具有非对称性双官能团的弯曲核反应性介晶及其的制备方法与流程
本发明涉及具有非对称性双官能团的弯曲核反应性介晶、包含上述反应性介晶的取向膜组合物、利用上述反应性介晶的液晶单元及包含其的液晶显示装置。
背景技术:
反应性液晶单体(RM,reactive mesogens)是指液晶性或非液晶性化合物,主要可通过与可表达液晶性的介晶(mesogen)进行光反应来实现高分子化且具有一种以上的可进行聚合的末端基的化合物。通常,作为用于表达液晶性的介晶,使用用于表达向列(nematic)液晶相的棒状(calamitic)介晶。并且,作为可进行聚合的末端基,多使用容易进行自由基聚合的丙烯基或甲基丙烯基,除此之外,也可使用可进行聚合的任何官能团。由于液晶如液体具有流动性,因而可均匀地涂敷于大面积的基板上,并如结晶具有取向性,从而具有使分子容易进行排列的优点。若在液晶相中使反应性液晶单体分子进行取向来聚合,则具有维持排列并可取得交联的高分子网状物的优点。并且,相比于使用相同结构的液晶高分子具有在液晶相中相对低的粘度,因此可取得具有取向得更好的结构的大面积的区域,从而在应用领域多样的方面上与反应性液晶单体相关的技术引起更大的关注。
在过去数十年中,弯曲核液晶在液晶研究领域中受到很大的关注,主要对具有对称形的香蕉型液晶研究得多。
本发明人对弯曲核型反应性介晶的制备方法进行探索中,制备在介晶的两侧末端基具有丙烯酸烷基酯,且具有氟取代基的非对称性弯曲核反应性介晶,从而完成了本发明。
技术实现要素:
技术问题
本发明的目的在于提供具有呈现快的响应时间及优秀的黑色可视性的新型非对称性双官能团的反应性介晶及其的用途。
解决问题的方案
本发明提供具有非对称性双官能团的弯曲核反应性介晶。
并且,本发明提供包含上述反应性介晶的取向膜组合物。
并且,本发明提供利用上述反应性介晶的液晶单元及包含上述液晶单元的液晶显示装置。
发明的效果
本发明的具有非对称性双官能团的弯曲核反应性介晶通过调节中心的苯基及末端烷基的数量,从而可调节分子量,利用上述反应性介晶的液晶单元的机械性及热性稳定,并具有呈现优秀的黑色可视性及高的液晶响应速度的效果。并且,半永久性地固定分子的取向状态结构,从而具有可实现调节液晶的预倾角的效果。
附图说明
图1为以图示化的方式表示垂直取向模式用液晶单元的制备过程的图。
图2为表示对本发明的垂直取向模式用液晶单元实施光固化前后的光学组织的变化的图。
图3为以图示化的方式表示本发明的垂直取向模式用液晶单元的电压-透过率曲线图及响应速度-透过率曲线图的图。
具体实施方式
本发明提供由以下化学式1表示的的具有非对称性双官能团的弯曲核反应性介晶。
化学式1
在上述化学式1中,X1、X2、X3及X4分别独立地为-H或-F,并排除均为-H的情况,m为0至1的整数,n为1至20的整数,优选为3至12。
本发明的反应性介晶的特征在于,将形成基本骨架结构的苯基的数量设置为2个或4个来限定为偶数个,并取代一个以上的氟来提高其极性。
优选地,上述反应性介晶可由以下化学式2至化学式17表示,但以下化学式示出用于表示本发明的技术的化学式的例,本发明的技术并不局限于此。
化学式2
化学式3
化学式4
化学式5
化学式6
化学式7
化学式8
化学式9
化学式10
化学式11
化学式12
化学式13
化学式14
化学式15
化学式16
化学式17
本发明的反应性介晶在两侧末端基具有丙烯酸烷基酯或甲基丙烯酸烷基酯,这是上述反应性介晶化合物实际上借助光被活性化的光反应基。上述反应性介晶化合物借助上述光反应基的活性化来进行聚合。
并且,本发明提供包含由以下化学式1表示的具有非对称性双官能团的弯曲核反应性介晶的取向膜组合物。
化学式1
在上述化学式1中,X1、X2、X3及X4分别独立地为-H或-F,并排除均为-H的情况,m为0至1的整数,n为1至20的整数,优选为3至12。
优选地,将由上述化学式1表示的反应性介晶和通常可取得的取向剂相混合来制备上述取向膜,但是并不局限于此。
相对于取向剂的总重量,可添加0.1至30重量份的上述反应性介晶。上述弯曲核反应性介晶被添加进行光反应后,不大改变主机液晶的粘度,并可有效地改善响应速度。
并且,本发明提供包含由以下化学式1表示的具有非对称性双官能团的弯曲核反应性介晶和极性及非极性液晶化合物的液晶组合物。
化学式1
在上述化学式1中,X1、X2、X3及X4分别独立地为-H或-F,并排除均为-H的情况,m为0至1的整数,n为1至20的整数,优选为3至12。
相对于100重量份的具有负介电常数的液晶组合物,可添加0.1至10重量份的上述反应性介晶。
并且,本发明提供液晶单元,上述液晶单元的特征在于,包括:第一基板,分别涂敷有氧化铟锡膜(ITO);第二基板,与上述第一基板相向;液晶组合物,注入于上述第一基板及第二基板之间;以及取向膜组合物,在上述氧化铟锡膜上涂敷由以下化学式1表示的反应性介晶和垂直取向剂的混合物而形成,向分别涂敷有上述氧化铟锡膜的第一基板及第二基板之间施加电压,并照射紫外线来进行光固化,从而形成上述液晶单元。
化学式1
在上述化学式1中,X1、X2、X3及X4分别独立地为-H或-F,并排除均为-H的情况,m为0至1的整数,n为1至20的整数,优选为3至12。
优选地,上述液晶为向列液晶,但是并不局限于此。
作为上述向列液晶,可使用联苯类化合物、三联苯类化合物、苯基环己基类化合物、联苯基环己基类化合物、苯基二环己基类化合物、苯甲酸苯酯类化合物,环己基苯甲酸苯酯类化合物,苯基苯甲酸苯酯类化合物,联环己基羧酸苯酯类化合物,甲亚胺类化合物,偶氮及偶氮氧类化合物,二苯乙烯类化合物,联环己烷类化合物,苯基嘧啶类化合物,联苯嘧啶类化合物,嘧啶类化合物,联苯乙炔类化合物等,但是并不局限于此。
上述涂敷包括旋转涂敷(spin coating)、辊涂敷(roll coating)、浸渍涂敷(dip coating)、丝网涂敷(screen coating)、喷射涂敷(s pray coating)、丝网印刷(screen printing)、墨水喷射(ink jet)等,但并不局限于此。
优选地,分别涂敷有上述氧化铟锡膜的第一基板及第二基板之间的电压为1至20V,更优选为3至10V,但并不局限于此。
优选地,作为为了上述光固化而被照射的紫外线,使用200至800nm范围的紫外线,更优选地,将300至400nm波长的紫外线照射5至10J/cm2,但并不局限于此。
本发明的液晶单元呈现优秀的黑色可视性,并实施光固化之后具有光学组织稳定化的时间显著变快的特征。
并且,本发明提供包含上述液晶单元的液晶显示装置。
可使用在本发明所属技术领域中使用的公知的方法来制备上述液晶显示装置。
本发明的反应性介晶可适用于利用与光聚合性单体和液晶的复合类的高分子稳定-图像垂直调整(PS-PVA,Polymer stabilized PVA)模式及利用与光聚合性单体和取向膜的复合类的表面控制-图像垂直调整(SC-PVA,Surface controlled-PVA)模式,并能够以与苯基的个数,烷基的个数,F的个数及位置无关的方式适用。
并且,利用本发明的具有非对称性双官能团的弯曲核反应性介晶而制备的液晶单元的机械性及热性稳定,并具有呈现优秀的黑色可视性及高的液晶响应速度的特征,并且,半永久性地固定液晶分子的取向状态结构,从而具有可实现调节液晶的预倾角的效果。
以下,为了便于理解本发明而公开优选的实施例。但是以下实施例为了更容易理解本发明而提供,本发明的内容并不限定于实施例。
化学式1至化学式9的反应性介晶的制备
适用以下反应式1合成了具有上述化学式2至化学式9的具有非对称性双官能团的弯曲核介晶。
反应式1
实施例1.化学式3化合物的合成
中间化合物1-1-b(4-(苄氧基)-2,3-二氟苯甲酸(4-(benzyloxy)-2,3-difluorobenzoic acid))的合成
将作为碱催化剂的过量的56.14g的碳酸铯(172.3mmol)和15g的甲基2,3-二氟-4-羟基苯甲酸(methyl 2,3-difluoro-4-hydroxybenzo ic acid)(86.2mmol)放入于N,N-二甲基甲酰胺进行搅拌并溶解。之后利用冰浴器(ice bath)进行冷却,并滴加10.3ml的苄基溴(86.2mmol),并将上述混合溶液在常温条件下反应24小时。放入过量的蒸馏水结束反应,滴落HCl直到pH成为2为止并进行了沉淀。对沉淀物进行过滤来获取了作为粉末状固体生成物的化合物1-1-b(收率:74.6%)。
确认获取的化合物2的1H-NMR,并且获取的化合物2的1H-NM R如下。
1H NMR(300MHz,acetone-d6)δ7.92-7.90(d,1H),7.48-7.45(d,2H),7.40-7.37(t,2H),7.32-7.30(t,1H),6.92-6.90(d,1H),5.16(s,2H)。
中间化合物1-2-b(3-(苄氧基)苯基4-(苄氧基)-2,3-二氟苯甲酸酯(3-(benzyloxy)phenyl 4-(benzyloxy)-2,3-difluorobenzoate))的合成
将在上述步骤中合成的9.00g的4-(苄氧基)-2,3-二氟苯甲酸(34.06mmol)放入于烧瓶并在50ml的二氯甲烷中溶解。在常温条件下,进行搅拌并慢慢地滴加10ml的氯化亚砜。此时,提高温度来回流5小时。反应结束后,进行减压蒸馏去除多余的氯化亚砜,在50ml的无水二氯甲烷中添加6.82g的3-苄氧基苯酚(3-benzyloxyphenol)(34.06m mol)并进行溶解。此时,在混合溶液中慢慢地滴加0.2ml的吡啶,并使未溶解的部分完全溶解,在常温条件下,反应了48小时。反应结束后,对溶剂进行减压蒸馏来去除,并利用5%的HCl水溶液洗净多次。在接到的有机溶剂中放入硫酸镁来去除水分之后,进行过滤并对溶剂进行减压蒸馏,来取得淡黄色的液状物质。在二氯甲烷中溶解上述物质,利用乙酸乙酯和己烷混合溶剂并通过柱层析进行纯化。冷却干燥对经纯化的溶液的溶剂进行减压蒸馏后生成的透明的液状物质,来获取了作为白色结晶性固体生成物的化合物1-2-b(收率:89.7%)。
确认获取的化合物3的1H-NMR,并且获取的化合物3的1H-NMR如下。
1H NMR(300MHz,acetone-d6)δ7.93-7.90(d,1H),7.50-7.48(d,4H),7.46-7.43(t,1H),7.40-7.37(t,4H),7.32-7.30(t,2H),6.90-6.86(d,1H),6.84-6.82(m,2H),6.80-6.77(d,1H),5.28(s,2H),5.16(s,2H)。
中间化合物1-3-b(3-羟基苯基2,3-二氟-4-羟基苯甲酸酯(3-hy droxyphenyl 2,3-difluoro-4-hydroxybenzoate))的合成
将在上述步骤中合成的9.00g的3-(苄氧基)苯基4-(苄氧基)-2,3-二氟苯甲酸酯(22.40mmol)放入于四氢呋喃(THF),在常温条件下搅拌并溶解。放入0.22g的10%的Pd/C催化剂,并将反应温度提高60℃来供给氢12小时。反应结束后,对10%的Pd/C催化剂进行过滤,并进行减压蒸馏来去除了溶剂。在少量的氯仿中溶解残留物之后,在正己烷中进行沉淀来获取了作为粉末状固体生成物的化合物1-3-b(收率:91.1%)。
确认获取的化合物4的1H-NMR,并且获取的化合物4的1H-NM R如下。
1H NMR(300MHz,acetone-d6)δ7.76-7.74(d,1H),7.29-7.26(t,1H),6.76-6.74(d,1H),6.72-6.71(d,2H),6.69(s,1H),5.71(s,1H)。
中间化合物1-4-b(3-(3-羟基丙氧基)苯基2,3-二氟-4-(3-羟基丙氧基)苯甲酸酯(3-(3-hydroxypropoxy)phenyl 2,3-difluoro-4-(3-hydr oxypropoxy)benzoate))的合成
在氮条件下在乙醇和蒸馏水混合溶剂中溶解10.78g的氢氧化钾(192.12mmol)和少量的碘化钾之后,添加在上述步骤中合成的10g的3-羟基苯基2,3-二氟-4-羟基苯甲酸酯(37.57mmol),并进行搅拌来完全溶解。之后,在常温条件下,滴加7.10g的3-氯-1-丙醇(75.13mmol),并提高反应温度来回流了48小时。
反应结束后,对溶剂进行减压蒸馏,并在蒸馏水中溶解剩下的物质,利用二乙醚清洗之后,滴加HCl直到pH成为2为止。此时,沉淀出白色固体,并对上述白色固体进行过滤后,利用蒸馏水清洗多次来进行中性化。利用乙醇对经过滤的沉淀物进行重结晶来获取了作为白色粉末状固体生成物的化合物1-4-b(收率:64.6%)。
确认获取的化合物5的1H-NMR,并且获取的化合物5的1H-NMR如下。
1H NMR(300MHz,acetone-d6)δ7.93-7.90(d,1H),7.46-7.40(t,1H),6.90-6.86(d,1H),6.84-6.82(m,2H),6.80-6.77(d,1H),4.29-4.26(t,4H),3.69-3.66(t,4H),2.03-2.00(m,4H),1.89(s,2H)。
最终化合物1-5-b(3-(3-(丙烯酰氧基)丙氧基)苯基4-(3-(丙烯酰氧基)丙氧基)-2,3-二氟苯甲酸酯(3-(3-(acryloyloxy)propoxy)p henyl 4-(3-(acryloyloxy)propoxy)-2,3-difluorobenzoate))的合成
在氮条件下,将在上述步骤中合成的13.47g的3-(3-羟基丙氧基)苯基2,3-二氟-4-(3-羟基丙氧基)苯甲酸酯(56.53mmol)添加于二氯甲烷进行搅拌并溶解。之后利用冰浴器进行冷却,并滴加过量的10.6ml的三乙胺和丙烯酰氯(130.76mmol),之后,在常温条件下搅拌了6小时。反应结束后,在二氯甲烷中溶解对溶剂进行减压蒸馏来生成的物质,利用乙酸乙酯和己烷混合溶剂并通过柱层析进行了纯化。在少量的氯仿中溶解经纯化的物质之后,在甲醇中进行沉淀来获取了作为固体生成物的化合物1-5-b(收率:69.2%)。
确认了获取的化合物6的1H-NMR,并且获取的化合物6的1H-N MR如下。
1H NMR(300MHz,acetone-d6)δ7.93-7,90(d,1H),7.46-7.40(t,1H),6.90-6.86(d,1H),6.84-6.82(m,2H),6.80-6.76(d,1H),6.41-6.38(d,2H),6.12-6.10(t,2H),5.83-5.80(d,2H),4.29-4.25(t,4H),4.20-4.16(t,4H),2.12-2.09(m,4H)。
实施例2.化学式2化合物的合成
以相同的摩尔比的4-(苄氧基)-2-氟苯甲酸(4-(benzyloxy)-2-flu orobenzoic acid)替代4-(苄氧基)-2,3-二氟苯甲酸来使用,除此之外,以与实施例1相同的方法进行了合成。
实施例3.化学式4化合物的合成
以相同的摩尔比的4-(苄氧基)-2,6-二氟苯甲酸(4-(benzyloxy)-2,6-difluorobenzoic acid)替代4-(苄氧基)-2,3-二氟苯甲酸来使用,除此之外,以与实施例1相同的方法进行了合成。
实施例4.化学式5化合物的合成
以相同的摩尔比的4-(苄氧基)-2,3,5,6-四氟苯甲酸(4-(ben zyloxy)-2,3,5,6-tetrafluorobenzoic acid)替代4-(苄氧基)-2,3-二氟苯甲酸来使用,除此之外,以与实施例1相同的方法进行了合成。
实施例5.化学式6化合物的合成
以相同的摩尔比的6-氯己醇(6-chlorohexanol)替代3-氯丙醇(3-chloropropanol)来使用,除此之外,以与实施例2相同的方法进行了合成。
实施例6.化学式7化合物的合成
以相同的摩尔比的6-氯己醇替代3-氯丙醇来使用,除此之外,以与实施例1相同的方法进行了合成。
实施例7.化学式8化合物的合成
以相同的摩尔比的6-氯己醇替代3-氯丙醇来使用,除此之外,以与实施例3相同的方法进行了合成。
实施例8.化学式9化合物的合成
以相同的摩尔比的6-氯己醇替代3-氯丙醇来使用,除此之外,以与实施例4相同的方法进行了合成。
化学式10至化学式17的反应性介晶的制备
适用以下反应式2来合成了具有上述化学式10至化学式17的非对称性双官能团的弯曲核介晶。
反应式2
实施例9.化学式17化合物的合成
中间化合物1-3-d(3-羟基苯基2,3,5,6-四氟-4-羟基苯甲酸酯(3-hydroxyphenyl 2,3,5,6-tetrafluoro-4-hydroxybenzoate))的合成
以相同的摩尔比的4-(苄氧基)-2,3,5,6-四氟苯甲酸(4-(ben zyloxy)-2,3,5,6-tetrafluorobenzoic acid)替代4-(苄氧基)-2,3-二氟苯甲酸来使用,除此之外,以与实施例1相同的方法进行了4-(3-羟基苯基2,3,5,6-四氟-4-羟基苯甲酸酯)的合成。
中间化合物2-1(4-(苄氧基)-2,3-二氟苯甲酸)的合成
在氮条件下,在乙醇和蒸馏水混合溶剂中溶解10.78g的氢氧化钾(potassium hydroxide)(192.12mmol)和少量的碘化钾(potassium i odide)之后,添加10g的4-羟基苯甲酸(4-hydroxybenzoic acid)(72.40mmol),并进行搅拌来完全溶解。之后,在常温条件下,滴加9.7ml的6-氯-1-己醇(6-chloro-1-hexanol)(72.40mmol)),并提高反应温度来回流了48小时。
反应结束后,对溶剂进行减压蒸馏,并在蒸馏水中溶解剩余的物质,利用二乙醚清洗之后,滴加HCl直到水溶液的pH成为2为止。此时,若沉淀出白色固体,则对白色固体进行过滤后,利用蒸馏水清洗多次来进行中性化。利用乙醇对经过滤的沉淀物进行重结晶来获取了作为白色粉末状固体生成物的化合物2-1(收率:78.1%)。
确认了获取的化合物7的1H-NMR,并且获取的化合物7的1H-N MR如下。
1H NMR(300MHz,acetone-d6)δ7.90-7.86(d,2H),7.14-7.11(d,2H),4.11-4.09(t,2H),3.62-3.59(t,2H),1.77-1.75(m,2H),1.58-1.55(m,2H),1.52-1.49(m,2H),1.47-1.44(m,2H),1.24(s,1H)。
中间化合物2-2(4-(6-(丙烯酰氧基)己氧基)苯甲酸(4-(6-(a cryloyloxy)hexyloxy)benzoic acid))的合成
在氮条件下,将在上述步骤中合成的13.47g的4-(6-羟基己氧基)苯甲酸(4-(6-hydroxyhexyloxy)benzoic acid)(56.53mmol)、2.23g的作为酸催化剂的对甲苯磺酸(13mmol)、2.3g的作为聚合抑制剂的对苯二酚(20.9mmol)及过量的15ml的丙烯酸(207.2mmol)放入于苯进行溶解之后,在与迪安-斯脱克分水器(Dean stark trap)相连接的烧瓶中进行反应,并回流了5小时直到去除所生成的水为止。反应结束后,在二乙醚中溶解对溶剂进行减压蒸馏来生成的物质,利用蒸馏水洗净直到pH成为4~5左右为止。向获取的有机溶剂层放入硫酸镁来去除水分之后,进行过滤,并利用异丙醇(Isopropanol)对减压蒸馏后生成的物质进行重结晶,从而获取了呈现淡粉红色的作为粉末状固体生成物的化合物2-2(收率:70.0%)。
确认了获取的化合物8的1H-NMR,并且获取的化合物8的1H-N MR如下。
1H NMR(300MHz,acetone-d6)δ7.90-7.87(d,2H),7.14-7.11(d,2H),6.41-6.39(d,1H),6.12-6.10(t,1H),5.83-5.80(d,1H),4.11-4.09(t,2H),3.97-3.95(t,2H),1.77-1.74(m,2H),1.60-1.57(m,2H),1.51-1.48(m,2H),1.47-1.44(m,2H)。
最终化合物2-3(3-(4-(6-(丙烯酰氧基)己氧基)苯甲酰氧基)苯基4-(4-(6-(丙烯酰氧基)己氧基)苯甲酰氧基)-2,3,5,6-四氟苯甲酸酯(3-(4-(6-(acryloyloxy)hexyloxy)benzoyloxy)phenyl 4-(4-(6-(acryloyloxy)hexyloxy)benzoyloxy)-2,3,5,6-tetrafluorobenzoate))的合成
将在上述步骤中合成的8.59g的4-(6-(丙烯酰氧基)己氧基)苯甲酸(29.39mmol)放入于烧瓶并在50ml的二氯甲烷中溶解。在常温条件下,进行搅拌并慢慢地滴加6ml的氯化亚砜。此时,提高温度来回流了5小时。反应结束后,进行减压蒸馏来去除多余的氯化亚砜,放入4.44g的3-羟基苯基2,3,5,6-四氟-4-羟基苯甲酸酯(14.69mm ol),并在50ml的无水二氯甲烷中进行溶解。此时,在混合溶液中慢慢地滴加0.2ml的吡啶,并使未溶解的部分完全溶解,在常温条件下,反应了48小时。反应结束后,对溶剂进行减压蒸馏来去除,并利用5%的HCl水溶液洗净多次。在接到的有机溶剂中放入硫酸镁来去除水分之后,进行过滤并对溶剂进行减压蒸馏,来取得淡黄色的液状物质。在二氯甲烷中溶解上述物质,利用乙酸乙酯和己烷混合溶剂并通过柱层析进行纯化。冷却干燥对经纯化的溶液的溶剂进行减压蒸馏后生成的透明的液状物质,来获取了作为白色结晶性固体生成物的化合物2-3(收率:50.3%)。
确认了获取的化合物9的1H-NMR,并且获取的化合物9的1H-N MR如下。
1H NMR(300MHz,acetone-d6)δ8.10-8.06(d,4H),7.53-7.50(t,1H),7.46-7.43(s,1H),7.10-7.08(d,4H),7.06-7.04(d,2H),6.41-6.39(d,2H),6.12-6.10(t,2H),5.83-5.80(d,2H),4.11-4.09(t,4H),3.97-3.96(t,4H),1.77-1.74(m,4H),1.60-1.57(m,4H),1.52-1.49(m,4H),1.47-1.44(m,4H)。
实施例10.化学式13化合物的合成
以相同的摩尔比的4-(苄氧基)-2-氟苯甲酸(4-(benzyloxy)-2-flu orobenzoic acid)替代4-(苄氧基)-2,3,5,6-四氟苯甲酸来使用,除此之外,以与实施例9相同的方法进行了合成。
实施例11.化学式14化合物的合成
以相同的摩尔比的4-(苄氧基)-2,6-二氟苯甲酸(4-(benzyloxy)-2,6-difluorobenzoic acid)替代4-(苄氧基)-2,3-二氟苯甲酸来使用,除此之外,以与实施例9相同的方法进行了合成。
实施例12.化学式15化合物的合成
以相同的摩尔比的4-(苄氧基)-2,3,5,6-四氟苯甲酸替代4-(苄氧基)-2,3-二氟苯甲酸来使用,除此之外,以与实施例9相同的方法进行了合成。
实施例13.化学式9化合物的合成
以相同的摩尔比的3-氯丙醇替代6-氯己醇使用,除此之外,以与实施例10相同的方法进行了合成。
实施例14.化学式10化合物的合成
以相同的摩尔比的3-氯丙醇替代6-氯己醇来使用,除此之外,以与实施例11相同的方法进行了合成。
实施例15.化学式11化合物的合成
以相同的摩尔比的3-氯丙醇替代6-氯己醇来使用,除此之外,以与实施例12相同的方法进行了合成。
实施例16.化学式12化合物的合成
以相同的摩尔比的3-氯丙醇替代6-氯己醇来使用,除此之外,以与实施例9相同的方法进行了合成。
实施例17.复合类单元(cell)的制作
为了制作复合类试片,作为取向剂,使用了具有垂直取向特性的日本合成橡胶公司(JSR)的AL60702,作为液晶,使用了具有负介电异向性的默克公司(Merck)公司的MLC-6608(Δn:0.083,Δε:-4.1,TNI:90℃)。并且,在制作单元之前,为了均匀的分散,将在上述实施例1中制备的2重量百分比的反应性介晶(化学式3的化合物)和垂直取向剂相混合并进行搅拌24小时。
单元的制备工序如下:首先,以规定的大小(2cm×2.5cm)对分别涂敷有氧化铟锡膜的第一基板及第二基板进行切割并清洗后,将上述均匀地分散而成的垂直取向剂(AL60702)及在上述实施例1中制备的反应性介晶(2重量百分比)旋涂于氧化铟锡膜上。之后,为了去除溶剂,在100℃温度下,预烘(pre-bake)10分钟之后,在180℃温度下,进行后热处理(硬性烘烤处理(hard-bake)1小时,以进行亚胺化反应(imidization)。制作单元(厚度:3.01μm),并在作为清亮点(c learing point)的90℃下,将具有负介电异向性的向列液晶注入于第一基板及第二基板之间。之后,在常温条件下,向第一基板及第二基板之间施加10V的电压,并照射365nm波长的紫外线来进行光固化30分钟。在图1中以图示化的方式示出基于上述过程的液晶单元的制备过程。
在常温条件下,利用偏光显微镜对利用上述说明的方法制备而成的单元,通过对10V的1kHz的交流方波(AC square wave)进行导通/截止(on/off)来确认了肌理(Texture)的变化。
并且,利用在上述实施例2~实施例16中制备的化学式2、化学式4~化学式16的化合物,以与上述单元的制作方法相同的方法制作了单元。
实验例1.垂直取向模式用液晶单元的电及光学特性评价
对在上述实施例17中制备而成的液晶单元进行电及光学特性评价,作为比较例,以4-(3-丙烯酰氧基丙氧基)苯甲酸2-甲基-1,4-苯酯(RM257)(巴斯夫公司(BASF))替代具有非对称性双官能团的弯曲核反应性介晶来使用,除此之外,与此相同地制备液晶单元之后进行了评价。
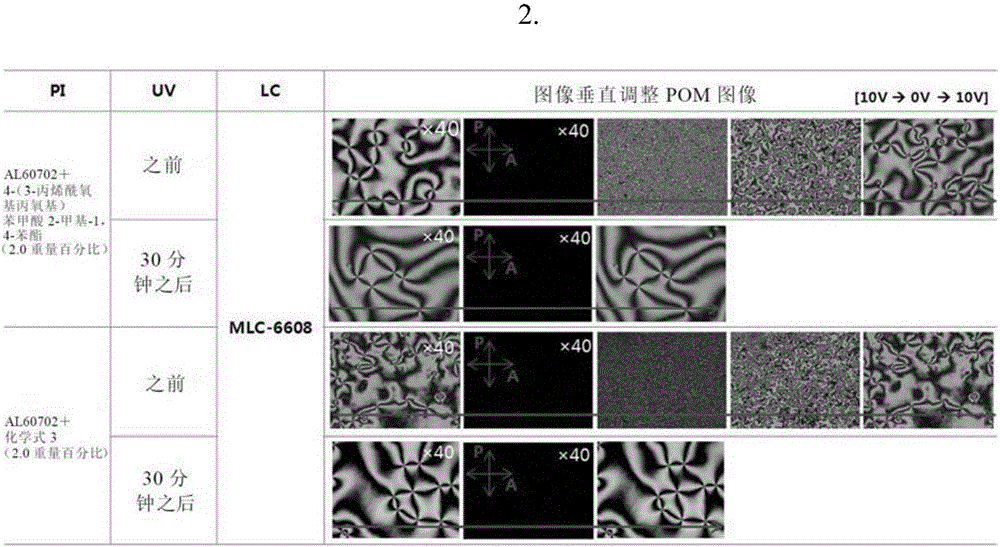
首先,对上述液晶单元,导通/截止进行光固化前及光固化后的10V的1kHz的交流电压来确认了取向的液晶的光学组织的变化。代表性地,利用由化学式3的化合物制备而成的液晶进行了确认,在图2中表示其结果。
如图2所示,在进行光固化之前,在包含化学式3的化合物的取向膜中,均确认到当每次导通/截止电压时,本发明的弯曲核反应性介晶的光学组织发生变化。但在进行光固化之后,确认到当每次导通/截止电压时,呈现相同的形状的光学组织,从而因反应性介晶的影响指向矢被固定。
并且,在上述实施例17中,利用由化学式3(当苯基为2个时的代表性化合物)和化学式17(当苯基为4个时的代表性化合物)制备而成的液晶单元,向液晶单元施加10V的交流(AC)的驱动电压并将受光量从10%达到90%的时间作为上升时间(rising time),将受光量从90%达到10%的时间作为下降时间(falling time)来进行测定,并相加两个值来测定了响应速度。之后,测定电压-透过率(Voltage-T ransmittance)及响应速度-透过率(Response Time-Transmittance),并在表1中表示其结果,将呈现最优秀的性能的化学式3的化合物的电光学特性与4-(3-丙烯酰氧基丙氧基)苯甲酸2-甲基-1,4-苯酯的电光学特性进行比较,并在图3中以曲线图的方式表示。
如在表1及图3中表示,观察到了已制备的具有非对称性双官能团的弯曲核反应性介晶与现有的介晶相比响应速度得到改善。尤其,下降时间得到约30%的改善。判断为这是因为具有非对称性双官能团的弯曲核反应性介晶借助光固化进行高分子网状化,并维持预倾角,并形成高的表面固定能量。
表1
- 还没有人留言评论。精彩留言会获得点赞!