一种二芳基乙烯类可光致变色杀虫化合物及其制备方法和用途与流程
本发明属于农药领域。具体地,本发明涉及一种含新烟碱杀虫剂单元的二芳基乙烯类可光致变色化合物及其制备方法和用途。
技术背景
以吡虫啉为代表的新烟碱类杀虫剂因杀虫活性高,杀虫谱广,对哺乳动物和水生动物毒性低,且有良好的系统物性及适当的田间稳定性和环境友好性,成为新农药创制的重要热点领域。后来又相继开发出噻虫啉、噻虫胺、噻虫嗪、啶虫脒、烯啶虫胺、呋虫胺等一系列烟碱类杀虫剂。
但是由于吡虫啉过量频繁使用造成较为严重的抗性问题以及由于结构相似性带来的新烟碱杀虫剂之间的交互抗性,在一定程度上限制了该类化合物的应用,制约了此类化合物发展,同时新烟碱类杀虫剂主要对同翅目和鞘翅目害虫高效,其相对较窄的杀虫谱也限制了虫害防治方面的用药选择性。
因此,如何对具有高活性的硝基亚甲基化合物进行结构改造,以产生新的、更有效的杀虫剂,解决新烟碱类杀虫剂的抗性问题,扩大杀虫谱,使其应用于杀虫剂组合物就成为本领域迫切需要解决的技术问题。
技术实现要素:
本发明的目的在于提供一类高效防治害虫的化合物(如式(A)化合物和式(B)化合物)及其制备方法和用途。
本发明第一方面提供了一种具有式(A)所示结构的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐:
式中:
R1和R2各自独立地为含氮、氧和/或硫的五元或六元杂环基,或卤代的含氮、氧和/或硫的五元或六元杂环基,取代或未取代的苯基;
R3,R4各自独立地为H、C1-4烷基、烯丙基、苄基、C1-4烷氧基-C1-4烷基、C1-4烷基-羰基、C1-4烷氧基-羰基或苯氧羰基;或者R3和R4和它们相连的原子共同构成-CH2-CH2-或-CH2-CH2-CH2-;
R5,R6各自独立地为H、C1-4烷基、烯丙基、苄基、C1-4烷氧基-C1-4烷基、C1-4烷基-羰基、C1-4烷氧基-羰基或苯氧羰基;或者R5和R6和它们相连的原子共同构成-CH2-CH2-或-CH2-CH2-CH2-;
R7为氢、C1-4烷基、卤素、取代或未取代的5-6元芳环基、或取代或未取代的5-6元芳杂环基;
R8、R9、R10、R11、R12和R13各自独立地为氢、卤素、C1-4烷基、取代或未取代的5-6元芳环基、或取代或未取代的5-6元芳杂环基;或R8、R9和R10、R11的结合构成取代或未取代的5-6元芳环基、或取代或未取代的5-6元芳杂环基;或R12、R13和R10、R11的结合构成取代或未取代的5-6元芳环基、或取代或未取代的5-6元芳杂环基;
R14为C1-4烷基或C1-4烷氧基;
X为硝基、氰基、三氟甲基、三氟乙酰基或三氟甲磺酰基;
Y为N或CH;
Z为S、O或NH;
其中,所述取代是指被选自下组中的一个或多个取代基所取代:卤素、C1-4烷基或卤代C1-4烷基、C1-4烷氧基或卤代C1-4烷氧基、C2-6烯基、C2-6炔基、卤代的C2-6烯基、卤代的C2-6炔基、C1-4烷氧基-C1-4烷基、C1-4烷氧基-羰基、C2-6炔基-羰基、C2-6烯基-羰基。
在另一优选例中,R1和R2各自独立地为吡啶基、噻唑基、嘧啶基、四氢呋喃基、噁唑基,或其卤代物。
在另一优选例中,R1和R2各自独立地为卤代的吡啶基、卤代的噻唑基、卤代的嘧啶基、卤代的四氢呋喃基、或卤代的噁唑基。
在另一优选例中,所述卤代物为氯代物。
在另一优选例中,R1和R2各自独立地为
在另一优选例中,R3,R4各自独立地为H或C1-3烷基;或者R3和R4和它们相连的原子共同构成-CH2-CH2-或-CH2-CH2-CH2-;和/或R5,R6各自独立地为H或C1-3烷基;或者R5和R6和它们相连的原子共同构成-CH2-CH2-或-CH2-CH2-CH2-。
在另一优选例中,R3,R4各自独立地为氢、甲基或乙基;或者R3和R4和它们相连的原子共同构成-CH2-CH2-或-CH2-CH2-CH2-。
在另一优选例中,R5,R6各自独立地为氢、甲基或乙基;或者R5和R6和它们相连的原子共同构成-CH2-CH2-或-CH2-CH2-CH2-。
在另一优选例中,R7为氢、C1-3烷基或卤素。
在另一优选例中,R7为氢。
在另一优选例中,R8、R9、R10、R11、R12和R13各自独立地为氢、氟、C1-4烷基。
在另一优选例中,R8、R9、R10、R11、R12和R13各自独立地为氢或氟。
在另一优选例中,R14为C1-2烷基或C1-2烷氧基。
在另一优选例中,R14为甲基。
在另一优选例中,X为硝基或氰基。
在另一优选例中,X为硝基。
在另一优选例中,Y为N或CH。
在另一优选例中,Y为N。
在另一优选例中,Z为S、O或NH。
在另一优选例中,Z为S。
本发明第二方面提供了一种杀虫剂组合物,其包含:
(a)0.001-99.99重量%的本发明第一方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐;
(b)农药学上可接受的载体和/或赋形剂。
在另一优选例中,组分(a)占所述杀虫剂组合物的0.01-99.9重量%,优选0.05-90重量%。
在另一优选例中,所述杀虫剂组合物用于杀灭或预防选自如下的害虫:鞘翅目、鳞翅目、半翅目、直翅目、等翅目、或双翅目昆虫。
在另一优选例中,所述害虫为等翅目或鳞翅目昆虫。
在另一优选例中,所述害虫具有刺吸式或锉吸式口器。
在另一优选例中,所述害虫为蚜虫、孑孓、飞虱、粉虱、叶蝉、蓟马、棉铃虫、菜青虫、小菜蛾、斜纹夜蛾、粘虫。
在另一优选例中,所述杀虫剂还包含其它活性物质,所述其它活性物质选自:杀虫剂、饵剂、杀菌剂、杀螨剂、杀线虫剂、杀真菌剂或生长控制剂。
本发明第三方面提供了一种杀虫和/或防虫方法,所述方法包括步骤:将本发明第二方面所述的杀虫剂组合物或本发明第一方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐施加于遭受或可能遭受虫害的植物体、其周围的土壤或环境中。
本发明第四方面提供了一种杀虫和/或防虫方法,所述方法包括步骤:将本发明第二方面所述的杀虫剂组合物或本发明第一方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐施加于遭受或可能遭受虫害的植物体、其周围的土壤或环境中,并用波长为310-380nm的光照射该植物体、其周围的土壤或环境。
本发明第五方面提供了一种提高杀虫活性的方法,包括步骤:在使用本发明第二方面所述的杀虫剂组合物或本发明第一方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐时,用波长为310-380nm的光照射所述的杀虫剂组合物或所述的化合物或其光学异构体或顺反异构体。
本发明第六方面提供了一种本发明第一方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐在制备杀虫剂组合物中的用途。
本发明第七方面提供了一种本发明第一方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐在杀虫和/或防虫中的用途。
本发明第八方面提供了一种本发明第一方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐的制备方法,
(A)所述方法包括步骤:在惰性溶剂中,将式(1g)化合物与式01化合物和式02化合物进行反应,从而形成式(A)化合物;
或(B)所述方法包括步骤:在惰性溶剂中,将式(2f)化合物与式01化合物和式02化合物进行反应,从而形成式(A)化合物;
各式中,R1、R2、R3、R4、R5、R6、R7、R8、R9、R10、R11、R12、R13、R14、X、Y、Z定义同前。
本发明第九方面提供了一种具有式(B)所示结构的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐:
式中:
R1和R2各自独立地为含氮、氧和/或硫的五元或六元杂环基,或卤代的含氮、氧和/或硫的五元或六元杂环基,取代或未取代的苯基;
R3,R4各自独立地为H、C1-4烷基、烯丙基、苄基、C1-4烷氧基-C1-4烷基、C1-4烷基-羰基、C1-4烷氧基-羰基或苯氧羰基;或者R3和R4和它们相连的原子共同构成-CH2-CH2-或-CH2-CH2-CH2-;
R5,R6各自独立地为H、C1-4烷基、烯丙基、苄基、C1-4烷氧基-C1-4烷基、C1-4烷基-羰基、C1-4烷氧基-羰基或苯氧羰基;或者R5和R6和它们相连的原子共同构成-CH2-CH2-或-CH2-CH2-CH2-;
R7为氢、C1-4烷基、卤素、取代或未取代的5-6元芳环基、或取代或未取代的5-6元芳杂环基;
R8、R9、R10、R11、R12和R13各自独立地为氢、卤素、C1-4烷基、取代或未取代的5-6元芳环基、或取代或未取代的5-6元芳杂环基;或R8、R9和R10、R11的结合构成取代或未取代的5-6元芳环基、或取代或未取代的5-6元芳杂环基;或R12、R13和R10、R11的结合构成取代或未取代的5-6元芳环基、或取代或未取代的5-6元芳杂环基;
R14为C1-4烷基或C1-4烷氧基;
X为硝基、氰基、三氟甲基、三氟乙酰基或三氟甲磺酰基;
Y为N或CH;
Z为S、O或NH;
其中,所述取代是指被选自下组中的一个或多个取代基所取代:卤素、C1-4烷基或卤代C1-4烷基、C1-4烷氧基或卤代C1-4烷氧基、C2-6烯基、C2-6炔基、卤代的C2-6烯基、卤代的C2-6炔基、C1-4烷氧基-C1-4烷基、C1-4烷氧基-羰基、C2-6炔基-羰基、C2-6烯基-羰基。
本发明第十方面提供了一种本发明第九方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐的用途,用于高通量筛选杀虫剂。
本发明第十一方面提供了一种高通量筛选杀虫剂的方法,所述方法包括步骤:
(a)提供一含有已知浓度的本发明第十方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐和已知浓度的烟碱乙酰胆碱受体的溶液;
(b)在步骤(a)的溶液中加入梯度浓度的烟碱乙酰胆碱受体的竞争性配体;
(c)根据荧光偏振技术测定所述竞争性配体的IC50,从而筛选出有活性或活性高的配体。
在另一优选例中,所述烟碱乙酰胆碱受体的浓度高于所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐的浓度和烟碱乙酰胆碱受体的竞争性配体的浓度。
在另一优选例中,所述化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐的浓度通过荧光偏振结合实验确定。
本发明第十二方面还提供了一种将本发明第一方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐和本发明第十方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐互相转换的方法,包括步骤:
将本发明第一方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐在光1照射下发生光致异构反应,从而形成本发明第十方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐,其中,所述光1为波长为310-380nm的光;
将本发明第十方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐在光2照射下发生光致异构反应,从而形成本发明第一方面所述的化合物或其光学异构体或顺反异构体,或其农药学上可接受的盐,其中,所述光2为波长为420-780nm的光;
各式中,R1、R2、R3、R4、R5、R6、R7、R8、R9、R10、R11、R12、R13、R14、X、Y、Z定义同前。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
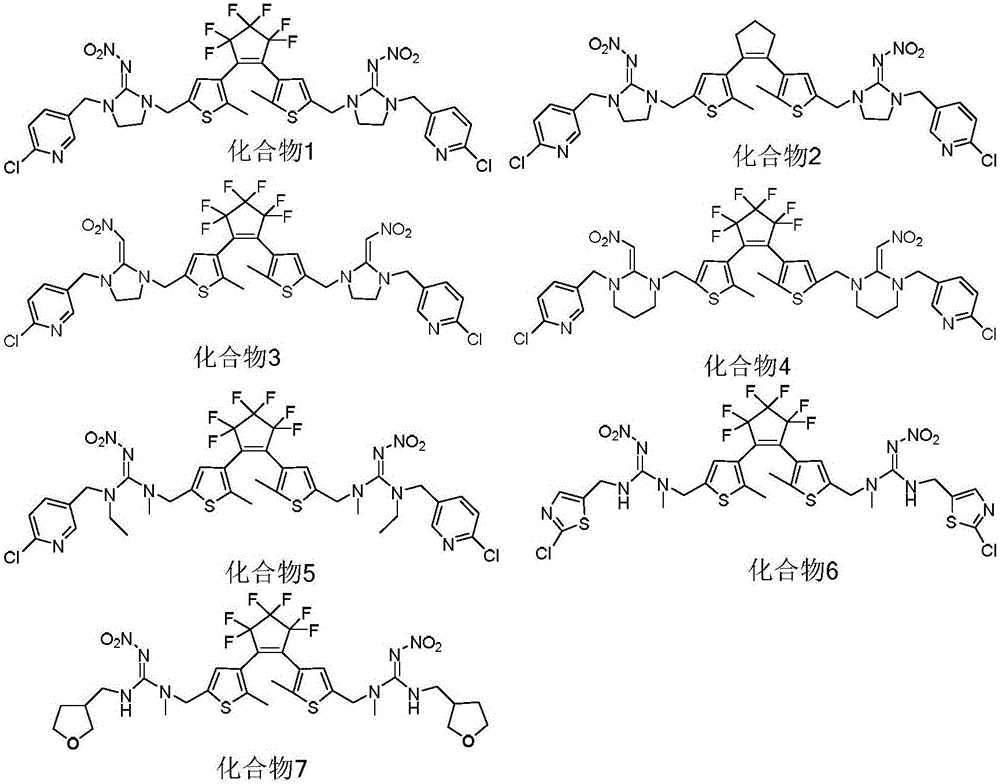
图1为本发明的光致变色二芳基乙烯化合物的分子式。
图2为化合物1在乙腈溶液中紫外光照射前后的吸收光谱图。
图3为化合物1在特定波长处的抗疲劳性。
图4显示了关环化合物1与乙酰胆碱受体结合的最优浓度。
具体实施方式
本发明人通过长期而深入的研究,基于现有的新烟碱杀虫剂的结构和二芳基乙烯光控开关的结构,将二芳基乙烯片段引入已经商品化的新烟碱类的杀虫剂,合成了一类含新烟碱杀虫剂单元的光致变色二芳基乙烯类化合物,该化合物光照关环产物杀虫活性高,并具有荧光。在此基础上,发明人完成了本发明。
基团定义
如本文所用,术语“C1-4烷基”指具有1-4个碳原子的直链或支链烷基,例如甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、或类似基团。
术语“C1-4烷氧基”指具有1-4个碳原子的直链或支链烷氧基,例如甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、异丁氧基、仲丁氧基、叔丁氧基、或类似基团。
术语“卤素”指氟、氯、溴、或碘。术语“卤代的”指被相同或不同的一个或多个上述卤原子取代的基团,例如三氟甲基、五氟乙基、或类似基团。
术语“五元或六元杂环基”指含一个或多个选自氮、氧或硫的杂原子的五元或六元环,例如吡啶基、噻唑基、嘧啶基、四氢呋喃基、或噁唑基等。
术语“5-6元芳环基”是指5元或6元的芳香基,例如苯基等。
术语“5-6元芳杂环基”是指5元或6元的带有杂原子(如氮、氧、硫)的芳香基,例如吡啶基等
术语“杂环”指形成所述杂环骨架的原子中至少一个原子不是碳,为氮、氧或硫。通常,杂环包含不超过4个氮、不超过2个氧和/或不超过2个硫。除非另外指明,杂环可以是饱和的、部分不饱和的或完全不饱和的环。
术语“烷基”是指烷烃分子中少掉一个氢原子而成的基团;术语“亚烷基”是指烷烃分子中少掉两个氢原子而成的基团。类似地,“亚烯基”、“亚炔基”、“亚环烷基”、“亚环烯基”、“亚苯基”、“亚萘基”、“亚杂环基”或“亚杂芳二环或三环环系”的定义类似。
术语“光致变色”是指一个化合物(A),在收到一定波长的光照射时,可进行特定的化学反应或物理效应,获得产物(B),由于结构的改变导致其(可见部分的)吸收光谱发生明显的变化。而在另一波长的光照射或热的作用下,产物(B)又能恢复到原来的形式。
术语“关环产物”是指二芳基乙烯类化合物经特定波长紫外光照射后,发生分子内周环反应关环后得到的化合物。
术语“荧光偏振”是指当荧光分子受平面偏振光激发时,如果分子在受激发时期(对于荧光素约持续4纳秒)保持静止,发射光将位于同样的偏振平面。如果在受激发时期,分子旋转或翻转偏离这一平面,发射光将位于与激发光不同的偏振面。如果用垂直的偏振光激发荧光素,可以在垂直的和水平的偏振平面检测发射光光强(发射光从垂直平面偏向水平平面的程度与荧光素标记的分子的迁移率有关)。如果分子很大,激发时发生的运动极小,发射光偏振程度较高。如果分子小,分子旋转或翻转速度快,发射光相对于激发光平面将去偏振化。
术语“抗疲劳性”是指基于化合物经历光致变色的呈色和消色之间循环次数的评价指标。
术语“高通量筛选”是指技术是指以分子水平和细胞水平的实验方法为基础,以微板形式作为实验工具载体,以自动化操作系统执行试验过程,以灵敏快速的检测仪器采集实验结果数据,以计算机分析处理实验数据,在同一时间检测数以千万的样品,并以得到的相应数据库支持运转的技术体系,它具有微量、快速、灵敏和准确等特点。
术语“惰性溶剂”指的是不与原料发生反应的各种溶剂,包括各种直链、支链或环状的醇,醚或酮,卤代烷,1,4-二氧六环,乙腈,四氢呋喃,N,N-二甲基甲酰胺(DMF),二甲基亚砜(DMSO)等。
本文所用的“组合物”指任何混合物。可以是溶液、混合液、液体、粉末、油膏、水性的、非水性的或他们的任何组合。
这里使用的任何保护性基团,氨基酸和其他化合物的缩写,与他们通用的、公认的缩写或IUPAC-IUB委员会颁布的生化命名一致,除非特别说明。
术语“荧光”,又作"萤光",是指一种光致发光的冷发光现象。当某种常温物质经某种波长的入射光(通常是紫外线或X射线)照射,吸收光能后进入激发态,并且立即退激发并发出比入射光的的波长长的出射光(通常波长在可见光波段);而且一旦停止入射光,发光现象也随之立即消失。具有这种性质的出射光就被称之为荧光。
本发明的化合物可以含有一个或多个不对称中心,并因此以消旋体、外消旋混合物、单一对映体、非对映异构体化合物和单一非对映体的形式出现。可以存在的不对称中心,取决于分子上各种取代基的性质。每个这种不对称中心将独立地产生两个旋光异构体,并且所有可能的旋光异构体和非对映体混合物和纯或部分纯的化合物包括在本发明的范围之内。本发明包括化合物的所有异构形式。
本发明化合物的制备方法
本发明通式(A)所示化合物可通过如下的方法制得,然而该方法的条件,例如反应物、溶剂、酸碱、所用化合物的量、反应温度、反应所需时间等并不限于下面的解释。本发明化合物还可以任选地通过将在本说明书中描述的或本领域已知的各种合成方法组合起来而方便的制得,这样的组合可由本发明所属领域的技术人员容易地进行。
本发明通式(A)所示化合物可通过如下的方法(A)合成,所述方法包括步骤:
(1)在惰性溶剂中,将式(1a)化合物与溴化试剂(如溴)进行反应,从而形成式(1b)化合物;
(2)在惰性溶剂中,将式(1b)化合物与乙二醇进行反应(如在对甲基苯磺酸存在下),从而形成式(1c)化合物;
(3)在惰性溶剂中,将式(1c)化合物与进行反应,从而形成式(1d)化合物:
(4)在惰性溶剂中,将式(1d)化合物进行脱保护反应(如在对甲苯磺酸存在下),从而形成式(1e)化合物;
(5)在惰性溶剂中,将式(1e)化合物进行还原反应(如在硼氢化钠存在下),从而形成式(1f)化合物:
(6)在惰性溶剂中,将式(1f)化合物与氯化试剂(如氯化亚砜)进行反应,从而形成式(1g)化合物:
(7)在惰性溶剂中,将式(1g)化合物与式01化合物和式02化合物进行反应,从而形成式(A)化合物;
本发明通式(A)所示化合物可通过如下的方法(B)合成,所述方法包括步骤:
(1)在惰性溶剂中,将式(2a)化合物与取代的戊二酰氯进行傅克酰化反应(如在三氯化铝存在下),从而形成式(2b)化合物;
(2)在惰性溶剂中,将式(2b)化合物进行关环反应(如在锌和四氯化钛存在下),从而形成式(2c)化合物;
(3)在惰性溶剂中,将式(2c)化合物与二甲基甲酰胺进行反应,从而形成式(2d)化合物;
(4)在惰性溶剂中,将式(2d)化合物进行还原反应(如在硼氢化钠存在下),从而形成式(2e)化合物;
(5)在惰性溶剂中,将式(2e)化合物与氯化试剂(如氯化亚砜)进行反应,从而形成式(2f)化合物;
(6)在惰性溶剂中,将式(2f)化合物与式01化合物和式02化合物进行反应,从而形成式(A)化合物;
各式中,R1、R2、R3、R4、R5、R6、R7、R8、R9、R10、R11、R12、R13、R14、X、Y、Z定义同前。
本发明活性物质的杀虫活性
术语“本发明的活性物质”或“本发明的活性化合物”是指本发明化合物、其光学异构体、顺反异构体或农药学上可接受的盐,其具有显著提高的杀虫活性,以及扩大的杀虫谱。
术语“农药学上可接受的盐”意指该盐的阴离子在形成杀虫剂药学上可接受的盐时为已了解的和可接受的。该盐较好的为水溶性的。合适的、由式(A)化合物形成的酸加成盐包括有无机酸形成的盐,例如盐酸盐、磷酸盐、硫酸盐、硝酸盐;及包括有机酸形成的盐,如醋酸盐,苯甲酸盐。
本发明的活性物质能用作控制和消灭广泛的农林植物害虫、贮藏谷类的害虫、公共卫生害虫以及危害动物健康的害虫等。在本说明书中,“杀虫剂”是具有防治上述提到的所有害虫的作用的物质的统称。害虫的例子包括但不限于:鞘翅目昆虫:玉米象(Sitophilus zeamais),赤拟谷盗(Tribolium castaneum),马铃薯瓢虫(Henosepilachna vigintioctomaculata),二十八星瓢虫(Henosepilachna sparsa),细胸叩头虫(Agriotes fuscicollis),红脚绿金龟(Anomala cupripes),四纹丽金龟(Popillia quadriguttata),马铃薯叶甲(Monolepta hieroglyphica),松天牛(Monochamus alternatus),稻根象(Echinocnemus squameus),泡桐叶甲(Basiprionota bisignata),星天牛(Anoplophora chinensis),桑天牛(Apripona germari),脐腹小蠹(Scolytus schevy),或细胸金针虫(Agriotes fuscicollis)。鳞翅目昆虫:舞毒娥(Lymantria dispar),天幕毛虫(Malacosoma neustria testacea),黄杨绢野螟(Diaphania perspectalis),大袋蛾(Clania variegata),黄刺蛾(Cnidocampa flauescens),马尾松毛虫(Dendrolimus punctatus),古毒蛾(Orgyia gonostigma),白杨透翅蛾(Paranthrene tabaniformis),斜纹夜蛾(Spodoptera litura),二化螟(Chilo suppressalis),玉米螟(Ostrinia nubilalis),粉斑螟(Ephestia cautella),棉卷蛾(Adoxophyes orana),栗子小卷蛾(laspyresia splendana),小地老虎(Agrotis fucosa),大蜡螟(Galleria mellonella),菜蛾(Plutella xylostella),桔潜蛾(Phyllocnistis citrella),或东方粘虫(Mythimna separata)。同翅目昆虫:黑尾叶蝉(Nephotettix cincticeps),稻褐飞虱(Nilaparvata lugens),康氏粉蚧(Pseudococcus comstocki),矢尖蚧(Unaspis yanonensis),桃蚜(Myzus persicae),棉蚜(Aphis gossydii),萝卜蚜(Lipaphis erysimi pseudobrassicae),梨班网蝽(Stephanitis nashi),或粉虱(Bemisia tabaci)。直翅目昆虫:德国小蠊(Blattella germanica),美国大蠊(Periplaneta american),非洲蝼蛄(Gryllotalpa africana),或亚洲飞蝗(Locus migratoria)。等翅目昆虫:入侵红火蚁(Solenopsis invicta),或家白蚁(Coptotermes formosanus)。双翅目昆虫:家蝇(Musca domestica),埃及伊蚊(Aedes aegypti),种蝇(Delia platura),库蚊(Culex sp.),或中华按蚊(Anopheles sinensis)。
本发明涉及的化合物尤其对刺吸式、锉吸式口器害虫如:蚜虫、叶蝉、飞虱、蓟马、粉虱等农林害虫有特效以及对卫生害虫蚊子幼虫有特效。
含本发明活性物质的杀虫剂组合物
可将本发明的活性物质以常规的方法制备成杀虫剂组合物。这些活性化合物可做成常规的制剂,例如溶液剂,乳剂,混悬剂,粉剂,泡沫剂,糊剂,颗粒剂;气雾剂,用活性物质浸渍的天然的和合成的材料,在多聚物中的微胶囊,用于种子的包衣复方,和与燃烧装置—块使用的制剂,例如烟熏药筒,烟熏罐和烟熏盘,以及ULV冷雾(Cold mist)和热雾(Warm mist)制剂。
这些制剂可用已知的方法生产,例如,将活性化合物与扩充剂混合,这些扩充剂就是液体的或液化气的或固体的稀释剂或载体,并可任意选用表面活性剂即乳化剂和/或分散剂和/或泡沫形成剂。例如在用水作扩充剂时,有机溶剂也可用作助剂。
用液体溶剂作稀释剂或载体时基本上是合适的,如:芳香烃类,例如二甲苯,甲苯或烷基萘;氯化的芳香或氯化的脂肪烃类,例如氯苯,氯乙烯或二氯甲烷;脂肪烃类,例如环己烷或石蜡,例如矿物油馏分;醇类,例如乙醇或乙二醇以及它们的醚和脂类;酮类,例如丙酮,甲乙酮,甲基异丁基酮或环已酮;或不常用的极性溶剂,例如二甲基甲酰胺和二甲基亚砜,以及水。液化气的稀释剂或载体是指在常温常压下将成为气体的液体,例如气溶胶推进剂,如卤化的烃类以及丁烷,丙烷,氮气和二氧化碳。
固体载体可用研磨的天然的矿物质,例如高岭土,粘土,滑石,石英,活性白土,蒙脱土,或硅藻土,和研磨合成的矿物质,例如高度分散的硅酸,氧化铝和硅酸盐。供颗粒用的固体载体是碾碎的和分级的天然告石,例如方解石,大理石,浮石,海泡石和白云石,以及无机和有机粗粉合成的颗粒,和有机材料例如锯木屑,椰子壳,玉米棒子和烟草梗的颗粒等。
非离子的和阴离子的乳化列可用作乳化剂和/或泡沫形成剂。例如聚氧乙烯-脂肪酸酯类,聚氧乙烯-脂肪醇醚类,例如烷芳基聚乙二醇醚类,烷基磺酸酯类,烷基硫酸酯类,芳基磺酸酯类以及白蛋白水解产物。分散剂包括,例如木质素亚硫酸盐废液和甲基纤维素。
在制剂中可以用粘合剂,例如羧甲基纤维素和以粉末,颗粒或乳液形式的天然和合成的多聚物,例如阿拉伯胶,聚乙烯基醇和聚乙烯醋酸酯。可以用着色剂例如无机染料,如氧化铁,氧化钻和普鲁士蓝;有机染料,如有机染料,如偶氯染料或金属钛菁染料;和用痕量营养剂,如铁,猛,硼,铜,钴,铝和锌的盐等。
本发明的这些活性化合物可与其它活性化合物制成混合物存在于商品制剂中或从这些制剂制备的使用剂型中,所述其它的活性化合物包括但不限于:杀虫剂,合饵,杀菌剂,杀螨剂,杀线,杀真菌剂,生长控制剂等。杀虫剂包括,例如磷酸酯类,氨基甲酸酯类,除虫菊酯类,氯化烃类,苯甲酰脲类,沙蚕毒素类以及由微生物产生的物质,如阿维菌素。
此外,本发明的这些活性化合物也可与增效剂制成一种混合物存在于它们的商品制剂中成从这些制剂制备的使用剂型中。增效剂是提高活性化合物作用的化合物,由于活性化合物本身有活性,也可不必加增效剂。
这些制剂通常含有占所述杀虫剂组合物0.001-99.99重量%,优选0.01-99.9重量%,更优选0.05-90重量%的本发明的活性化合物。从商品制剂制成使用剂型中的活性化合物的浓度可在广阔的范围内变动。使用剂型中的活性化合物的浓度可从0.0000001-100%(g/v),最好在0.0001与1%之间。
本发明的优点主要包括:
发明人创新地将二芳基乙烯片段引入已经商品化的新烟碱类的杀虫剂中,合成了一类含新烟碱杀虫剂单元的光致变色二芳基乙烯类化合物,全面探索了这类化合物的两种异构体在光物理学方面和杀虫活性方面的差异,实现了通过光来调控杀虫剂性能的目的。
本发明提供的一类含新烟碱杀虫剂单元的光致变色二芳基乙烯类化合物,光致变色明显,热稳定性好,抗疲劳性好,关环产物杀虫活性高,可用作杀虫剂,防治农业害虫;同时关环产物具有荧光,可利用荧光偏振技术进行药物的高通量筛选。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。其中,r.t.或rt代表室温。
实施例1化合物1的合成
1.1制备4-溴-5-甲基-2-噻吩甲醛
在100mL圆底烧瓶中加入5-甲基-2-噻吩甲醛(6.3g,50.0mmol)和50mL乙酸,然后用恒压滴液漏斗称取Br2(8.78g,55.0mmol)溶于适量乙酸,室温下慢慢滴加溴的乙酸溶液,室温搅拌24h,TLC跟踪反应结束后,加入水停止反应。反应混合物用无水碳酸钠中和至中性。产物用乙醚萃取三次,有机相用无水硫酸钠干燥,过滤,减压除去溶剂得黑色液体,柱层析,用石油醚:乙酸乙酯=40:3(v:v)洗脱。蒸干溶剂得到白色固体3.1g,产率为30.1%。1H NMR(400MHz,CDCl3)δ9.78(s,1H),7.26(s,1H),2.49(s,3H)。13C NMR(101MHz,CDCl3)δ181.60,145.84,140.12,138.73,111.24,15.93。
1.2制备4-溴-5-甲基-2-(1,3-二氧戊环)噻吩
在100mL圆底烧瓶中加入4-溴-5-甲基-2-噻吩甲醛(2.0g,9.75mmol)、乙二醇(1.23g,19.51mmol)和对甲苯磺酸(0.15g,97.53mmol),加入60mL苯溶解,装上分水器,搅拌回流反应4h,TLC跟踪反应结束后,将反应液温度降至室温,用3.0mol/L的氢氧化钠溶液洗三次,再用水洗涤三次,有机相用无水硫酸钠干燥,过滤,减压除去溶剂得黄色液体,柱层析,用石油醚:乙酸乙酯=20:1(v:v)洗脱。蒸干溶剂得到白色油状物2.4g,产率为90.8%。1H NMR(400MHz,CDCl3)δ6.96(s,1H),6.00(s,1H),4.18-3.88(m,4H),2.38(s,3H)。13C NMR(101MHz,CDCl3)δ138.70,135.35,128.79,108.45,99.69,65.19,14.86。
1.3制备1,2-二[2-甲基-5-(1,3-二氧戊环)-3-噻吩基]八氟环戊烯
在氮气及-78℃条件下,在50mL低温反应瓶中加入4-溴-5-甲基-2-(1,3-二氧戊环)噻吩(2.4g,9.63mmol),加入30mL无水四氢呋喃,搅拌,慢慢用注射器加入2.5mol/L n-BuLi/正己烷(7.7mL,19.27mmol),继续低温搅拌反应40min,用预先冷却的注射器慢慢加入八氟环戊烯(1.23g,5.78mmol),继续低温搅拌反应2h,TLC跟踪反应结束后,将反应液自然升至室温,用浓度为2M的盐酸(15mL)淬灭反应,分液,水层用无水乙醚萃取三次,合并有机层,然后用水洗涤三次,有机层用无水硫酸钠干燥,过滤,减压除去溶剂得油状液体2.0g,此粗产物不需要进一步的分离,直接投入下一步脱保护反应。
1.4制备1,2-二(2-甲基-5-甲酰基-3-噻吩基)八氟环戊烯
在250mL圆底烧瓶中加入粗产物1,2-二[2-甲基-5-(1,3-二氧戊环)-3-噻吩基]八氟环戊烯(2.0g,3.90mmol)和对甲苯磺酸(0.4g,2.22mmol),加入混合溶剂水(30mL)和丙酮(80mL)溶解,然后向混合物中加入2.0mL吡啶,搅拌回流反应24h。TLC跟踪反应结束后,将反应液温度降至室温,反应液分别饱和碳酸氢钠溶液和水洗涤,分液,水层用二氯甲烷萃取,合并有机相,用无水硫酸钠干燥,过滤,减压除去溶剂得黄色液体,柱层析,用石油醚:乙酸乙酯=1:5(v:v)洗脱。蒸干溶剂得到蓝色固体0.15g,产率为8.7%。1H NMR(400MHz,CDCl3)δ9.86(s,2H),7.74(s,2H),2.03(s,6H)。19F NMR(376MHz,CDCl3)δ-110.30(t,J=5.1Hz,4F),-130.57--133.99(m,2F)。
1.5制备1,2-二(2-甲基-5-羟甲基-3-噻吩基)八氟环戊烯
在氮气及冰浴条件下,在50mL圆底烧瓶中加入1,2-二(2-甲基-5-甲酰基-3-噻吩基)八氟环戊烯(0.45g,1.06mmol),加入35mL甲醇溶解,然后分批加入硼氢化钠(0.16g,4.24mmol)。继续冰浴下搅拌反应1h,TLC跟踪反应结束后,加入适量的水淬灭反应,然后反应混合物用乙醚(30mL×2)萃取,合并有机相,有机相用饱和氯化钠溶液洗涤三次,无水硫酸钠干燥,过滤,减压除去溶剂得黄色固体,柱层析,用石油醚:乙酸乙酯=1:1(v:v)洗脱。蒸干溶剂得到白色固体0.45g,产率为99%。1H NMR(400MHz,CDCl3)δ6.95(s,2H),4.76(s,4H),1.88(s,8H)。19F NMR(376MHz,CDCl3)δ-110.10(t,J=10.9Hz,4F),-131.48--134.27(m,2F)。
1.6制备1,2-二(2-甲基-5-氯甲基-3-噻吩基)八氟环戊烯
在氮气及冰浴条件下,在100mL圆底烧瓶中加入1,2-二(2-甲基-5-羟甲基-3-噻吩基)八氟环戊烯(0.25g,0.58mmol)和无水吡啶(1.0mL),加入40mL无水四氢呋喃溶解,搅拌。将氯化亚砜(0.28g,2.33mmol)称在恒压滴液漏斗里,慢慢滴加到反应体系中,继续冰浴搅拌反应2h。TLC跟踪反应结束后,将反应液慢慢倒入40mL冰水混合物中,搅拌。然后反应混合物用乙醚(30mL×2)萃取,合并的有机相用30mL饱和碳酸氢钠洗涤,无水硫酸钠干燥,过滤,减压除去溶剂得淡黄色固体0.35g,产率为92%。产物不进行纯化立即投入下一步反应中。
1.7制备化合物1
在氮气及50℃油浴条件下,在50mL圆底烧瓶中加入吡虫啉(0.40g,1.55mmol)和碳酸铯(0.72g,2.19mmol),加入30mL二甲基甲酰胺溶解,50℃恒温搅拌反应半个小时。然后慢慢加入用5mL二甲基甲酰胺溶解的1,2-二(2-甲基-5-氯甲基-3-噻吩基)八氟环戊烯,继续恒温反应4h。TLC跟踪反应结束后,过滤除去沉淀物,减压除去溶剂得棕红色固体,柱层析,用二氯甲烷:甲醇=20:1(v:v)洗脱,蒸干溶剂得到白色固体0.35g,产率为60%。m.p.108.7-110.9℃。1H NMR(400MHz,DMSO-d6)δ8.35(d,J=2.0Hz,2H),7.78(dd,J=8.3,2.4Hz,2H),7.55(d,J=8.2Hz,2H),7.10(s,2H),4.53(d,J=28.1Hz,4H),4.46(s,4H),3.77-3.47(m,8H),1.83(d,J=13.4Hz,6H).13C NMR(101MHz,DMSO-d6)δ160.19,149.72,149.53,149.38,142.96,139.42,135.74,130.39,127.34,123.28,46.52,45.57,45.02,44.15,13.90。19F NMR(376MHz,DMSO-d6)δ-109.35(t,J=10.9Hz,4F),-131.11(m,4F)。HRMS(ESI-TOF):m/z C35H3035Cl2F6N10O4S2Na[M+Na]+计算值:925.1072,实测值:925.1080;m/z C35H3035Cl37ClF6N10O4S2Na[M+Na]+计算值:927.1042,实测值:927.1025;m/z C35H3037Cl2F6N10O4S2Na[M+Na]+计算值:929.1013,实测值:929.1030.
实施例2化合物2的合成
2.1制备1,5-二(5-氯-2-甲基噻吩-3-基)戊烷-1,5-二酮
在50mL圆底烧瓶中依次加入无水三氯化铝(12.1g,90.49mmol)以及硝基甲烷150mL,冰浴条件下,先滴加戊二酰氯(6.37,37.71mmol)的硝基甲烷溶液,再滴加2-氯-5-甲基噻吩(10g,75.41mmol)的硝基甲烷溶液。加完后,继续反应0.5h,然后撤去冰浴,室温条件下继续搅拌反应6h,TLC跟踪反应结束后,小心慢慢加入60mL冰水混合物,继续搅拌5min,分液分出有机相,水层用无水乙醚(50mL×2)萃取,合并有机相,用饱和碳酸氢钠溶液调至中性,然后用水洗涤,饱和氯化钠溶液洗涤,无水硫酸镁干燥过夜,旋蒸除去大量溶剂,将得到的粘稠黑色粗产品用石油醚和乙酸乙酯过硅胶柱(7:3,v/v),蒸干溶剂得到16g淡黄色固体粉末,产率为59%。1H NMR(400MHz,CDCl3)δ7.19(s,2H),2.93-2.78(t,J=18Hz,4H),2.66(s,6H),2.06(m,2H)。13C NMR(101MHz,CDCl3):δ194.7,147.6,134.7,126.7,125.2,40.4,18.1,16.0。
2.2制备1,2-二(5-氯-2-甲基噻吩-3-基)环戊烯
在氮气保护条件下,在250mL三口烧瓶中加入100mL无水四氢呋喃和锌粉(7.24g,110.71mmol),冰浴,剧烈搅拌条件下,缓慢滴加四氯化钛溶液(16.80g,88.57mmol),滴加完毕后继续冰浴搅拌5min,转移到油浴,升温至80℃,加热回流1小时,反应液颜色逐渐由黄绿色变成深黑色。自然冷却至室温,用针筒注入40mL含1,5-二(5-氯-2-甲基噻吩-3-基)戊烷-1,5-二酮(16g,44.29mmol)的无水四氢呋喃溶液,继续加热回流4小时,停止加热,冷却后将反应液倒入10%的碳酸钾水溶液中,析出大量固体,抽滤,滤饼用无水乙醚反复洗涤,用乙醚(50mL×2)萃取水层,合并有机相,无水硫酸钠干燥过夜,过滤,旋蒸除掉溶剂,粗品用石油醚为洗脱剂,过硅胶柱,蒸干溶剂后得到无色油状液体3.3g,低温静置成无色蜡状固体,产率为30%。1H NMR(400MHz,CDCl3)δ6.57(s,2H),2.71(t,J=7.5Hz,4H),2.07-1.96(m,2H),1.88(s,6H)。13C NMR(101MHz,CDCl3)δ134.82,134.41,133.28,126.67,125.18,38.32,22.81,14.16。
2.3制备1,2-二(5-甲酰基-2-甲基噻吩-3-基)环戊烯
在氮气及-78℃条件下,在50mL低温反应瓶中加入1,2-二(5-氯-2-甲基噻吩-3-基)环戊烯(1.50g,4.56mmol),加入10mL无水四氢呋喃,搅拌,慢慢用注射器加入2.5mol/L n-BuLi/正己烷(5.5mL),继续低温搅拌反应60min,然后用预先冷却的注射器慢慢加入DMF(2.29mL),继续低温搅拌反应1h,TLC跟踪反应结束后,将反应液自然升至室温,用浓度为2M的盐酸(15mL)淬灭反应,分液,水层用无水乙醚(20mL×2)萃取,合并有机层,然后用水洗涤三次,有机层用无水硫酸钠干燥,过滤,减压除去溶剂,将得到的粗产品用石油醚和乙酸乙酯过硅胶柱(5:1,v/v),蒸干溶剂得到1.5g蓝色固体粉末,产率为46%。1H NMR(400MHz,CDCl3)δ9.74(s,2H),7.43(s,2H),2.84(t,J=7.5Hz,4H),2.17-2.07(m,2H),2.05(s,6H)。13C NMR(101MHz,CDCl3)δ182.38,146.38,140.23,137.42,137.07,134.98,38.41,22.89,15.38。
2.4制备1,2-二(5-羟甲基-2-甲基噻吩-3-基)环戊烯
在氮气及冰浴条件下,在50mL圆底烧瓶中加入1,2-二(5-甲酰基-2-甲基噻吩-3-基)环戊烯(0.87g,2.75mmol),加入35mL甲醇溶解,然后分批加入硼氢化钠(0.42g,11.0mmol)。继续冰浴下搅拌反应1h,TLC跟踪反应结束后,加入适量的水淬灭反应,然后反应混合物用乙醚(30mL×2)萃取,合并有机相,有机相用饱和氯化钠溶液洗涤三次,无水硫酸钠干燥,过滤,减压除去溶剂得黄色固体,柱层析,用石油醚:乙酸乙酯=6:1(v:v)洗脱。蒸干溶剂得到白色固体0.86g,产率为98%。1H NMR(400MHz,CDCl3)δ6.61(s,2H),4.61(s,4H),2.75(t,J=7.4Hz,4H),2.32(s,2H),2.06-1.98(m,2H),1.95(s,6H)。13C NMR(101MHz,CDCl3)δ139.35,135.33,134.93,134.56,126.91,59.91,38.25,22.98,14.36。
2.5制备1,2-二(5-氯甲基-2-甲基噻吩-3-基)环戊烯
在氮气及冰浴条件下,在100mL圆底烧瓶中加入1,2-二(5-羟甲基-2-甲基噻吩-3-基)环戊烯(0.86g,2.86mmol)和无水吡啶(1.0mL),加入40mL无水四氢呋喃溶解,搅拌。将氯化亚砜(1.28g,10.73mmol)称在恒压滴液漏斗里,慢慢滴加到反应体系中,继续冰浴搅拌反应2h。TLC跟踪反应结束后,将反应液慢慢倒入40mL冰水混合物中,搅拌。然后反应混合物用乙醚(30mL×2)萃取,合并的有机相用30mL饱和碳酸氢钠洗涤,无水硫酸钠干燥,过滤,减压除去溶剂得棕色固体。产物不进行纯化立即投入下一步反应中。
2.6制备化合物2
在氮气及50℃油浴条件下,在50mL圆底烧瓶中加入吡虫啉(1.37g,5.37mmol)和碳酸铯(2.48g,7.61mmol),加入30mL二甲基甲酰胺溶解,50℃恒温搅拌反应半个小时。然后慢慢加入用5mL二甲基甲酰胺溶解的1,2-二(5-氯甲基-2-甲基噻吩-3-基)环戊烯(0.80g,2.24mmol),继续恒温反应4h。TLC跟踪反应结束后,过滤除去沉淀物,减压除去溶剂得棕红色固体,柱层析,用二氯甲烷:甲醇=20:1(v:v)洗脱,蒸干溶剂得到白色固体0.61g,产率为34%。m.p.193.4-196.5℃。1H NMR(400MHz,DMSO-d6)δ8.34(d,J=2.0Hz,2H),7.77(dd,J=8.2,2.3Hz,2H),7.55(d,J=8.2Hz,2H),6.73(s,2H),4.45(s,8H),3.68-3.60(m,4H),3.60-3.52(m,4H),2.72(t,J=7.4Hz,4H),1.98(m,2H),1.86(s,6H)。13C NMR(101MHz,DMSO-d6)δ160.16,149.71,149.37,139.42,134.92,134.74,134.10,132.27,130.40,128.99,124.26,46.51,45.50,44.76,44.22,37.68,22.35,13.82。HRMS(ESI-TOF):m/z C35H3635Cl2N10O4S2Na[M+Na]+计算值:817.1637,实测值:817.1635;m/z C35H3635Cl37ClN10O4S2Na[M+Na]+计算值:819.1608,实测值:819.1606;m/z C35H3637Cl2N10O4S2Na[M+Na]+计算值:821.1578,实测值:821.1581。
实施例3化合物3的合成
目标化合物的合成与实施例1的步骤1.1-1.6相同,不同之处在最后一步。
在氮气及50℃油浴条件下,在50mL圆底烧瓶中加入(E)-1-((6-氯吡啶-3-基)甲基)-2-(硝基亚甲基)咪唑烷(0.40g,1.55mmol)和碳酸铯(0.77g,2.19mmol),加入30mL二甲基甲酰胺溶解,50℃恒温搅拌反应半个小时。然后慢慢加入用5mL二甲基甲酰胺溶解的1,2-二(2-甲基-5-氯甲基-3-噻吩基)八氟环戊烯,继续恒温反应4h。TLC跟踪反应结束后,过滤除去沉淀物,减压除去溶剂得棕红色固体,柱层析,用二氯甲烷:甲醇=20:1(v:v)洗脱,蒸干溶剂得到白色固体0.40g,产率为65%。m.p.101.7-103.9℃.1H NMR(400MHz,DMSO-d6)δ8.36(d,J=2.0Hz,2H),7.72(dd,J=8.3,2.4Hz,2H),7.53(d,J=8.2Hz,2H),7.11(s,2H),4.53(d,J=28.1Hz,4H),4.40(s,4H),4.20(s,2H),3.67-3.47(m,8H),1.80(d,J=13.4Hz,6H)。13C NMR(101MHz,DMSO-d6)δ161.19,142.72,142.53,144.38,145.96,138.42,137.74,131.39,126.34,122.28,45.52,45.57,43.02,41.15,12.90。19F NMR(376MHz,DMSO-d6)δ-108.35(t,J=11.9Hz,4F),-130.11(m,4F).HRMS(ESI-TOF):m/z C37H3235Cl2F6N8O4S2Na[M+Na]+计算值:923.1269,实测值:923.1272;m/z C37H3235Cl37ClF6N8O4S2Na[M+Na]+计算值:925.1253,实测值:925.1264;m/z C37H3237Cl2F6N8O4S2Na[M+Na]+计算值:927.1246,实测值:927.1260。
实施例4化合物4的合成
目标化合物的合成与实施例1的步骤1.1-1.6相同,不同之处在最后一步。
在氮气及50℃油浴条件下,在50mL圆底烧瓶中加入(E)-1-((6-氯吡啶-3-基)甲基)-2-(硝基亚甲基)六氢嘧啶(0.50g,1.55mmol)和碳酸铯(0.91g,2.19mmol),加入30mL二甲基甲酰胺溶解,50℃恒温搅拌反应半个小时。然后慢慢加入用5mL二甲基甲酰胺溶解的1,2-二(2-甲基-5-氯甲基-3-噻吩基)八氟环戊烯,继续恒温反应4h。TLC跟踪反应结束后,过滤除去沉淀物,减压除去溶剂得棕红色固体,柱层析,用二氯甲烷:甲醇=20:1(v:v)洗脱,蒸干溶剂得到白色固体0.80g,产率为46%。m.p.110.7-112.9℃.1H NMR(400MHz,DMSO-d6)δ8.31(d,J=2.0Hz,2H),7.72(dd,J=8.3,2.4Hz,2H),7.51(d,J=8.2Hz,2H),7.11(s,2H),4.52(d,J=28.1Hz,4H),4.42(s,4H),4.22(s,4H),3.77-3.47(m,8H),1.85(d,J=13.4Hz,6H)。13C NMR(101MHz,DMSO-d6)δ159.19,143.72,140.53,141.38,140.96,137.42,133.74,129.39,127.34,120.28,45.52,44.57,43.03,40.32,15.55。19F NMR(376MHz,DMSO-d6)δ-112.45(t,J=10.9Hz,4F),-135.11(m,4F).HRMS(ESI-TOF):m/z C39H3635Cl2F6N8O4S2Na[M+Na]+计算值:951.1582,实测值:951.1562;m/z C39H3635Cl37ClF6N8O4S2Na[M+Na]+计算值:953.1437,实测值:953.1440;m/z C39H3637Cl2F6N8O4S2Na[M+Na]+计算值:955.1256,实测值:955.1235。
实施例5化合物5的合成
目标化合物的合成与实施例1的步骤1.1-1.6相同,不同之处在最后一步。
在氮气及50℃油浴条件下,在50mL圆底烧瓶中加入烯啶虫胺(0.50g,1.55mmol)和碳酸铯(0.90g,2.19mmol),加入30mL二甲基甲酰胺溶解,50℃恒温搅拌反应半个小时。然后慢慢加入用5mL二甲基甲酰胺溶解的1,2-二(2-甲基-5-氯甲基-3-噻吩基)八氟环戊烯,继续恒温反应4h。TLC跟踪反应结束后,过滤除去沉淀物,减压除去溶剂得棕红色固体,柱层析,用二氯甲烷:甲醇=20:1(v:v)洗脱,蒸干溶剂得到白色固体0.60g,产率为35%。m.p.120.7-121.5℃.1H NMR(400MHz,DMSO-d6)δ8.20(d,J=2.0Hz,2H),7.56(dd,J=8.3,2.4Hz,2H),7.46(d,J=8.2Hz,2H),7.03(s,2H),4.45(d,J=28.1Hz,4H),4.37(s,4H),3.5(q,4H),3.27-2.90(m,8H),2.80(t,6H),2.60(s,6H),1.78(d,J=13.4Hz,6H)。13C NMR(101MHz,DMSO-d6)δ169.35,159.21,143.98,142.36,142.78,138.78,137.25,136.20,125.23,116.36,49.37,46.36,45.27,42.28,19.01。19F NMR(376MHz,DMSO-d6)δ-120.36(t,J=10.9Hz,4F),-140.19(m,4F).HRMS(ESI-TOF):m/z C37H3835Cl2F6N10O4S2Na[M+Na]+计算值:957.1803,实测值:957.1805;m/z C37H3835Cl37ClF6N10O4S2Na[M+Na]+计算值:959.1489,实测值:959.1428;m/z C37H3837Cl2F6N10O4S2Na[M+Na]+计算值:961.8954,实测值:961.8937。
实施例6化合物6的合成
目标化合物的合成与实施例1的步骤1.1-1.6相同,不同之处在最后一步。
在氮气及50℃油浴条件下,在50mL圆底烧瓶中加入噻虫胺(0.50g,1.55mmol)和碳酸铯(0.97g,2.19mmol),加入30mL二甲基甲酰胺溶解,50℃恒温搅拌反应半个小时。然后慢慢加入用5mL二甲基甲酰胺溶解的1,2-二(2-甲基-5-氯甲基-3-噻吩基)八氟环戊烯,继续恒温反应4h。TLC跟踪反应结束后,过滤除去沉淀物,减压除去溶剂得棕红色固体,柱层析,用二氯甲烷:甲醇=20:1(v:v)洗脱,蒸干溶剂得到白色固体0.60g,产率为34%。m.p.123.1-124.5℃.1H NMR(400MHz,DMSO-d6)δ8.35(d,J=2.0Hz,2H),8.25(d,J=1.5Hz,2H),8.10(s,2H),3.77-3.47(m,8H),2.30(s,6H)2.25(d,J=12.4Hz,6H),1.87(d,J=13.4Hz,6H)。13C NMR(101MHz,DMSO-d6)δ158.11,143.31,141.34,140.38,140.29,139.22,137.25,134.23,129.35,128.28,42.68,41.91,40.37,40.41,17.49。19F NMR(376MHz,DMSO-d6)δ-124.17(t,J=10.9Hz,4F),-135.17(m,4F).HRMS(ESI-TOF):m/z C29H2635Cl2F6N10O4S2Na[M+Na]+计算值:903.0301,实测值:903.0311;m/z C29H2635Cl37ClF6N10O4S2Na[M+Na]+计算值:905.1239,实测值:905.1254;m/z C29H2637Cl2F6N10O4S2Na[M+Na]+计算值:907.8375,实测值:907.8371。
实施例7化合物7的合成
目标化合物的合成与实施例1的步骤1.1-1.6相同,不同之处在最后一步。
在氮气及50℃油浴条件下,在50mL圆底烧瓶中加入噻虫胺(0.50g,1.55mmol)和碳酸铯(1.20g,2.19mmol),加入30mL二甲基甲酰胺溶解,50℃恒温搅拌反应半个小时。然后慢慢加入用5mL二甲基甲酰胺溶解的1,2-二(2-甲基-5-氯甲基-3-噻吩基)八氟环戊烯,继续恒温反应4h。TLC跟踪反应结束后,过滤除去沉淀物,减压除去溶剂得棕红色固体,柱层析,用二氯甲烷:甲醇=20:1(v:v)洗脱,蒸干溶剂得到白色固体0.80g,产率为42%。m.p.99.7-100.1℃.1H NMR(400MHz,DMSO-d6)δ8.29(d,J=2.0Hz,2H),8.24(d,J=1.5Hz,2H),3.56-3.43(m,8H),3.52-3.41(m,4H),3.31-3.23(m,4H),3.12-3.01(m,4H),2.29(s,6H)2,12(d,J=12.4Hz,6H),1.90(m,2H),1.79(d,J=13.4Hz,6H)。13C NMR(101MHz,DMSO-d6)δ149.12,143.31,141.34,140.31,140.23,139.89,137.48,134.29,129.93,128.19,43.68,41.91,38.37,39.41,11.39。19F NMR(376MHz,DMSO-d6)δ-119.35(t,J=10.9Hz,4F),-141.12(m,4F).HRMS(ESI-TOF):m/z C31H38F6N8O6S2Na[M+Na]+计算值:819.2268,实测值:819.2245。
图1显示了上述光致变色二芳基乙烯化合物的分子式。
实施例8本发明化合物的杀虫活性测试
(1)对蚜虫的杀虫活性
蚜虫俗称腻虫或蜜虫等,属于同翅目害虫,是一种常见的农作物害虫。蚜虫主要分布在北半球温带地区和亚热带地区,热带地区分布很少。蚜虫为刺吸式口器的害虫,常群集于叶片、嫩茎、花蕾、顶芽等部位,刺吸汁液,使叶片皱缩、卷曲、畸形,严重时引起枝叶枯萎甚至整株死亡。蚜虫分泌的蜜露还会诱发煤污病、病毒病并招来蚂蚁危害等。蚜虫繁殖和适应力强,各种防治方法都很难取得根治的效果。对于蚜虫防治,重要的是抓紧防治,避免蚜虫大量发生。以豆蚜(Aphis craccivora)为测试对象,采用浸渍法测试。
操作过程:选取健壮的豆蚜无翅成虫(虫体呈黑绿色,有光泽,腹部饱满。各足褪节、胫节、跗节均暗黑色,其余部分黄白色)若干。避光,在黑暗条件下饥饿处理0.5-1h。选取长势良好,高约3cm的蚕豆豆苗,洗去沙土(注意尽量保持豆皮完整),掐去豆根,插入带水海绵上,使豆苗保持水分供给的充足。将经过饥饿处理的豆蚜抖落在豆苗周围,在阳光下让蚜虫爬苗,并刺入口针(耗时约2-3h),最终使每棵豆苗爬有蚜虫约20-30头。配制所需浓度的化合物溶液,分为两组,第一组不经紫外光的照射(未光照组),第二组用310nm紫外灯照射一定的时间(光照组,放在黑暗条件下),溶液中DMSO含量不超过5%,溶液中含有少量表面活性剂曲拉通(500ml清水中含曲拉通2-3滴)。采用浸苗法处理蚜虫,将爬有豆蚜的豆苗在已配好的两组溶液中浸泡2-3s,连续3次。然后用吸水纸吸干豆苗上多余的溶液,每个浓度3个重复。然后将豆苗插入带水海绵上,罩上马灯罩,避光,在25℃恒温条件下放置48h。48h后观察并统计光照组和未光照组蚜虫死亡情况,并根据公式计算死亡率(%):死亡率(%)=(对照活虫数-处理活虫数)/对照活虫数×100%。结果见表1。
(2)对白纹伊蚊4龄幼虫的杀虫活性
白纹伊蚊(Aedes albopictus)俗称黑蚊子、花蚊子、花斑蚊、花脚蚊,生性凶猛、攻击力强,也被称为"亚洲虎蚊",属于长角亚目蚊科,中等大小的黑色蚊虫,上面具有白色鳞片形成的斑纹,白纹伊蚊源于东南亚,是东南亚和中国的常见蚊种。白纹伊蚊幼虫(孑孓)无脊椎动物,昆虫纲,双翅目蚊类的幼虫。它是由雌蚊在淡水中产的卵孵化而成,其体细长,胸部较头部及腹部宽大,游泳时身体一屈一伸,通称跟头虫。白纹伊蚊喜欢在小面积的积水上产卵,而且其幼虫具有"嗜静"的特性。水环境安静、阴凉,不易受打扰,最适合这种伊蚊产卵以及孑孓存活。孑孓身体细长,呈深褐色,在水中上下垂直游动,以水中的细菌和单细胞藻类为食,呼吸空气。幼虫主要孳生于人房附近的竹筒、树洞、石穴、废轮胎以及缸罐、瓦钵、泡菜缸等容器积水,也见于菠萝等植物叶腋。孑孓经4次蜕皮后发育成蛹,初期蛹仍可活动,将羽化时则基本不可动,而后羽化为成蚊。白纹伊蚊既是一种攻击性很强的蚊子,也是一种重要的病毒媒介,它可以传播很多病原体,包括登革热、罗斯河病毒和西尼罗病毒。蚊虫的综合治理方法包括环境治理、生物防治、化学防治、法规防治和遗传防治等。化学防治是通过对孳生地投药,对白纹伊蚊的成蚊直接喷洒起到了较好作用,但是在投入较大的人力、物力、财力的同时对环境造成了不同程度的污染,化学药物的应用也使蚊虫产生抗药性。因此研究高效、低毒的杀虫剂来杀灭蚊子的幼虫,可以有效的控制蚊子数量的增加,抑制人类疾病的传播。
操作过程:选取大小一致的4龄白纹伊蚊幼虫若干。将待测化合物以DMSO助溶(溶液中DMSO含量不超过5%),用除氯水稀释,准确配制成800、400、200、100、50、25、12.5、6.25、3.13、1.57ppm溶液,设置两组实验,第一组不经紫外光的照射(未光照组),第二组用310nm紫外灯照射一定的时间(光照组,放在黑暗条件下)。然后取配制好的各浓度的化合物2mL加入到4mL的离心管中,再分别转移30只孑孓进入相应的离心管,最后补加2mL的除氯水,使溶液体积为4mL,最终化合物浓度为400、200、100、50、25、12.5、6.25、3.13、1.57、0.79ppm,并以除氯水溶液处理作空白对照,以吡虫啉作为阳性对照,每个浓度设3个重复。将光照组和未光照组都放入黑暗条件下,在25℃恒温条件下放置24h。24h后观察并统计光照组和未光照组孑孓死亡情况,并根据公式计算死亡率(%):死亡率(%)=(对照活虫数-处理活虫数)/对照活虫数×100%。结果见表1。
表1.式(A)化合物的杀虫活性
实施例9化合物1的光致变色现象
光致变色现象(photochromism)是指化合物A,在受到特定波长光照射后,能够进行特定的化学反应,进而生成产物B,而在另一波长光照射或者热的作用下,又可恢复到原来的状态的现象。
二芳基乙烯类化合物也是一类基于周环反应的有机光致变色化合物。此类化合物的母体结构是一个6π电子共轭的己三烯。在紫外光激发下,处于反平行状态的分子发生分子内的顺旋关环反应,形成共轭的闭环态分子而呈色。而在可见光的照射后,闭环态分子开环恢复至原来状态。经过适当的分子结构修饰,此类化合物的闭环态可以呈现出从黄色到绿色的全部颜色。
以化合物1为例说明:采用310nm的紫外光照射化合物1乙腈溶液,在可见光区520nm处出现一个很宽的吸收峰,且随着照射时间的延长而吸收峰的强度增强,关环产物浓度逐渐增大。大约经过90s照射后,化合物1达到开环闭环的平衡态(图2),可以观察到溶液的颜色很快由无色变为红色,这说明化合物1在紫外光的作用下发生了闭环的光致变色反应。然后用430nm的蓝光照射关环化合物1,可以观察到溶液颜色由红色变为无色,说明关环化合物1在可见光下发生了开环的光致变色反应,反应式如下:
结合实施例8和实施例9可知:关环化合物的杀虫效果更好。
实施例10化合物1的耐疲劳性
耐疲劳性是基于化合物经历光致变色的呈色和消色之间循环次数的评价指标。光致变色反应总是伴随着化学键的改变,在此过程中,副反应的发生不可避免。这无疑会导致化合物的部分降解,降低化合物的耐疲劳度。二芳基乙烯化合物的耐疲劳性除了受到环境因素的影响外,其自身分子结构的差异也会造成其耐疲劳度的不同。在分子结构方面,芳基上取代基的类型会对其耐疲劳性造成很大的影响。
以化合物1为例说明:当采用紫外光和可见光交替照射化合物1的乙腈溶液时,它表现出良好的耐疲劳性。当用310nm紫外光照射时,化合物1的乙腈溶液由无色变为红色,并且在520nm附近处出现了新的特征吸收峰,这表明它形成了闭环体的化合物。而当大于400nm光照射时,溶液红色消失,闭环态的化合物发生开环反应变成开环态的化合物。如图3所示,整个成色和漂白过程可以重复6次以上而未发生大的衰减,这表明化合物1具有良好的耐疲劳性。
实施例11荧光偏振实验及对新烟碱杀虫剂进行高通量筛选
本发明的化合物1关环后有荧光,并且与烟碱乙酰胆碱受体有很好的结合力,我们利用荧光偏振实验确定了化合物及蛋白结合时的最优浓度,并通过竞争性实验首次对新烟碱杀虫剂进行高通量筛选。
实验方法:
制备烟碱乙酰胆碱受体溶液
解剖:选取美洲大蠊雄性成虫(数量根据实验需要而定),75%酒精体表消毒,背部向上固定在解剖蜡盘中。在解剖镜下,沿背中线剪开体壁,去掉背板,剔除消化道、气管、脂肪体等杂质,剖离出神经节用漂洗2次,整个过程在室温下操作。解剖过程中要不断更换生理溶液,使神经索始终浸泡于新鲜生理溶液A中(放在冰上)。
匀浆:将上述含有神经元细胞的溶液用聚四氟乙烯匀浆器匀浆。
离心:将上述匀浆在4℃、25000*g离心30min,沉淀用缓冲溶液A清洗两遍。然后用缓冲溶液A重新悬浮沉淀(放在冰上30min),再次用相同的条件离心,用pH7.4缓冲液B(50mM Tris-HCl、120mM NaCl)重悬浮沉淀,-80℃保存备用。
测定关环化合物1与蛋白结合时的最优浓度
标准曲线测定:
考马斯亮蓝法测定蛋白标准曲线(8个浓度,每个浓度3个重复,每孔加297μL考马斯亮蓝液和3μL标准蛋白液)。酶标仪检测。
蛋白浓度检测:
考马斯亮蓝法测定蛋白的浓度,96孔板,3个平行,每个平行加入297μL考马斯亮蓝液和3μL蛋白液。酶标仪检测。
荧光偏振结合实验:
实验在全黑96孔发光板(上海卧宏生物科技有限公司)中进行,每个浓度3个重复。配置1.95*100、3.91*100、7.81*100、15.625*100、31.25*100、62.50*100和80*100μM梯度浓度的关环化合物1,将制备的蛋白浓度稀释到229mg/L。首先,在全黑96孔发光板中加入99μl蛋白液(终浓度227mg/L),然后每孔加入1μl梯度浓度的关环化合物1(终浓度为1.95、3.91、7.81、15.625、31.25、62.50和80μM)。将全黑96孔板放入22℃恒温培养箱中培育1h。Synergy H1酶标仪(Bio-Tek)检测荧光偏振值,激发波长和发射波长分别设定为420nm和590nm。
实验结果见表2和图4:
表2
从图4可以看出当化合物浓度达到40μM时,FP值达到平衡。
通过竞争性实验对新烟碱杀虫剂进行高通量筛选
实验在全黑96孔发光板(上海卧宏生物科技有限公司)中进行,每个浓度3个重复。配置0.001*101、0.003*101、0.01*101、0.03*101、0.1*101、0.3*101、1*101、3*101和10*101μM梯度浓度的吡虫啉、噻虫啉、啶虫脒、烯啶虫胺、噻虫嗪、噻虫胺、呋虫胺、烟碱、地棘蛙素、杀螟丹、多杀菌素、氟虫腈、毒死蜱和高效氯氰菊酯,配置4.04mM关环化合物1,将制备的蛋白液浓度稀释到232mg/L。首先,在全黑96孔发光板中加入99μl蛋白液(终浓度为227mg/L),然后每孔加入1μl梯度浓度的关环化合物1(终浓度为40μM),最后加入配置好的上述商品化农药(终浓度为0.001、0.003、0.01、0.03、0.1、0.3、1、3和10μM)。将全黑96孔板放入22℃恒温培养箱中培育1h。Synergy H1酶标仪(Bio-Tek)检测荧光偏振值,激发波长和发射波长分别设定为420nm和590nm。
竞争性药物的IC50值见表3。
表3.烟碱乙酰胆碱受体配体的IC50值
实施例12含有本发明化合物的杀虫剂组合物的制备
(a)油状悬浮液
按比例准备以下组分:25%(重量百分比,下同)化合物1-7中任一种化合物;5%聚氧乙烯山梨醇六油酸酯;70%高级脂肪族烃油。将各组分在沙磨中一起研磨,直到固体颗粒降至约5微米以下为止。所得的粘稠悬浮液可直接使用,但也在水中乳化后使用。
(b)水悬浮液
按比例准备以下组分:25%化合物1-7中任一种化合物;3%水合硅镁土(hydrate attapulgit);10%木质素磺酸钙;0.5%磷酸二氢钠;61.5%水。将各组分在球磨机中一起研磨,直到固体颗粒降至约10微米以下为止。该水悬浮液可直接使用。
(c)饵剂
按比例准备以下组分:0.1-10%化合物1-7中任一种化合物;80%小麦面粉;19.9-10%糖蜜。将这些组分完全混合,按需要形成饵形状。可食用饵可以分散到卫生害虫所侵染的场所,例如家居或工业场所,诸如厨房、医院或商店或户外区域,以通过口服摄入来防治害虫。
本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!