和NaHCO3(840mg,10mmol)在THF中的混合物(13mL),在室温下搅拌3小时,得到含淡黄色固体的悬浮液。减压浓缩混合物,残留物用CH2Cl2和H2O萃取。有机相用MgSO4干燥,过滤,并用EtOAc/正己烷(1:20)洗脱液进行硅胶柱快速层析法纯化,得到(噻吩-2-基)甲基氨基甲酸叔丁酯(C10H15NO2S,1.62g,76%收率)。
将(噻吩-2-基)甲基氨基甲酸叔丁酯(106mg,0.5mmol)、N-氯代琥珀酰亚胺(73mg,0.55mmol)在苯中的混合物(0.3mL),在80℃下搅拌。2小时后,加入醋酸(0.3mL,5mmol),并进一步反应21小时。混合物用CH2Cl2和H2O萃取。有机相用MgSO4干燥,过滤,并用EtOAc/正己烷(1:20)洗脱液进行硅胶柱快速层析法纯化,得到(5-氯噻吩-2-基)甲基氨基甲酸叔丁酯(C10H14ClNO2S,82.6mg,67%收率)。
将上述制备的化合物(65mg,0.26mmol)和TFA(1mL,13mmol)在CH2Cl2中的混合物(1mL),在室温下搅拌3小时。减压浓缩溶液,得到(5-氯噻吩-2-基)甲胺(~100%收率)。C5H6NSCl;淡黄色固体;1H NMR(400MHz,CD3OD)δ7.07(1H,d,J=4.0Hz),6.95(1H,d,J=3.6Hz),4.26(2H,s);13C NMR(100MHz,CD3OD)δ134.9,132.8,130.7,128.0,38.8;ESI–HRMS计算值C5H7ClNS:147.9988,测得值:m/z 147.9995[M+H]+。
N6-[(5-氯噻吩-2-基)甲基]腺苷(JMF3818)
(5-氯噻吩-2-基)甲胺(35.4mg,0.24mmol)、6-氯嘌呤呋喃核糖苷(0.12mmol)和二异丙基乙胺(0.36mL,2mmol)在乙醇(1mL)中的混合物,在封管中,通过微波辐照在80℃下搅拌30分钟。将混合物冷却至室温,减压浓缩,得到淡黄色油状物,依次经H2O和MeOH洗涤,得到标题化合物JMF3818。C15H16ClN5O4S;白色固体;1H NMR(400MHz,CD3OD)δ8.29(1H,s),8.27(1H,s),6.88(1H,d,J=3.6Hz),6.79(1H,d,J=3.6Hz),5.96(1H,d,J=6.8Hz),4.74–4.77(1H,m),4.32–4.34(1H,m),4.17(1H,d,J=2.8Hz),3.89(1H,dd,J=12.4,2.0Hz),3.75(1H,dd,J=12.4,2.8Hz);13C NMR(100MHz,CD3OD)δ156.0,153.6,142.8,142.0,129.9,127.0,126.6,121.7,91.5,88.4,75.6,72.9,63.7,40.4;ESI–HRMS计算值C15H17ClN5O4S:398.0690,测得值:m/z 398.0692[M+H]+。
药代动力学研究。化合物以生理盐水的水溶液进行给药。雄性ICR小鼠购自BioLASCO Taiwan Co.,Ltd。为了测量受试化合物(T1-11和JMF3464)口服生物利用度,在口服(10mg/kg)或静脉内(1mg/kg)给药受试化合物之后,从雄性ICR小鼠(6周龄;20-25克)中采集血液样本。对于T1-11,血液样本在静脉内给药后2、10、30、60、120、360分钟采集,以及在口服给药后15、30、45、60、120、360分钟采集。对于JMF3464,血液样本在静脉内给药后2、10、30、60、120、240、360、480分钟采集,以及在口服给药后15、30、60、90、120、240、360、480分钟采集。血液样本通过含0.1%甲酸的甲醇萃取,然后将10μL的萃取样本注入UPLC–MSMS中进行定量。
运用药代动力学程序WinNonlin,将数据拟合到非室模型,获得药代动力学参数。药代动力学参数通过时间与对数浓度的线性回归估算,其中包括血浆浓度与时间曲线下面积(AUC),到最后采样时间的曲线下面积(AUC0–120),到无限时间的曲线下面积(AUC0–∞),终末期半衰期(T1/2),血浆中化合物最大浓度(Cmax),Cmax的时间(Tmax),以及与曲线末端部分相关的一级速率常数(k)。总血浆清除率(CL)通过剂量/AUCi.v.计算。受试化合物口服给药的口服生物利用度(F),根据口服剂量的AUC0–∞除以静脉注射剂量的AUC0–∞计算出。
实施例2
放射性配体结合试验。放射性配体结合试验由MDS Pharma Services Taiwan(台北,台湾),使用标准结合方案进行。对于A2AR结合试验,从过表达人A2AR的HEK293细胞中收集的膜蛋白,在含有3H-CGS21680(50nM)的反应缓冲液[50mM Tris-HCl(pH 7.4)、10mM MgCl2、1mM EDTA、和2U/mL腺苷脱氨酶]中,在25℃下孵育90分钟。在50μM腺苷-5'-N-乙基甲酰胺存在下,对非特异性结合进行评估。为了测量T1-11对A3R的结合亲和力,从过表达人A3R的中国仓鼠卵巢(CHO)-Kl细胞中收集的膜蛋白与3H-AB-MECA(0.5nM),在25℃下在含有25mM HEPES(pH 7.4)、5mM MgCl2、1mM CaCl2、和0.1%牛血清白蛋白的反应缓冲液中孵育60分钟。在1μM IB-MECA(Tocris Bioscience,Ellisville,MO,USA)存在下,对非特异性结合进行评估。针对腺苷转运体的结合试验,按照前面所描述的进行操作。从Duncan Hartley衍生的豚鼠的大脑皮层收集的膜组分,与3H-标记的6-[(4-硝基苄基)硫代]-9-β-D-呋喃核糖基嘌呤(NBTI,0.5nM),在25℃下在含有50mM Tris-HCl(pH 7.4)的孵育缓冲液中孵育30分钟。在5μM NBTI(一种有效的平衡型核苷转运体抑制剂)存在下,对非特异性结合进行评估。请注意,NBTI是一种高亲和力的ENT1抑制剂,并在0.5nM下仅抑制人(h)ENT1。通过GF/B玻璃纤维过滤以及经反应缓冲液洗涤,来终止反应。
细胞培养和瞬时转染
购自美国典型培养物保藏中心(ATCC;Manassas,VA,USA)的大鼠PC12细胞,维持于在补加10%马血清和5%FBS的DMEM中并在CO2孵育器(5%)中在37℃孵育。按照制造商的方案,LIPOFECTAMINETM 2000(Invitrogen)被用作将质粒转入细胞的载体。质粒由Pulst博士(神经科,犹他大学,USA)友好提供。通常,5μg DNA与5μl LIPOFECTAMINETM2000混合,并添加于6孔板的每孔中。板接种数为(1~1.5)x106细胞/孔。转染6小时后,细胞用试剂再处理24小时。通过Zeiss Axiovert 200M倒置荧光显微镜(哥廷根,德国)进行成像。
MTT分析。存活率通过3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四氮唑溴盐(MTT)代谢试验进行评估。简单地说,处理后,将MTT加到培养基(0.5mg/ml)并在37℃下孵育2~3小时。在96孔板中的接种数为1x104细胞/孔。丢弃培养基后,将DMSO加到孔中以溶解甲臜晶体,通过微量-酶联免疫吸附试验(ELISA)读取器测定每孔中在570和630nm下的吸光度。
实施例3
动物和给药。雄性R6/2小鼠(Mangiarini等人,1996Cell.87:493-506)和同窝对照,最初来自Jackson实验室(Bar Harbor,ME,USA),并与雌性对照小鼠(B6CBAFI/J)交配。后代通过对基因组DNA的聚合酶链反应(PCR)基因分型技术加以鉴定,以确保CAG重复元件的数量保持在大约150,其中该基因组DNA是从尾组织中提取的,并使用位于转基因中的引物(5'-CCGCTCAGGTTCTGCTTTTA-3';SEQ ID NO:1,和5'-GGCTGAGGAAGCTGAGGAG-3';SEQ ID NO:2)进行PCR。动物安置在生物医学科学动物护理设施研究所(Institute of Biomedical Sciences Animal Care Facility),接受12/12小时的光亮/黑暗循环。每日记录一次小鼠体重。在经台湾省台北市中央研究院机构动物护理和利用委员会(Academia Sinica Institutional Animal Care and Utilization Committee)批准的协议下,进行动物实验。
转动棒行为(Rotarod performance)。采用转动棒装置(UGO BASILE,Comerio,Italy),2分钟内以恒定速度(12rpm),对动作协调性进行评估(Carter等,1999J Neurosci.19:3248-3257)。所有小鼠在4周龄时训练2天以使它们熟悉转动棒装置。然后对4~12周龄的动物每周测试三次。对于每项测试,动物在旋转启动前被放置在装置中。落下延迟(Latency to fall)被自动记录。每只小鼠进行三次试验,每次试验最多2分钟。
实施例4
动物。C57BL/6小鼠购自国家实验动物中心(台湾)。转基因小鼠(ATAXN2Q127)由Pulst博士提供。小鼠被放在一个隔音房间里,处于12/12小时光亮/黑暗循环和受控的温度(22±2℃)。食物和水可随意获得。根据实验动物护理和使用的NIH指南的原则和指示,尽所有努力减少动物使用数量以及它们的痛苦和不适。这些实验也经中国医药国立研究所的动物护理和使用委员会机构审查和批准(批准号:100-15)。
过滤阻滞试验。此方法按照Wanker等描述的(1999Methods Enzymol.309:375-386)方法并稍作修改。简单地说,将收获的细胞在裂解缓冲液(50mM Tris-HCl(pH 8.8),100mM NaCl,5.0mM MgCl2,1mM EDTA,和含l x蛋白酶抑制剂混合物的0.5%(w/v)的IPGEAL(Roche Diagnostics,印第安纳波利斯,IN,USA))中再悬浮,并超声处理10s(1脉冲/秒)。每组中的同等蛋白浓缩物(15~20μg/孔),使用Bio-Dot SF装置(Bio-Rad,Hercules,CA,USA),通过2%十二烷基硫酸钠(SDS)预平衡的醋酸纤维素膜(0.2μm;Whatman,梅德斯通,肯特,UK)进行过滤。抽吸时,每个孔用200μl 0.1%SDS洗涤两次。印迹在含3%脱脂奶粉的TBS(100mM Tris-HC1和150mM NaC1;pH 7.4)中,在室温下被封闭1小时,然后与抗-多聚谷氨酰胺(1:5000;MAB1574)抗体在含3%牛血清白蛋白(BSA)和0.02%NaN3中孵育(4℃过夜),以探查正常和突变的ATAXN2。随后的方法与上文所描述的相同。
转动棒行为。这项研究中使用5周龄的野生型和转基因小鼠。采用转动棒装置(UGO BASILE,科梅廖,意大利),5分钟内以增加的速度(10至28rpm)对动作协调性进行评估。所有小鼠在5周龄内训练2天以使它们熟悉转动棒装置。此外,这周内,部分转基因小鼠通过将T1-11溶解在它们日常饮用水中开始食用T1-11。然后从6周龄起每周正式测试动物2次。对于每项测试,动物在旋转启动前被放置在装置中。落下延迟(Latency to fall)被自动记录。每只小鼠每次进行2次试验。允许小鼠在试验之间休息20~30分钟。
实施例5
行为测试。小鼠的平衡和协调功能通过进行转动棒测试加以确定。小鼠被放置在转动棒装置的移动鼓状物(drum)(加速模型,Ugo Basile Biological Research Apparatus)上,然后加速,直到小鼠从鼓状物落到板上,并使计时器停止。在4天的过程中在四个日常试验中测量落下延迟。
免疫组化染色。麻醉野生型小鼠或SCA3转基因小鼠,并心脏灌注含4%多聚甲醛的PBS。大脑在Tissue-Tek包埋介质中平衡并在液态氮中冷冻。通过低温恒温器切割制备的冠状切片(20μm),在0.1%Triton X-100中渗透化(permeabilized),并与稀释的抗-NeuN单克隆抗血清(Chemicon)在4℃下孵育48小时。随后,清洗切片,并与生物素化的马抗鼠IgG孵育,接着与亲和素-生物素-辣根过氧化物酶复合物一起孵育。然后清洗切片并在二氨基联苯胺溶液中显影。在StereoInvestigator软件(MBF Bioscience)的协助下,通过配有Retiga-2000R CCD照相机(Qlmaging)和3轴计算机控制的MAC 600电动平台(Ludl Electronics)的Leica DM2500显微镜,对NeuN阳性神经元进行显现和计数。每个分析包括对每只小鼠的15个脑切片的处理。
实施例6
动物和药物传送。转基因小鼠B6SJL-Tg(Prnp-TARDBP)4Jlel/J购自Jackson实验室(Bar Harbor,缅因州,USA),在台南国家实验动物中心饲养。转基因小鼠通过带有正向引物5'-GGT GGT GGG ATG AAC TTT GG-3'(SEQ ID NO:3)和反向引物5'-GTG GAT AAC CCC TCC CCC AGC CTA GAC-3'(SEQ ID NO:4)的PCR筛选。野生型小鼠是非转基同窝仔畜(littermates)。6周龄小鼠接受手术,在腹部腹外侧皮下嵌入含DMSO或指定的JMF1907的1004型ALZET微型渗透泵(DURECT Corporation,库比蒂诺,CA,USA)。每28天更换泵。
握力。通过握力计量仪(TSE Systems,Inc.,MO,USA)测量握力。简单地说,一只小鼠通过它的尾巴被用手抓出并使它抓握一个高度可调的安装在力传感器上的握杆(grip)。拉力通过小鼠尾巴施加到小鼠上。当小鼠松开握杆时,最大力量显示在一个连接控制单元的数字显示面板上。每只小鼠重复测试3次。
旋转杆表现。运用装置(UGO BASILE,科梅廖,意大利),以恒定速度(40rpm)对野生型和转基因小鼠的运动协调性评估120秒。所有小鼠训练2周,每周训练2天。然后对小鼠从9周龄起每周正式测试2次。对于每项测试,动物在旋转启动前被放置在装置中。落下延迟(Latency to fall)被自动记录。每只小鼠每次进行3次试验。允许小鼠在试验之间休息20分钟。
细胞培养和转染。运动神经元细胞系(NSC34)是来自Neil Cashman博士(大脑研究中心,不列颠哥伦比亚大学,加拿大)的慷慨礼物,在含10%胎牛血清(FCS)、2mM L-谷氨酰胺和1%青霉素/链霉素(Invitrogen GibcoBRL,卡尔斯巴德,CA,USA)的高葡萄糖Dulbecco氏修饰的Eagle培养基(DMEM)中,在37℃和5%CO2下培养。
实施例7
小鼠。8-12周龄的雌性C57BL6N小鼠购自于BioLASCO(宜兰,台湾)。
酸诱导的慢性弥漫性疼痛模型。纤维肌痛模型是在Sluka组(Sluka等.,2003Pain.106:229-239)建立的酸诱导的慢性疼痛模型上改进的。小鼠短暂地被2%气化的异氟醚麻醉后,在左腓肠肌接受20μL染料木黄酮(1μM)肌内注射。3分钟后,在相同部位给予20μL酸的盐水(pH 4.0)注射。小鼠然后发展成2周以上的持久机械性痛觉过敏。在小鼠发展成机械性痛觉过敏后4天,对T1-11(用0.9-mm/7-cm的管进行灌胃口服)的镇痛效果测试。通过测试小鼠后爪对0.2-mN von Frey细丝刺激的回缩反应,来测定机械性痛觉过敏。
间歇冷应激模型。纤维肌痛模型由Ueda组开发,其中小鼠经间歇冷应激处理2天(Nishiyori和Ueda,2008Mol Pain.4:52)。经间歇冷应激处理的小鼠发展成持久的(>2周)机械和热痛觉过敏。间歇冷应激后5天,在这些小鼠中对JMF3464(腹腔内或口服)的镇痛效果进行测试。通过测试小鼠后爪对0.2-mN von Frey细丝刺激的回缩反应,来测定机械性痛觉过敏。
本发明示例性实施例的上述描述仅是为了示意和说明而呈现,并非意味是穷尽的或将本发明限制于所公开的确切形式。根据上述教导,许多修改和变化都是可能的。
被选取和描述的实施例和例子是为了解释本发明的原理及其实际应用,以使本领域其它技术人员能够利用本发明和各种实施例以及适合特定用途的各种变动形式。在不脱离本发明精神和范围下,其他实施例对于本发明所属技术领域的那些技术人员而言是明显的。
本说明书中所引用和讨论的所有文献通过引用全部并入本文,如同每篇文献单独通过引用并入一般。
- 还没有人留言评论。精彩留言会获得点赞!
- SeZn7MT3的组成、制备方法及其在防治阿尔茨海默症中的应用与流程
- 纤维支架材料、其制备方法、及亚甲蓝负载量的调节方法与流程
- 大驳骨在制备预防和/或治疗神经退行性疾病的药物中的应用及药物的制造方法与工艺
- 花叶假杜鹃在制备预防和/或治疗神经退行性疾病的药物中的应用及药物的制造方法与工艺
- 大脑内稳态调节蛋白的组成、制备方法及其在防治阿尔茨海默症中的应用与流程
- 一种药物组合物及其制备方法和在制备防治神经退行性疾病的药物中的应用与流程
- 片层碳‑四氧化三铁复合体在抑制Aβ聚集中的应用的制造方法与工艺
- 一种干细胞球切割收集装置及干细胞球的切割分离传代方法与流程
- 一种具有改善记忆功效的多肽及其分离制备方法和应用与制造工艺
- 治疗或预防神经退行性病症的组合物和方法与制造工艺
- SeZn7MT3的组成、制备方法及其在防治阿尔茨海默症中的应用与流程
- 纤维支架材料、其制备方法、及亚甲蓝负载量的调节方法与流程
- 大驳骨在制备预防和/或治疗神经退行性疾病的药物中的应用及药物的制造方法与工艺
- 花叶假杜鹃在制备预防和/或治疗神经退行性疾病的药物中的应用及药物的制造方法与工艺
- 大脑内稳态调节蛋白的组成、制备方法及其在防治阿尔茨海默症中的应用与流程
- 一种药物组合物及其制备方法和在制备防治神经退行性疾病的药物中的应用与流程
- 片层碳‑四氧化三铁复合体在抑制Aβ聚集中的应用的制造方法与工艺
- 一种干细胞球切割收集装置及干细胞球的切割分离传代方法与流程
- 一类抗氧化超短肽及其应用的制造方法与工艺
- 一种具有改善记忆功效的多肽及其分离制备方法和应用与制造工艺