新的光致变色化合物和用于生产光致变色化合物的中间化合物的制作方法
本发明涉及新的双环桥接[1,3]氧氮杂卓衍生物,其具备增强的光致变色特性且可用作分子光开关。
本发明还公开生产所述新的光致变色化合物的方法以及用于制备该化合物的中间物。
背景技术:
已知各类光致变色化合物:吲哚啉螺吡喃、吲哚啉螺恶嗪、苯并吡喃和萘并吡喃、俘精酸酐、二芳基乙烯、醌、呸啶螺环己二烯酮[v.malatesta,photodegradationoforganicphotochromesinorganicphotochromicandthermochromiccompounds.第2卷.j.c.crano及r.j.guglielmetti编.kluweracademic/plenumpublishers,newyork,1991,第473页]、吲哚并[2,1-b][1,3]苯并恶嗪。使螺吡喃和螺恶嗪溶液暴露于uv辐射阻断c-o键且形成着色的部花青,其由于分子的热运动或在微秒及毫秒内受可见光辐射后而恢复至初始状态。已知的光致变色化合物–吲哚啉并螺吡喃例如在专利ep0411884a1、gb1270928a、jp2006249622a、us5155230、us5241075及其他专利中描述。二氢吲哚啉与吲哚啉并螺吡喃类似,在受uv辐射后转变成着色形式且保温后恢复至初始状态,但着色形式的寿命与吲哚啉螺吡喃相比更长,且长达数百毫秒、分钟或甚至小时。[wo2012172093;r.zemribo、j.fotins、u.a.holgerkubas等人,photochromismofdihydroindolizines:第xiv部分。synthesisandphotophysicalbehaviorofphotochromicdihydroindolizinetripodallinkerstowardanchoringsensitizerstosemiconductornanoparticles.j.phys.org.chem.2011,24173-184;t.b.shrestha、j.melin、y.liu等人,newinsightsinthephotochromicspiro-dihydroindolizine/betaine-system.photochem.photobiol.sci.,2008,7,1449-1456]。已知的另一类光致变色化合物–醌类也未展示非着色形式与着色形式之间的极快互换,其着色转换耗时数微秒至数百微秒。[n.p.gritsan、l.s.klimenko.j.photochem.photobiol.a:chem.1993,70,103-117]。
呸啶环己二烯酮的特征在于着色形式的寿命极长,由于z-e构形交换而达到数天(与部花青中的e-z类似)。相对近期发现的光致变色化合物苯并吡喃和萘并吡喃不具备足够的光稳定性。由于uv辐射的作用,吡喃环转变成呋喃环且初始化合物丧失光致变色特性。[c.d.gabbutt、b.m.heron、s.b.kolla等人。ringcontractionduringthe6p-electrocyclisationofnaphthopyranvalencetautomers.orgbiomolchem2008;6:3096-3104;us6410754b1]。俘精酸酐和俘精酰亚胺的特征在于在暴露于uv辐射时发生电环化反应,此时形成具有着色的1,3-环己二烯片段的化合物。后者热稳定且只在受可见光辐射后可转变成初始状态。同样热稳定的二芳基乙烯的作用类似。
最新的一些种类的光致变色化合物-吲哚[2,1-b][1,3]苯并恶嗪具有热不稳定的着色形式,但它们比吲哚啉螺吡喃快得多且其寿命比吲哚啉螺吡喃的寿命短[us8252209]。新的一类光致变色化合物6-硝基-1,3,3‘,4-四氢螺[苯并吡喃-2,2‘-吲哚]在lithuanian专利lt6024(wo2014/035244)中描述,其着色形式的寿命仅为22-41ns。
当吲哚啉螺吡喃暴露于紫外线辐射时,发生吡喃环破裂且出现着色的部花青形式[v.i.minkin,photoswitchablemolecularsystemsbasedonsipropyransandspirooxazines,第37-80页.inmolecularswitches/由b.l.feringa和w.r.browne编.weinheim,wiley-vch,2011,第1-2卷;spiropyrans.r.c.bertelson,第11-84页.inorganicphotochromicandthermochromiccompounds,第1-2卷,j.c.crano,r.j.guglielmetti编,plenumpress.newyorkandlondon,1999,第1-2卷]。所形成的部花青热转变为螺吡喃由于反-顺再异构化阶段而显著延迟,这在着色形式恢复成初始的螺吡喃形式时发生且耗时数毫秒。
来自吲哚啉并螺吡喃产生着色形式相对较快,但由于热化学及光化学降解而仅维持较少个数循环,而且对空气中的氧也无抵抗力[v.malatesta,photodegradationoforganicphotochromesinorganicphotochromicandthermochromiccompounds.第2卷,j.c.crano和r.j.guglielmetti编。kluweracademic/plenumpublishers,newyork,1991,第473页]。除此之外,部花青形式的螺吡喃可易于水解[stafforst,t.和hilvert,d.kineticcharacterizationofspiropyransinaqueousmedia.chem.commun.2009,287-288]。
新的一类光致变色化合物,即6-硝基-1,3,3‘,4-四氢螺[苯并吡喃-2,2‘-吲哚],具有吲哚啉螺吡喃的碳骨架(其中吡喃环中的双键被氢化),在上文提到的lithuanian专利lt6024(wo2014/035244)中描述。
所述1‘,3,3‘,4-四氢螺[苯并吡喃-2,2‘-吲哚](a)在受uv辐射辐射之后,形成着色的4-硝基酚盐片段(b),其由于分子热运动而在数十纳秒内恢复至初始状态。这是由于不存在双键,因此不必要从反异构形式异构化为顺异构形式。然而,化合物a对碱性介质极为敏感。在不对称的碳原子处进行直接取代且形成具有4-硝基苯酚片段的着色化合物c,如果溶液中存在任意量的氢氧根离子,光致变色作用则消失。
因此,重要的是具有反应较快且在碱性溶液中不丧失其光致变色特性的光致变色化合物。
附图简要说明
本发明由以下图示进行说明,其中:
图1展示化合物6a在添加naoh(1mmolnaoh)后的乙腈中的动力学光谱;
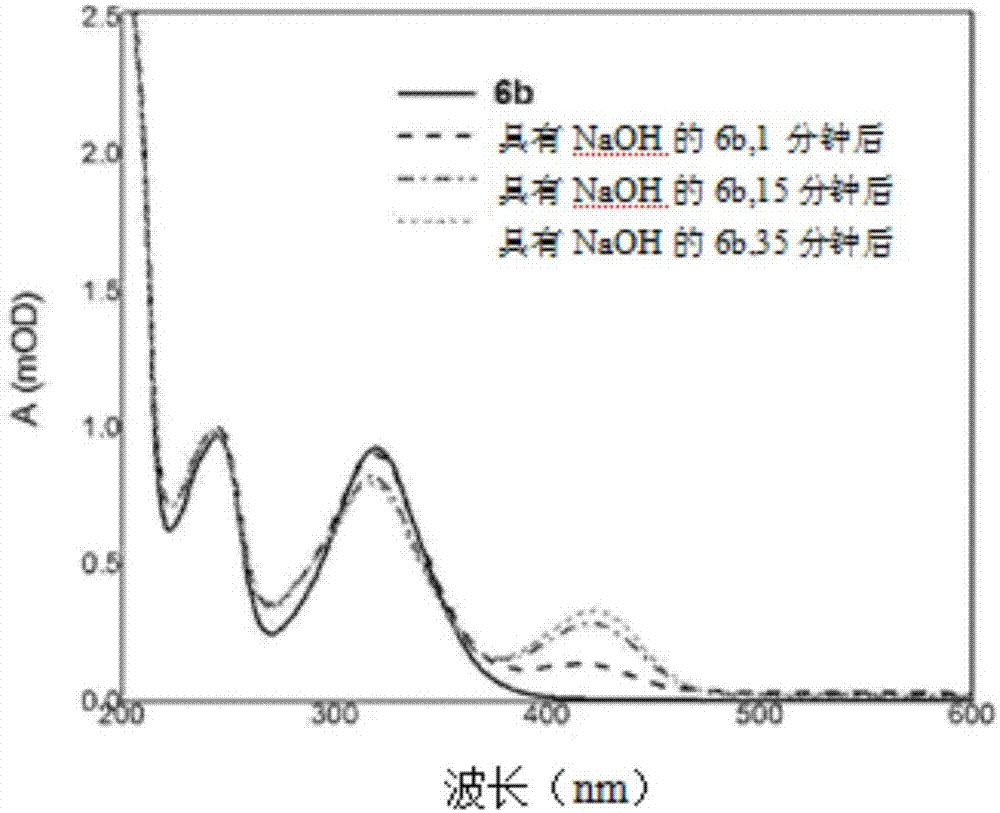
图2展示化合物6b在添加naoh(1mmolnaoh)后的乙腈中的动力学光谱。
技术实现要素:
本发明的目标在于获得新的光致变色化合物,它们通过甲醇苯并[f][1,3]氧氮杂卓并[3,2-a]吲哚中的3,4-二氢吡喃环的打开和闭合来起作用,其打开/闭合的速率高于已知光致变色体的速率,而且在中性和碱性介质中都具有活性。
为实现此目标,本发明提出通式(i)和(ii)的化合物
其中
r1和r2各自独立地为甲基、乙基或为螺环的部分:-(ch2)4-、-(ch2)5-;
r3为c1-c6-烷基、ω-官能化烷基、苄基、苯基、烯丙基。
此处,ω-官能化烷基具有可聚合基团(乙烯基、丙烯酸酯基、环氧基)或硫醇基,能连接至各种基质。
更具体而言,本发明涵盖通式(i)或(ii)之化合物,其中r1和r2为甲基且r3为乙基或苄基。
本发明的优选目标化合物为:
(5ar*,12r*,13s*)-n-乙基-6,6,8-三甲基-2-硝基-12,13-二氢-6h-5a,13-甲醇吲哚并[2,1-b][1,3]苯并氧氮杂卓-12-甲酰胺(6a);
(5ar*,12s*,13s*)-n-乙基-6,6,8-三甲基-2-硝基-12,13-二氢-6h-5a,13-甲醇吲哚并[2,1-b][1,3]苯并氧氮杂卓-12-甲酰胺(7);
(5ar*,12r*,13s*)-n-苄基-6,6,8-三甲基-2-硝基-12,13-二氢-6h-5a,13-甲醇吲哚并[2,1-b][1,3]苯并氧氮杂卓-12-甲酰胺(6b)。
本发明的目标化合物的特征在于光致变色特性。其可用作光致变色开关。
本发明的另一目标为通式(5)的化合物,
其中r3为乙基或苄基,且所述化合物为用于获得本发明的光致变色化合物的中间物。
本发明的又一目标为通式(4)的化合物,其用于合成化合物(5):
其中r3为乙基或苄基。
本发明的另一目标为式(3)的化合物,其用于合成化合物(4):
本发明的又一目标为式(2)的3h-吲哚盐,其用于合成式(3)的化合物:
具体实施方式
研发出新种类的光致变色化合物,包含通式(i)和(ii)的衍生物,顺-和反-甲醇苯并[f][1,3]氧氮杂卓并[3,2-a]吲哚衍生物
与最近公开的光致变色化合物1',3',3'-三甲基-6-硝基-1',3,3',4-四氢螺[苯并吡喃-2,2'-吲哚]iii和iv类似,这些衍生物具有由吲哚(a)和色满(b)杂环系统形成的1',3,3',4-四氢螺[苯并吡喃-2,2'-吲哚]的骨架。
然而,本发明的化合物带有另一c-c键(上文中以黑体显示),其降低了吡喃环的构象不稳定性。
发现本发明化合物的结构的独特之处在于在紫外线辐射对i型化合物之作用下,3,4-二氢[2h]吡喃环中之c-o键发生断裂,产生着色的两性离子v,其具有着色的4-硝基酚盐片段。此片段仅通过一个键连接,能绕其转动。同时,当已知的高速光致变色体a被uv辐射触发时,产生两性离子化合物b,其中4-硝基酚盐部分通过三个可能绕着旋转的单键与吲哚盐部分连接。与结构b(3个键)相比,在结构v(1个键)中由于可能绕c-c单键旋转而导致自由度降低的有利之处在于二氢吡喃环比5ns更快地恢复,而且化合物v恢复为初始结构i。
如上文提到的,本发明包括对于获得目标化合物而言必要的新颖中间物。
在发明人的知识范围内,未描述中间物(2)、(3)、(4)、(5),但它们可以通过已知的合成方法来制备。通过使通式(4)的化合物例如与2-羟基-5-硝基苯甲醛缩合获得通式(5)的中间物。通过用碘乙烷或苄基氯使7-甲氧基-9,9,9a-三甲基-9h-咪唑并[1,2-a]吲哚-2-酮烷基化来制备通式(4)的化合物。通过使3h-吲哚盐环化来获得式(3)的化合物,且通过用氯乙酰胺使2,3,3a-三甲基-5-甲氧基-3h-吲哚烷基化来获得式(2)的后一种盐。对于有机合成中的技术人员而言应了解如何来制备本发明的其它化合物。
通过处理发现,当本发明中提议的化合物的溶液被单色紫外线(例如,nd:yag激光(eksplanl30);三次谐波;波长355nm;脉冲持续时间-5ns;脉冲能量3mj)辐射时,连接具有氧原子的手性中心(*标记)的键发生异裂,导致形成两性离子化合物,包含连接3h-吲哚盐阳离子和着色4-硝基酚盐阴离子的片段的两个碳原子的构象限制链。在切断辐射之后,着色形式的化合物热恢复至初始的无色状态,之后酚盐阴离子的氧原子连接至吲哚环系统的α-碳原子。本发明的化合物甲醇苯并[f][1,3]氧氮杂卓并[3,2-a]吲哚的特征在于环闭合/打开的总速度较高,除此之外它们也在氧环境中进行操作。顺-甲醇苯并[f][1,3]氧氮杂卓并[3,2-a]吲哚对环境的ph的敏感性较小,尤其对于碱性介质而言,而6-硝基-1',3,3',4-四氢螺[苯并吡喃-2,2'-吲哚]对碱极为敏感。本专利申请中描述的化合物通过在回流下用乙醇中的氢氧化钾进行处理使相应的吲哚啉并螺吡喃发生再环化反应来合成。
所述甲醇苯并[f][1,3]氧氮杂卓并[3,2-a]吲哚可根据下文的通用流程来获得:
实施本发明的方式
下文提供关于实际实施方案实例的信息,公开本发明化合物的制备方法及其特性。提供此信息仅出于说明目的而提供且不限制本发明的范畴。
实施例1
5-甲氧基-2,3,3-三甲基-3h-氯化吲哚盐(初始化合物)
将5-甲氧基-2,3,3-三甲基-3h-吲哚12g(0,063mol)溶解于甲苯(20ml)中,将冷凝器连接至烧瓶且逐滴添加乙酰氯9ml(ρ=1.104g/ml,9,94g)。在将混合物冷却至周围温度后,将混合物在5℃下储存12h。过滤形成的晶体、用丙酮和冷的乙醚洗涤。产量为6.6g微黄色晶体状的氯化吲哚盐(46.12%)。熔点201-203℃。1hnmr(400mhz,dmso-d6):δ1.45(s,6h,2×ch3),2.62(s,3h,2-ch3),3.82(s,3h,5-och3),7.02(dd,j=8.8hz,j=2.4hz,1h,6-h),7.37(d,j=2.4hz,1h,4-h),7.54(d,j=8.4hz,1h,7-h)。13cbmr(100mhz,dmso-d6):δ14.3,22.0(2×c),53.8,55.9,109.4,113.9,117.2,134.9,145.3,159.7,193.0.ir(kbr),ν(cm-1):3134(n-h),3021,2973,1623,1479,1404,1289,1022,797。
实施例2
1-胺甲酰基甲基-5-甲氧基-2,3,3-三甲基[3h]氯化吲哚盐(中间化合物2)
将根据实施例1获得的5-甲氧基-2,3,3-三甲基-3h-氯化吲哚盐(6g,0.026mol)溶解于最小量的水中、用碳酸钠中和并用乙醚(2×30ml)萃取。将合并的萃取物经无水硫酸钠干燥且在旋转蒸发仪上除去溶剂。向获得的残余物(4.8g,0,025mol)中添加α-氯乙酰胺(2.6g,0.027mol)并将混合物在140℃下用冷凝器在邻二甲苯(7ml)中加热2h。冷却后,过滤形成的晶体并用冷的乙醚洗涤。从乙醇再结晶后,获得5g产物,产率为69.7%的深棕色晶体,熔点240-242℃。1hnmr(400mhz,dmso-d6):δ1.54(s,6h,2×ch3),2.73(s,3h,ch3),3.85(s,3h,o-ch3),5.36(s,2h,ch2co),7.14(dd,j=8.8hz,j=2.4hz,1h,6-h),7.51(d,j=2.4hz,1h,4-h),7.79-7.81(m,2h,7-h,1/2nh2),8.51(pl.s,1h,1/2nh2)。13cbmr(100mmhz,dmso-d6):δ14.4,22.6(2×c),50.1,54.5,56.6,109.9,114.9,116.3,135.2,143.9,161.1,165.1,196.4.ir(kbr),ν(cm-1):3290(n-h),3097,2977,1689(c=o),1298,1030,833。
实施例3
7-甲氧基-9,9,9a-三甲基-9,9a-二氢-1h-咪唑并[1,2-a]吲哚-2(3h)-酮(中间化合物3)
将根据实施例2的方法合成的1-胺甲酰基甲基-5-甲氧基-2,3,3-三甲基-3h-氯化吲哚盐(5g,0,018mol)溶解于最小量的水中、用碳酸钠中和并用乙醚(2×25ml)萃取。将醚萃取物合并、经无水硫酸钠干燥并在旋转蒸发仪上蒸发溶剂。向获得的中和混合物(5g,0,02mmol)中添加乙醇(20ml)和冰醋酸(9,2ml)并将混合物在100℃下加热1h。在将混合物冷却后,将其倒入水(50ml)中并用na2co3中和,直到开始形成白色晶体。将物质过滤、用水洗涤并从乙醇再结晶。产物的产量为2.5g(50%),呈白色晶体状,熔点为209-211℃。1hnmr(400mhz,cdcl3):δ1.17(s,3h,ch3),1.36(s,3h,ch3),1.50(s,3h,ch3),3.76(s,3h,o-ch3),3.77(ab-q,j=16.4hz,2h,ch2),6.62(d,j=2.8hz,1h,4-h),6.64(d,j=8.8hz,1h,7-h),6.70(dd,j=8.8hz,j=2.4hz,1h,6-h),7.86(pl.s,1h,nh)。13cbmr(100mhz,cdcl3):δ21.2,22.3,27.5,47.0,55.8,55.9,92.0,109.1,112.8,113.3,140.2,144.3,155.9,174.9(c=o)。ir(kbr,cm-1):3333(n-h),3056,2975,1700(c=o)。msm/z(%):247(m+h+,100)。
实施例4
1-乙基-7-甲氧基-9,9,9a-三甲基-9,9a-二氢-1h-咪唑并[1,2-a]吲哚-2(3h)-酮(中间化合物4a)
将根据实施例3所述的方法合成的7-甲氧基-9,9,9a-三甲基-9,9a-二氢-1h-咪唑并[1,2-a]吲哚-2(3h)-酮2,5g(0,01mol)溶解于二甲基甲酰胺(25ml)中并向此溶液中添加新鲜粉末状的koh(0,85g)。在碱溶解之后,逐滴添加碘乙烷(2,45ml)并继续搅拌3h。将混合物倒入水中并用乙醚(3×20ml)萃取。将醚萃取物合并、用5%的hcl溶液洗涤。用na2co3溶液中和、经无水硫酸钠干燥且在旋转蒸发仪上蒸发溶剂。产物产量为2.3g(82.7%),呈微黄色树脂状物质。1hnmr(400mhz,cdcl3):δ1.04(s,3h,ch3),1.29(t,j=7.2hz,3h,ch2ch3),1.40(s,3h,ch3),1.47(s,3h,ch3),2.98-3.07(m,1h,1/2ch2ch3),3.63-3.72(m,1h,1/2ch2ch3),3.76(s,3h,och3),3.83(ab-q,j=15.2hz,2h,co-ch2),6.59(d,j=2.8hz,1h,4-h),6.70-6.71(m,2h,ar-h)。13cnmr(100mhz,cdcl3):δ14.33,23.07,24.11,27.02,29.19,37.23,50.06,55.84,92.69,108.99,113.01,115.36,142.23,142.99,156.05,171.22(c=o)。ir(kbr),ν(cm-1):3029,2984,1703(c=o),1491,1273,1014,816。
实施例5
1-苄基-7-甲氧基-9,9,9a-三甲基-9,9a-二氢-1h-咪唑并[1,2-a]吲哚-2(3h)-酮(中间化合物4b)
将根据实施例3所述的方法合成的7-甲氧基-9,9,9a-三甲基-9,9a-二氢-1h-咪唑并[1,2-a]吲哚-2(3h)-酮2,0g(0,008mol)溶解于二甲基甲酰胺(18ml)中且向此溶液中添加0,7g(0,12mol)新鲜研磨的koh。在碱溶解之后,逐滴添加3.08苄基氯2.8ml(0,024mol,ρ=1.100g/ml,3.08g)且在室温下继续搅拌3hrs。将混合物倒入水中并用乙醚(3×20ml)萃取。将合并的萃取物用5%的hcl溶液洗涤并用na2co3溶液中和。将溶剂经无水硫酸钠干燥并用旋转蒸发仪蒸发。通过柱色谱(洗提液:己烷/丙酮3:1)纯化获得的树脂。产物的产量为1.4g(51.8%),呈浅橙色晶体状,熔点122-123℃。1hnmr(400mhz,cdcl3):δ1.01(s,3h,9a-ch3),1.25(s,3h,9-ch3),1.37(s,3h,9-ch3),3.73(ab-d,j=15.2hz,1h,1/2ch2co),3.73(s,3h,7-och3),4.14(dd,j=15.5hz,j=5.8hz,2h,ch2-ph),4.95(ab-d,j=15.6hz,1h,1/2ch2co),6.55(d,j=2.5hz,1h,8-h),6.69(dd,j=8.4hz,j=2.5hz,1h,6-h),6.74(d,j=8.4hz,1h,5-h),7.21-7.28(m,5h,ar-h)。13cnmr(100mhz,cdcl3):δ23.3,23.9,28.7,45.4,50.1,55.7,55.8,93.1,108.9,113.0,115.6,127.5,127.6(2×c),128.7(2×c),137.7,142.1,143.2,156.2,172.1(c=o)。ir(kbr),ν(cm-1):3023,2969,1706(c=o),1493,1278,1027,730。
实施例6
n-乙基-2-(5'-甲氧基-3',3'-二甲基-6-硝基螺[苯并吡喃-2,2'-吲哚基]-1'(3'h)-基)乙酰胺(中间化合物5a)
将根据实施例4所述的方法合成的1-乙基-7-甲氧基-9,9,9a-三甲基-9,9a-二氢-1h-咪唑并[1,2-a]吲哚-2(3h)-酮2,5g(0.009mol)和2-羟基-5-硝基苯甲醛1.52g(0.009mol)添加至冰醋酸(20ml)中并在100℃下加热3hrs。反应完成后,将混合物倒入5%的醋酸钠溶液(250ml)中并用醋酸乙酯(2×30ml)萃取。将合并的萃取物经无水硫酸钠干燥并在旋转蒸发仪上除去溶剂。通过柱色谱(洗提液:己烷/丙酮3:1)纯化获得的树脂。产物的产量为1.3g(33.8%),呈紫色非晶物质状。熔点为95-96℃。1hnmr(400mhz,cdcl3):δ1.09(t,j=7.2hz,3h,ch2ch3),1.26(s,3h,3’-ch3),1.3(s,3h,3’-ch3),3.25-3.34(m,2h,ch2ch3),3.59(ab-d,j=17.6hz,1h,1/2ch2co),3.80(s,3h,5’-och3),3.87(ab-d,j=17.6hz,1h,1/2ch2co),5.83(d,j=10.4hz,1h,ch=ch),6.43(d,j=8.4hz,1h,7’-h),6.49(t,j=5.2hz,1h,nh),6.72(dd,j=8.4hz,j=2.8hz,1h,6’-h),6.75-6.78(m,2h,4-h,4’-h),6.94(d,j=10.4hz,1h,ch=ch),8.00(d,j=2.8hz,1h,7-h),8.03(dd,j=8.8hz,j=2.8hz,1h,5-h)。13cnmr(100mhz,cdcl3):δ15.0,20.1,26.1,27.0,34.3,48.8,52.9,56.0,106.6(螺-c),108.3,110.0,111.7,115.6,118.3,120.4,123.1,126.3,129.4,137.8,140.0,141.5,155.4,158.8,169.3(c=o)。ir(kbr),ν(cm-1):3403(n-h),3065,2966,1666(c=o),1520,1481,1337,1273,1089,951。
实施例7
n-苄基-2-(5'-甲氧基-3',3'-二甲基-6-硝基螺[苯并吡喃-2,2'-吲哚基]-1'(3'h)-基)乙酰胺(中间化合物5b)
将根据实施例5所述的方法合成的1-苄基-7-甲氧基-9,9,9a-三甲基-9,9a-二氢-1h-咪唑并[1,2-a]吲哚-2(3h)-酮1.0g(0.003mol)和2-羟基-5-硝基苯甲醛(0.5g,0.003mol)添加至冰醋酸(10ml)中并在100℃下加热3hrs。反应完成后,将混合物倒入5%的醋酸钠溶液(100ml)中并用醋酸乙酯(2×30ml)萃取。将合并的萃取物经无水硫酸钠干燥并在旋转蒸发仪上除去溶剂。通过柱色谱(洗提液:己烷/丙酮3:1)纯化获得的树脂。产物的产量为0.41g(28.7%),呈紫色非晶物质状。熔点为84-85℃。1hnmr(400mhz,cdcl3):δ1.18(s,3h,3’-ch3),1.27(s,3h,3’-ch3),3.71(ab-d,j=17.6hz,1h,1/2ch2co),3.80(s,3h,5’-och3),3.91,(ab-d,j=17.6hz,1h,1/2ch2co),4.45(d,j=5.6hz,2h,ch2-ph),5.76(d,j=10.2hz,1h,ch=ch),6.45(d,j=8.0hz,1h,4-h),6.60(d,j=8.4hz,1h,6’-h),6.71(dd,j=8.4hz,j=2.4hz,1h,7’-h),6.74(d,j=2.4hz,1h,4’-h),6.83(t,j=5.8hz,1h,nh),6.92(d,j=10.2hz,1h,ch=ch),7.14-7.17(m,2h,ar-h),7.27-7.29(m,3h,ar-h),7.96-7.80(m,2h,5-h,7-h)。13cnmr(100mhz,cdcl3):δ20.14,26.10,43.43,48.75,52.97,56.06,106.65(螺-c),108.23,110.06,111.79,115.61,118.29,120.51,123.03,126.31,127.57(2×c),127.72,128.87(2×c),129.41,137.66,138.0,139.84,141.58,155.38,158.66,169.49(c=o)。ir(kbr),ν(cm-1):3395(n-h),3030,2930,1671(c=o),1519,1494,1481,1337,1272,1089,951。
实施例8
(5ar*,12s*,13s*)-和(5ar*,12r*,13s*)-n-乙基-8-甲氧基-6,6-二甲基-2-硝基-12,13-二氢-6h-5a,13-甲醇吲哚并[2,1-b][1,3]苯并氧氮杂卓并-12-甲酰胺(目标化合物6a、7)
将根据实施例6所述的方法合成的螺化合物5a(1g,0.002mol)溶解于乙醇(30ml)中,添加精细研磨的氢氧化钾(0.34g,0.006mol)且使混合物回流2hrs。反应完成后,在旋转蒸发仪上除去大部分的溶剂,并向残余物中逐滴添加水直到溶液变得浑浊。使混合物在5℃下保持10hrs。过滤形成的晶体、用小量冷的乙醚洗涤并从乙醇再结晶。将滤液用醋酸乙酯(2×30ml)萃取,将合并的萃取物经无水硫酸钠干燥并在旋转蒸发仪上除去溶剂。通过柱色谱(洗提液:己烷/丙酮5:1)纯化残余物。使获得的产物自乙醇再结晶。
顺-异构体(6a)
产率10.4%,黄色晶体,熔点241-242℃。1hnmr(400mhz,cdcl3):δ0.61(t,j=7.2hz,3h,ch2ch3),1.48(s,3h,6-ch3),1.50(s,3h,6-ch3),2.16(d,j=11.6hz,1h,14-ha),2.20(dd,j=11.6hz,j=3.6hz,1h,14-hb),2.81-2.91(m,1h,1/2ch2ch3),3.12-3.23(m,1h,1/2ch2ch3),3.78(s,3h,8-och3),3.80(t,j=3.6hz,13-h),3.88(d,j=4.8hz,1h,12-h),6.43(d,j=9.2hz,1h,ar-h),6.66-6.69(m,2h,ar-h),6.89-6.92(m,2h,ar-h,nh),7.98(d,j=2.8hz,1h,1-h),8.07(dd,j=9.2hz,j=2.8hz,1h,3-h)。13cnmr(100mhz,cdcl3):δ14.7,23.11,26.6,32.9,33.7,42.4,45.5,56.0,78.2,109.6,111.1,111.5,113.1,116.3,124.7,125.1,125.9,139.8,141.4,142.4,156.0,158.4,169.5(c=o)。ir(kbr),ν(cm-1):3288(n-h),3071,2982,1652(c=o),1513,1344,1269,1090,816。
反-异构体(7)
产率2.5%,微黄色晶体,熔点184-185℃。1hnmr(400mhz,cdcl3):δ1.09(t,j=7.2hz,3h,ch2ch3),1.37(s,3h,6-ch3),1.60(s,3h,6-ch3),2.05(d,j=11.6hz,1h,14-ha),2.69(dd,j=11.6hz,j=4.0hz,1h,14-hb),3.24-3.35(m,2h,ch2ch3),3.75(s,3h,8-och3),3.77(d,j=4.0hz,1h,13-h),4.31(s,1h,12-h),5.97(t,j=5.2hz,1h,nh),6.25(d,j=8.4hz,1h,10-h),6.59(dd,j=8.4hz,j=2.4hz,1h,9-h),6.75(d,j=2.4hz,1h,7-h),6.79(d,j=9.2hz,1h,4-h),8.03(dd,j=8.8hz,j=2.8hz,1h,3-h),8.16(d,j=2.8hz,1h,1-h)。ir(kbr),ν(cm-1):3298(n-h),3082,2970,1650(c=o),1510,1496,1338,1268,1086,917。
实施例9
(5ar*,12s*,13s*)-n-苄基-8-甲氧基-6,6-二甲基-2-硝基-12,13-二氢-6h-5a,13-甲醇吲哚并[2,1-b][1,3]苯并氧氮杂卓并-12-甲酰胺(目标化合物6b)
将根据实施例7所述的方法合成的螺化合物5b0.385g(0.8mmol)溶解于乙醇(10ml)中,添加精细研磨的氢氧化钾(0.13g,2.4mmol)且使混合物回流2hrs。反应完成后,在旋转蒸发仪上除去大部分的溶剂,并逐滴添加水直到溶液变得浑浊。使混合物在5℃下保持10hrs。过滤形成的晶体、用小量冷的乙醚洗涤并从乙醇再结晶。产率为23.4%,呈黄色晶体状。熔点218-219℃。1hnmr(400mhz,cdcl3):δ1.46(s,6h,2×6-ch3),2.13(d,j=11.6hz,1h,14-ha),2.21(dd,j=11.6hz,j=4.0hz,1h,14-hb),3.78(s,3h,8-och3),3.82(t,j=4.0hz,1h,13-h),3.93-3.97(m,2h,1/2ch2-ph,12-h),4.51(dd,j=14.8hz,j=8.0hz,1h,1/2ch2-ph),6.46(d,j=9.2hz,1h,4-h),6.68-6.70(m,2h,ar-h),6.73-6.75(m,2h,ar-h),6.79(d,j=8.8hz,1h,ar-h),7.10-7.16(m,3h,ar-h),7.28-7.31(m,1h,nh),7.93(d,j=2.8hz,1h,1-h),7.97(dd,j=9.2hz,j=2.8hz,1h,3-h)。13cnmr(100mhz,cdcl3):δ23.0,26.6,33.0,42.1,42.8,45.4,55.9,78.3,109.6,111.1,111.3,113.0,116.3,124.6,125.3,125.5,127.4(2×c),127.5,128.5(2×c),137.7,139.8,141.3,142.4,156.0,158.2,169.5(c=o)。ir(kbr),ν(cm-1):3377(n-h),3029,2975,1676(c=o),1517,1496,1340,1262,1086,909。
由乙腈中的溶液(10-5m/l)确定目标化合物6a、b和7的光致变色特性。在扫描分光计(shimadzuuv-3101pc)上记录稳定态的uv-可见光吸收光谱。用nd:yag激光3次谐波辐射(eksplanl30,脉冲能量为每次脉冲3mj,λ=355nm,脉冲持续时间5ns)进行闪光光解实验。通过对脉冲计数来确定脉冲数,同时开放形式的光吸收光谱中的最大值达到初始吸收最大值的0.8。使用光学上根据激发波长进行调整的苯甲酮在mecn中的溶液(0.5od,在355nm下)来测量量子产率。根据能态特征(不同吸光度信号的生长相对于激发脉冲能量)和摩尔消光系数,与对照溶液相比来评估光致变色转变成开放形式的量子产率。
表中提供测量结果。
表中呈现的关于光致变色特性的数据表明,本发明的化合物可用作分子光开关,其中在uv辐射下,发生3,4-二氢-2h-吡喃环打开且产生变色形式的化合物,接着热恢复成初始的未变色形式。形成变色形式并恢复回未变色形式的总的持续时间小于5ns。这个过程可以重复多次。本发明的化合物的特征在于光稳定性高,在操作5000次以上循环时无明显的降解。
本发明的化合物对碱性介质比早前提到的化合物a较不敏感。如果我们向类似物a在乙腈中的溶液中滴加氢氧化钠溶液,氢氧根离子将立即攻击α-c碳原子,3,4-二氢吡喃环将打开并且形成着色的硝基酚盐。
同时,图1中提供的光谱表明,在用氢氧化钠处理化合物6a之后,3,4-二氢吡喃环保持稳定而且不形成着色的4-硝基酚盐阴离子。化合物6b(图2)对碱性介质更敏感,但不像化合物a那么敏感。图2中提供化合物6b的动力学uv-可见光光谱。
总的来说,本发明与已知的现有技术相比具有以下主要优势:
1.本发明的顺-和反-甲醇苯并[f][1,3]氧氮杂卓并[3,2-a]吲哚形成着色形式以及恢复回初始的未着色状态的总的持续时间比吲哚啉并螺吡喃或1',3,3',4-四氢螺[苯并吡喃-2,2'-吲哚]中明显较短。
2.顺-甲醇苯并[f][1,3]氧氮杂卓并[3,2-a]吲哚的特征在于对介质变化尤其具有抗性,而且只有通过uv辐射的作用才会出现着色形式。碱类不易于打开[1,3]苯并恶嗪环而且不产生着色的4-硝基酚片段。
3.本发明的化合物的特征在于比1',3,3',4-四氢螺[苯并吡喃-2,2'-吲哚]对光破坏的抗性更高,而且可以承受5000次以上的打开-闭合循环。
4.这些化合物对空气中的氧不敏感,而且可以用在未除去氧的环境中。
权利要求书(按照条约第19条的修改)
1.一种通式(i)或(ii)的化合物
其中
r1和r2各自独立地为c1-c3-烷基或r1与r2一起形成-(ch2)4-、-(ch2)5-;
r3为c1-c6烷基、苄基、苯基、烯丙基。
2.根据权利要求1的通式(i)或(ii)的化合物,其中
r1为甲基、r2为甲基且r3为乙基或苄基。
3.根据权利要求1或2的化合物,其为(5ar*,12r*,13s*)-n-乙基-6,6,8-三甲基-2-硝基-12,13-二氢-6h-5a,13-甲醇吲哚并[2,1-b][1,3]苯并氧氮杂卓-12-甲酰胺(6a)。
4.根据权利要求1或2的化合物,其为(5ar*,12s*,13s*)-n-乙基-6,6,8-三甲基-2-硝基-12,13-二氢-6h-5a,13-甲醇吲哚并[2,1-b][1,3]苯并氧氮杂卓-12-甲酰胺(7)。
5.根据权利要求1或2的化合物,其为(5ar*,12s*,13s*)-(n-苄基-6,6,8-三甲基-2-硝基-12,13-二氢-6h-5a,13-甲醇吲哚并[2,1-b][1,3]苯并氧氮杂卓-12-甲酰胺(6b)。
6.根据前述权利要求中任一项的化合物,其展示光致变色特性,其特征弛豫时间小于5ns。
7.一种根据权利要求1-6中任一项的化合物的用途,其用作分子光开关。
8.一种通式(5)的化合物,
其中r3为乙基或苄基。
9.一种通式(4)的化合物,
其中r3为乙基或苄基。
10.一种式(3)的7-甲氧基-9,9,9a-三甲基-9,9a-二氢-1h-咪唑并[1,2-a]吲哚-2(3h)-酮
11.一种式(2)的1-胺甲酰基甲基-5-甲氧基-2,3,3-三甲基[3h]氯化吲哚盐
- 还没有人留言评论。精彩留言会获得点赞!