用于检测基因组中遗传剪刀的脱靶位点的方法与流程
本披露涉及用于检测基因组中可编程核酸酶的脱靶位点的方法,并且具体来说涉及用于通过数据分析检测脱靶位点的方法,所述方法包括通过用可编程核酸酶处理在体外分离的基因组(无细胞基因组dna)来裂解基因组,然后进行全基因组测序;并且涉及用于使用这种方法选择rgen的中靶位点的方法,所述方法将脱靶效应降到最低。
背景技术:
来源于ii型crispr/cas(规律间隔成簇重复序列/crispr相关)原核适应性免疫系统等的可编程核酸酶(例如zfn(锌指核酸酶)、talen(转录激活因子样效应因子核酸酶)和rgen(rna指导的工程化核酸酶))广泛用于培养细胞和完整生物体中的基因组编辑。使用可编程核酸酶的基因组编辑技术是非常有用的技术,其可用于生命科学、生物技术和医学领域中的多种用途。例如,用于种种遗传性或获得性疾病的基因/细胞疗法已通过在干细胞或体细胞中引发靶向遗传修饰而变得可能。然而,可编程核酸酶不仅可使中靶位点突变,还可使与其同源的脱靶位点突变(nucleicacidsresearch,2013,41(20):9584-9592)。
作为代表性例子,包括来源于酿脓链球菌(s.pyogenes)的cas9蛋白和小指导rna(sgrna)的rgen识别23-bp(碱基对)靶dna序列,所述靶dna序列由与该sgrna杂交的20-bp(碱基对)序列和由cas9识别的5'-ngg-3'前间区序列邻近基序(protospacer-adjacentmotif,pam)序列组成,但可容忍多达数个核苷酸序列处的错配(genomeres,2014,24:132-141)。另外,rgen也可裂解与该sgrna序列相比具有额外碱基序列(dna凸起)或缺少碱基(rna凸起)的脱靶dna序列。同样,zfn和talen二者也可裂解在一些碱基上不同的序列。这表明,在将可编程核酸酶应用于基因组的情形中,除了中靶位点以外,可能还存在大量的脱靶位点。
脱靶dna裂解可导致非预期基因(例如原癌基因和肿瘤抑制基因)突变,以及大范围(gross)基因组重组(例如易位、缺失和倒位),并且引起在研究和医学中使用可编程核酸酶的严重担忧(procnatlacadsci,2009,106:10620-10625)。就这一点而言,已报道多种策略可降低可编程核酸酶的脱靶效应,尚未报道在整个基因组规模中特异性地作用于中靶位点而没有脱靶效应的可编程核酸酶。为了解决这个问题,必须研发询问可编程核酸酶在基因组规模上的特异性的方法。
发明概述
[技术问题]
由于诸位发明人尽力研发能在基因组规模上检测并分析可编程核酸酶的靶位点和脱靶位点的系统,已研发出用于通过用可编程核酸酶裂解基因组后进行全基因组测序(wgs)来检测可编程核酸酶的脱靶位点的方法来完成本发明(消化基因组测序(digenome-seq),核酸酶裂解的基因组dna测序)。
[技术方案]
本披露的目的在于提供用于检测可编程核酸酶的脱靶位点的方法,其包括:(a)用靶特异性可编程核酸酶裂解分离的基因组dna;(b)进行所裂解dna的全基因组测序;和(c)测定通过该测序获得的序列读取中的经裂解位点。
本披露的另一目的在于提供用于在基因组编辑中降低脱靶效应的方法,其包括:使用质粒作为模板将体外转录的指导rna引入细胞中。
[效果]
本披露的消化基因组测序可在基因组规模上以高再现性检测出可编程核酸酶的脱靶位点,并且因此可用于生产和研究具有高靶特异性的可编程核酸酶。
附图说明
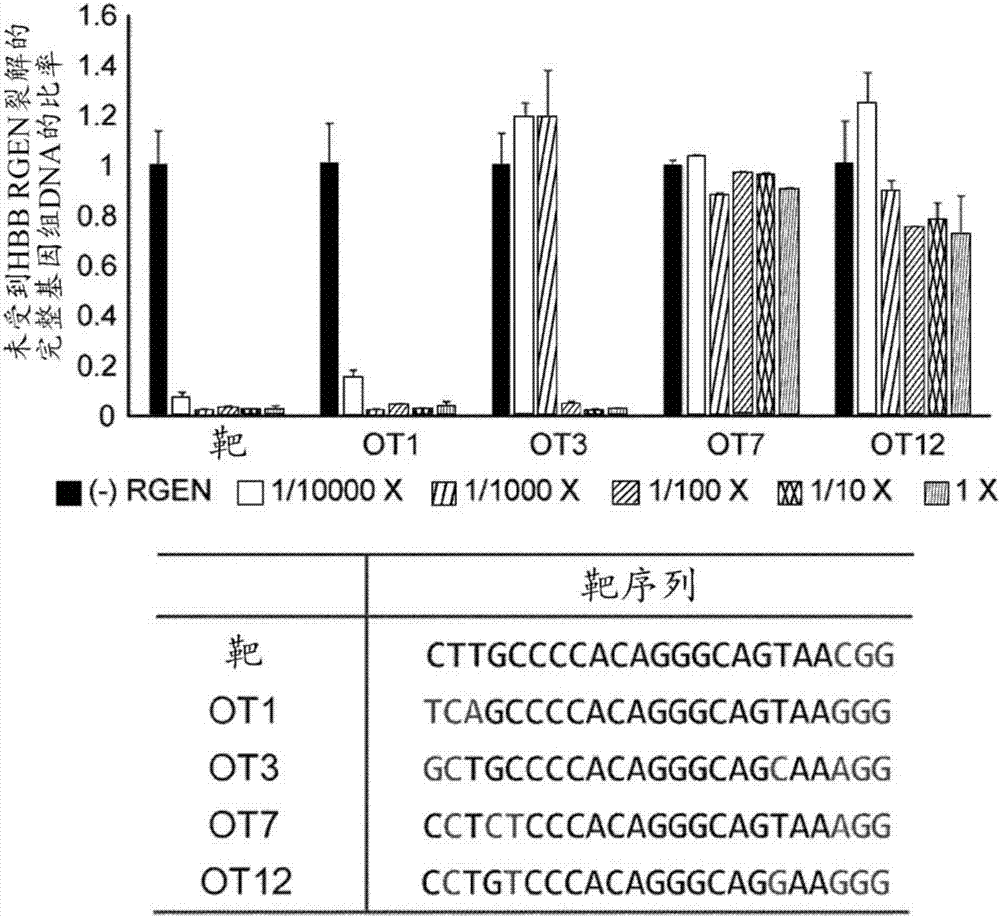
图1涉及rgen介导的体外基因组dna裂解。(a)其为rgen介导的体外基因组dna裂解的模拟图。(b)其鉴别基因组dna是否被靶向hbb的rgen在中靶位点和4个潜在脱靶位点处裂解。对于1x反应,使cas9蛋白(40μg,300nm)和sgrna(30μg,900nm)与8μghap1基因组dna反应8小时。将cas9和sgrna连续稀释10倍到10,000倍。通过qpcr测量未裂解的dna。(下图)其展示了中靶位点和4个潜在脱靶位点的dna序列。错配核苷酸以红色显示,并且pam序列以蓝色显示。(c)其用t7e1分析来测量中靶位点和潜在脱靶位点处由rgen导致的突变频率。(d)其进行靶向深度测序以测量插缺(indel)频率。
图2涉及用于鉴别脱靶位点的rgen诱导的消化基因组测序。(a)其为用于鉴别脱靶位点的核酸酶裂解的全基因组测序(wgs)的模拟图。将从未转化或经rgen转化的细胞中分离的基因组dna通过rgen裂解,并经历wgs。将序列读取与参考基因组(hg19)比对并使用igv程序可视化。正向和反向序列读取分别以橙色和天蓝色显示。红色三角形和垂直虚线指示裂解位置。(b)其为使用hbb特异性rgen在中靶位点处获得的代表性igv数据。插缺由箭头指示。(c)其显示了根据核苷酸位置具有相同5'端的序列读取的绝对数目和相对数目。
图3涉及用于鉴别脱靶位点的rgen诱导的消化基因组测序。(a-d)其为使用hbb特异性rgen在潜在脱靶位点ot1(a)、ot3(b)、ot7(c)和ot12(d)处获得的代表性igv数据。插缺由箭头指示(a)或显示于框中(b)。
图4展示了基因组上的特定位置处的5'端数目的图。(a)其显示了核酸酶裂解位点处的igv数据。(b、c)其展示了显示出在ot1(b)和ot3(c)位点处根据核苷酸位置具有相同5'端的序列读取的绝对数目和相对数目的5'端图。
图5展示了通过消化基因组测序鉴别并通过靶向深度测序验证的hbbrgen的脱靶位点。(a)其为显示出在未转化或经rgen转化的细胞中通过使用hbbrgen的消化基因组测序鉴别的中靶位点和脱靶位点的数目的维恩图(venndiagram)。(b)其展示了比较通过消化基因组测序鉴别的位点与中靶位点的热图。(c)其展示了使用通过消化基因组测序鉴别的位点处的dna序列通过weblogo获得的序列标志。(d)其为消化基因组测序和靶向深度测序的结果汇总。n.d.意味着未测得。(e)其展示了通过靶向深度测序验证的脱靶位点。蓝条和红条代表使用未转化hap1细胞和经hbbrgen转化的hap1细胞获得的插缺频率。(左图)其展示了中靶位点和脱靶位点的dna序列。错配碱基以红色显示,并且pam序列以蓝色显示。(右图)通过费雪精确检验(fisherexacttest)计算p值。
图6展示了在完整基因组序列中鉴别的假阳性位置。(a-c)其为假阳性位点周围的代表性igv数据,所述假阳性位点是由于hap1细胞中天然存在的插缺产生的。
图7展示了在新验证的脱靶位点处由hbbrgen诱导的插缺序列。(a,b)通过靶向深度测序检测脱靶插缺。插入的核苷酸以红色显示并且pam序列以蓝色显示。
图8展示了通过消化基因组测序鉴别的vegf-argen的脱靶位点。(a)其展示了在一个vegf-a脱靶位点处的5'端数目的图。(b)其为比较通过消化基因组测序鉴别的位点与中靶位点的热图。深红色和深蓝色对应给定位置处的100%和0%匹配。(c)其展示了使用通过消化基因组测序鉴别的位点处的dna序列通过weblogo获得的序列标志。(d)其为消化基因组测序和靶向深度测序的结果汇总。n.d.意味着未测得。(e)其展示了通过靶向深度测序验证的脱靶位点。蓝条和红条代表使用未转化hap1细胞和经vegf-argen转化的hap1细胞获得的插缺频率。(左图)其展示了中靶位点和脱靶位点的dna序列。错配碱基以红色显示,并且pam序列以蓝色显示。(右图)通过费雪精确检验计算p值。
图9展示了用于鉴别vegf-argen的脱靶位点的rgen诱导的消化基因组测序。(a-d)其展示了显示出根据中靶位点位点(a)和脱靶位点(b-d)中的核苷酸位置具有相同5'端的序列读取的绝对数目和相对数目的5'端图。
图10展示了在新验证的脱靶位点处由vegf-argen诱导的插缺序列。(a-d)通过靶向深度测序检测脱靶插缺。插入的核苷酸以红色显示并且pam序列以蓝色显示。
图11展示了用于消化基因组测序分析的体外dna裂解评分系统。
图12展示了经改良的消化基因组测序分析。(a)其展示了体外dna裂解分数的基因组规模circos图。使用人类基因组dna(红色)和经rgen裂解的基因组dna(绿色)进行全基因组测序(wgs)。(b)其展示了使用寡核苷酸双链或从质粒转录的sgrna的消化基因组测序的模拟图。(c)其展示了使用寡核苷酸双链或从质粒转录的sgrna获得的序列标志。
图13展示了体外dna裂解评分系统的再现性。
图14展示了使用从寡核苷酸双链转录的sgrna通过消化基因组测序鉴别的凸起型脱靶位点。
图15展示了多重消化基因组测序。(a)其展示了多重消化基因组测序的模拟图。(b)其展示了显示出通过单一和多重消化基因组测序分析鉴别的体外裂解位点数目的维恩图。(c)其展示了通过单一或多重消化基因组测序获得的x-染色体上的体外dna裂解分数。
图16展示了通过多重消化基因组测序鉴别的位点的分析。(a)通过消化基因组测序、guide-seq和htgts鉴别的位点数目显示于维恩图中。(b)其展示了根据错配总数(上图)和种子区域中的错配数目(下图),通过消化基因组测序鉴别的位点的百分比。(c)人类基因组中具有少于或等于6个核苷酸的错配的位点数目和通过消化基因组测序鉴别的位点数目以散点图显示(上图)。将11个rgen中靶位点分为两个组:g1(人类基因组中少于13,000个具有少于或等于6个核苷酸的错配的位点)和g2(人类基因组中多于或等于16,000个具有少于或等于6个核苷酸的错配的位点)(下图)。误差条代表sem。通过学生t检验计算p值。(d)通过guide-seq鉴别的位点数目和通过消化基因组测序鉴别的位点数目以散点图显示。
图17说明guide-seq阳性位点数目与人类基因组中具有少于或等于6个核苷酸的错配的同源位点数目之间缺乏相关性。
图18展示了两个emx1脱靶位点,其是通过htgts和guide-seq鉴别出的,但未通过消化基因组测序鉴别出。
图19展示了呈维恩图的通过消化基因组测序和chip-seq鉴别的位点数目。
图20展示了在经rnf2特异性sgrna转化的hela细胞中中靶位点和脱靶位点的插缺频率(对数标度)。
图21使用靶向深度测序在脱靶位点处鉴别插缺频率。(a)其以模拟方式展示了一般sgrna(gx19sgrna)和经修饰sgrna(ggx20sgrna)。(b-d)其展示了通过ngs验证的(b)emx1、(c)hek293-3和(d)rnf2sgrna的中靶位点和脱靶位点的插缺频率。(e-g)其展示了通过将(e)emx1、(f)hek293-3和(g)rnf2sgrna的中靶位点处的插缺频率除以脱靶位点处的插缺频率计算出的特异性比率。
图22展示了通过ngs验证的脱靶位点和未通过ngs验证的脱靶位点的分析。(a-c)其展示了根据(a)整个20-nt序列或(b和c)10-nt种子序列中显示的错配在脱靶位点处的相对插缺频率(对数标度)图。将通过ngs鉴别的位点(a)分为两组:经验证位点(b)和未验证位点(c)。
图23展示了对100个中靶位点进行的消化基因组测序的结果。(a)其以模拟方式展示了测试过程,并且(b)其展示了比较基于消化基因组测序预测脱靶位点的程序与其他程序(crop-it)的结果。
图24在基因组规模上通过消化基因组测序展示了zfn(锌指核酸酶)的脱靶效应。(a)其为在zfn-224处理之前和之后中靶位点的代表性igv照片。(b)其展示了在基因组规模上显示出未处理基因组dna(红色)、经zfn-224(wtfoki)裂解的dna(绿色)和经zfn-224(kk/elfoki)裂解的dna(蓝色)的体外dna裂解分数的circos图。(c-d)其展示了使用zfn-224(wtfoki)或zfn-224(kk/elfoki)中的候选脱靶位点获得的序列标志。
图25展示了在zfn的消化基因组测序中检测脱靶位点的结果。(a)通过在zfn-224(kk/elfoki)的候选脱靶位点处使用靶向深度测序测量插缺频率。(b-c)其为显示出(b)在体外检测的候选脱靶位点和(c)经验证的中靶位点的消化基因组测序、ildv和数目的维恩图。
发明详述
根据一个方面,为了实现本披露的这个目的,提供了用于检测基因组中的脱靶位点的方法,所述方法包括:(a)用靶特异性可编程核酸酶裂解分离的基因组dna;(b)进行所裂解dna的全基因组测序;和(c)测定通过该测序获得的序列读取中的经裂解位点。诸位发明人将所述方法命名为“消化基因组测序”,其是指核酸酶裂解的基因组dna测序。
基因组编辑/基因编辑技术是可将靶定向突变引入动物和植物细胞(包括人类细胞)的基因组碱基序列中的技术。其可敲除或敲入特定基因,或可将突变引入不产生蛋白质的非编码dna序列中。本披露的方法检测这种基因组编辑/基因编辑技术中使用的可编程核酸酶的脱靶位点,所述技术可用于有效地研发仅特异性地作用于中靶位点的可编程核酸酶。
步骤(a)是用靶特异性可编程核酸酶裂解分离的基因组dna的步骤,也就是在体外用特异性地作用于中靶位点的可编程核酸酶裂解该分离的基因组dna的步骤。然而,即使可编程核酸酶是针对靶特异性地产生的,其他位点(也就是脱靶位点)也可取决于特异性而被裂解。于是结果,通过步骤(a),所用的靶特异性可编程核酸酶裂解中靶位点位置和多个脱靶位点,从而获得特定位点被裂解的基因组dna,所述可编程核酸酶可对基因组dna具有活性。基因组dna的类型并无特别限制,并且可为野生型细胞或经转化细胞的基因组dna。另外,取决于消化基因组测序的目的,经转化细胞可被转化以表达特定的可编程核酸酶。
本披露中所用的术语“可编程核酸酶”是指所有形式的核酸酶,其能识别并裂解所需基因组上的特定位点。具体地,其可包括但不限于与来源于植物致病基因的转录激活因子样效应因子(tal)结构域(其是识别基因组上的特定靶序列的结构域)和裂解结构域融合的转录激活因子样效应因子核酸酶(talen)、锌指核酸酶、大范围核酸酶、来源于crispr(其为微生物免疫系统)的rgen(rna指导的工程化核酸酶)、cpf1、ago同系物(dna指导的内切核酸酶)等。
可编程核酸酶识别动物和植物细胞(包括人类细胞)的基因组中的特定碱基序列,以引发双链断裂(dsb)。双链断裂包括通过裂解dna双链导致的平端或粘端二者。dsb通过细胞内的同源重组或非同源末端连接(nhej)机制得到有效修复,从而容许研究者在此过程期间将所需突变引入中靶位点中。可编程核酸酶可为人工的或经操作非天然存在的。
本披露中所用的术语“中靶位点”意指通过使用可编程核酸酶将突变引入其中的位点,并且可取决于其目的而任意选择。所述位点可为非编码dna序列,其可存在于特定基因内且不产生蛋白质。
可编程核酸酶具有序列特异性,并且由此作用于中靶位点,但是可取决于靶序列而作用于脱靶位点。本披露中所用的术语“脱靶位点”是指如下位点,其中可编程核酸酶在具有与该可编程核酸酶的靶序列不同的序列的位点处具有活性。也就是说,脱靶位点是指由可编程核酸酶裂解的并非中靶位点的位点。具体地,本披露中的脱靶位点不仅包括特定可编程核酸酶的实际脱靶位点,而且还包括可能变为脱靶位点的位点。脱靶位点可为但不限于由可编程核酸酶在体外裂解的位点。
可编程核酸酶甚至在并非中靶位点的位点处具有活性的事实可能是由于多种原因可引起的现象所致。然而,具体地,在与中靶位点具有高序列同源性的脱靶序列具有针对该中靶位点设计的靶序列和核苷酸错配的情形中,可编程核酸酶有可能发挥作用。脱靶位点可为但不限于具有靶序列和一个或多个核苷酸错配的位点。
其可导致基因组中非预期基因的突变,并引起可编程核酸酶的使用的严重担忧。就这一点而言,精确检测并分析脱靶位点以及基因可编程核酸酶在中靶位点的活性的方法也可非常重要,并且可有效地用于研发仅特异性地作用于中靶位点而没有脱靶效应的可编程核酸酶。
可编程核酸酶可选自下组,该组由以下各项组成:大范围核酸酶、zfn(锌指核酸酶)、talen(转录激活因子样效应因子核酸酶)、rgen(rna指导的工程化核酸酶)、cpf1。只要可编程核酸酶识别靶基因的特定序列并具有核苷酸裂解活性并且可在靶基因中引起插入和缺失(插缺),其就可被包括于但不限于本披露的范围中。
大范围核酸酶可为但不限于天然存在的大范围核酸酶,其识别15至40个碱基对的裂解位点,所述大范围核酸酶通常归类为4个家族:laglidadg家族、giy-yig家族、his-cyst盒家族和hnh家族。示例性大范围核酸酶包括i-scei、i-ceui、pi-pspi、pi-scei、i-sceiv、i-csmi、i-pani、i-sceii、i-ppoi、i-sceiii、i-crei、i-tevi、i-tevii和i-teviii。
已在植物、酵母、果蝇(drosophila)、哺乳动物细胞和小鼠中使用来源于天然存在的大范围核酸酶、主要来自laglidadg家族的dna结合结构域促成了位点特异性基因组修饰。这种方法基于同源基因的修饰,其中大范围核酸酶靶序列是保守的(monet等人(1999)biochem.biophysicsres.common.255:88-93),并且对该靶序列引入其中的预工程化基因组的修饰有限制。因此,已尝试将大范围核酸酶工程化以展现在医学或生物技术相关位点处的新颖的结合特异性。另外,来源于大范围核酸酶的天然存在的或工程化的dna结合结构域可操作地连接至来源于异源核酸酶(例如,fokl)的裂解结构域。
zfn包括所选基因和经工程化以结合至裂解结构域或裂解半结构域的中靶位点的锌指蛋白。zfn可为包括锌指dna结合结构域和dna裂解结构域的人工限制酶。此处,锌指dna结合结构域可经工程化以结合至所选序列。例如,beerli等人(2002)naturebiotechnol.20:135-141;pabo等人(2001)ann.rev.biochem.70:313-340;isalan等人(2001)naturebiotechnol.19:656-660;segal等人(2001)curr.opin.biotechnol.12:632-637;choo等人(2000)curr.opin.struct.biol.10:411-416可作为参考材料被包括在本说明书中。与天然存在的锌指蛋白相比,工程化锌指结合结构域可具有新颖的结合特异性。工程化方法包括但不限于对各种类型的合理设计和选择。合理设计包括使用含有例如三联(或四联)核苷酸序列以及单个锌指氨基酸序列的数据库,其中每个三联或四联核苷酸序列与结合至特定三联或四联序列的锌指的一或多个序列相关。
靶序列的选择以及融合蛋白(及其编码多核苷酸)的设计与构建为本领域技术人员所熟知,并且详细描述于美国专利申请公开案第2005/0064474号和第2006/0188987号的全文中。所述公开案的全部披露内容都作为本披露的参考文献而被包括在本说明书中。另外,如在这些参考文献和相关领域的其他参考文献中所披露的,锌指结构域和/或多指锌指蛋白可通过包括任何适宜的接头序列的接头(例如长度为5个或更多个氨基酸的接头)连接在一起。长度为6个或更多个氨基酸的接头序列的例子披露于美国专利第6,479,626号;第6,903,185号;第7,153,949号中。本文中解释的蛋白质可包括该蛋白质的每一锌指之间的适宜接头的任何组合。
另外,核酸酶(例如zfn)含有核酸酶活性部分(裂解结构域、裂解半结构域)。众所周知,裂解结构域可与dna结合结构域异源,例如像,来自核酸酶的裂解结构域与锌指dna结合结构域不同。异源裂解结构域可从任何内切核酸酶或外切核酸酶获得。可得到裂解结构域的示例性内切核酸酶包括但不限于限制性内切核酸酶和大范围核酸酶。
类似地,裂解半结构域可来源于任何核酸酶或其一部分,其裂解活性需要二聚作用,如上文所指示的。如果融合蛋白包括裂解半结构域,通常两个融合蛋白需要裂解。可替代地,可使用包括两个裂解半结构域的单一蛋白质。两个裂解半结构域可来源于相同的内切核酸酶(或其功能性片段),或每一裂解半结构域可来源于不同的内切核酸酶(或其功能性片段)。另外,两个融合蛋白的中靶位点是以使得裂解半结构域通过两个融合蛋白与其相应的中靶位点结合而在空间上朝向彼此定向的方式来定位的。因此,优选的是布置裂解半结构域以能够通过二聚作用形成功能性裂解结构域。因此,在一个实施方案中,中靶位点的相邻边缘由3至8个核苷酸或14至18个核苷酸隔开。然而,可将任何整数的核苷酸或核苷酸对插入两个中靶位点之间(例如,2至50个或更多个核苷酸对)。通常,裂解位点位于中靶位点之间。
限制性内切核酸酶(限制酶)存在于许多物种中,可序列特异性地结合至dna(在中靶位点处),并且在结合位点处或结合位点附近直接裂解dna。一些限制酶(例如,iis型)在远离识别位点的位点处裂解dna,并且具有可分离的结合结构域和可裂解结构域。例如,iis型酶foki在一条链上离识别位点9个核苷酸处和在另一条链上离识别位点13个核苷酸处催化dna双链断裂。因此,在一个实施方案中,融合蛋白包括来自至少一种iis型限制酶的裂解结构域(或裂解半结构域)和一个或多个锌指结合结构域(可经或可未经工程化)。
本披露中所用的术语“talen”是指能识别并裂解dna靶区域的核酸酶。talen是指包括tale结构域和核苷酸裂解结构域的融合蛋白。在本披露中,术语“tal效应因子核酸酶”和“talen”可互换。tal效应因子作为在黄单胞菌属(xanthomonas)细菌被多种植物物种感染时由其iii型分泌系统分泌的蛋白质为人所知。该蛋白质可与宿主植物中的启动子序列组合以激活帮助细菌感染的植物基因的表达。该蛋白质通过由34个或更少的不同数目的氨基酸重复序列组成的中央重复结构域识别植物dna序列。因此,预期tale是基因组工程化工具的新平台。然而,为了构建具有基因组编辑活性的功能性talen,迄今尚未得知的一些关键参数应该如下定义。i)tale的最小dna-结合结构域,ii)构成一个靶区域的两个半指(half-digit)之间的间隔区的长度,和iii)连接foki核酸酶结构域与dtale的接头或融合接点。
本披露的tale结构域是指以序列特异性方式通过一或多个tale重复模块结合核苷酸的蛋白质结构域。tale结构域包括但不限于至少一个tale重复模块,并且更具体地,1至30个tale重复模块。在本披露中,术语“tal效应因子结构域”与“tale结构域”可互换。tale结构域可包括tale重复模块的一半。涉及此talen的国际专利公开案第wo/2012/093833号或美国专利申请公开案第2013-0217131号中披露的全部内容作为参考文献被包括在本说明书中。
本披露中所用的术语“rgen”意指包括靶dna特异性指导rna和cas蛋白作为组分的核酸酶。
在本披露中,rgen可应用于但不限于呈靶dna特异性指导rna形式的体外分离的基因组dna和分离的cas蛋白。
指导rna可在体外转录,并且具体地,其可以但不限于从寡核苷酸双链或质粒模板转录。
在本披露中,术语“cas蛋白”是crispr/cas系统的主要蛋白质组分,并且是能形成活化内切核酸酶或切口酶的蛋白质。
cas蛋白可与crrna(crisprrna)和tracrrna(反式激活crrna)形成复合物以展现该cas蛋白的活性。
cas蛋白或基因信息可从已知数据库获得,例如国家生物技术信息中心(nationalcenterforbiotechnologyinformation,ncbi)的genbank。具体地,该cas蛋白可为cas9蛋白。另外,该cas蛋白可为链球菌属(streptococcusgenus),更具体地,来源于酿脓链球菌(streptococcuspyogens)的cas蛋白,并且更具体地是cas9蛋白。另外,该cas蛋白可为奈瑟氏菌属(neisseriagenus),更具体地,来源于脑膜炎奈瑟氏菌(neisseriameningitidis)的cas蛋白,并且更具体地是cas9蛋白。另外,该cas蛋白可为巴斯德菌属(pasteurellagenus),更具体地,来源于多杀巴斯德菌(pasteurellamultocida)的cas蛋白,并且更具体地是cas9蛋白。另外,该cas蛋白可为弗朗西斯菌属(francisellagenus),更具体地,来源于新凶手弗朗西斯菌(francisellanovicida)的cas蛋白,并且更具体地是cas9蛋白。另外,该cas蛋白可为弯曲杆菌属(campylobactergenus),更具体地,来源于空肠弯曲杆菌(campylobacterjejuni)的cas蛋白,并且更具体地是cas9蛋白。然而,本披露并不限于上述例子。
另外,该cas蛋白在本披露中作为如下概念使用:包括能与指导rna合作用作活化的内切核酸酶或切口酶的天然蛋白质以及变体。该cas9蛋白的变体可为cas9的突变形式,其中催化性天冬氨酸残基变为任何其他氨基酸。具体地,其他氨基酸可为但不限于丙氨酸。
在本披露中,该cas蛋白可为重组蛋白。
在就例如细胞、核酸、蛋白质或载体等使用时,术语“重组”是指异源核酸或蛋白质的引入或天然核酸或蛋白质的修饰,或通过来源于经修饰细胞的细胞修饰的细胞、核酸、蛋白质或载体。因此,例如,重组cas蛋白可通过使用人类密码子表重构编码该cas蛋白的序列来制得。
该cas蛋白或其编码核酸可呈容许该cas蛋白在细胞核中发挥作用的形式。
所分离的cas蛋白也可呈易于引入细胞中的形式。例如,cas蛋白可连接至细胞穿透肽或蛋白质转导结构域。蛋白质转导结构域可为但不限于聚精氨酸或来源于hiv的tat蛋白。除了上述例子外,多种类型的细胞穿透肽或蛋白质转导结构域在相关领域中也为人所熟知,使得本领域技术人员可将但不限于多种例子应用于本披露。
另外,编码该cas蛋白的核酸可进一步包括核定位信号(nls)序列。因此,除了用于表达该cas蛋白的调节序列(例如启动子序列等)外,含有编码该cas蛋白的核酸的表达盒还可包括但不限于nls序列。
该cas蛋白可连接至有利于分离和/或纯化的标签。例如,小肽标签(例如his标签、flag标签或s标签等)或谷胱甘肽s-转移酶(gst)标签或麦芽糖结合蛋白质(mbp)标签可以但不限于取决于目的而被连接。
本披露中所用的术语“指导rna”意指靶dna特异性rna,其可结合至cas蛋白并将cas蛋白引导至靶dna。
在本披露中,该指导rna是包括两种rna、也就是crrna(crisprrna)和tracrrna(反式激活crrna)作为组分的双rna;或包括第一位点(包括与靶dna中的序列互补的序列)和第二位点(包括与cas蛋白相互作用的序列)的形式,且更具体地,单链指导rna(sgrna),其呈crrna和tracrrna的主要部分的融合物形式。
sgrna可包括具有与靶dna中的序列(还称为间隔区(spacerregion)、靶dna识别序列、碱基配对区等)互补的序列的部分和用于cas蛋白结合的发夹结构。更具体地,其可包括具有与靶dna中的序列互补的序列的部分、用于cas蛋白结合的发夹结构和终止子序列。上述结构可以但不限于以5'至3'的顺序连续存在。
本披露中也可使用任何类型的指导rna,只要该指导rna包括crrna和tracrrna的主要部分和靶dna的互补部分即可。
该crrna可与靶dna杂交。
rgen可由cas蛋白和双rna组成,或可以但不限于由cas蛋白和sgrna组成。
该指导rna、具体地该crrna或sgrna可包含与靶dna中的序列互补的序列,并且可在crrna或sgrna上游区域、具体地在sgrna或双rna的crrna的5'端包括一或多个另外的核苷酸。另外的核苷酸可为但不限于鸟嘌呤(g)。
出于本披露的目的,rgen可在体内和体外具有核酸酶活性。因此,rgen可用于在体外检测基因组dna的脱靶位点,并且当其在体内应用时,可预期其即使在与所检测脱靶位点相同的位点处也具有活性。
基因组dna可从经转化细胞中分离,使得未转化细胞或靶特异性可编程核酸酶具有核酸酶活性,并且可取决于检测可编程核酸酶的脱靶位点的目的在不限制其来源的情况下使用。
在本披露中,术语“cpf1”是新crispr系统的可编程核酸酶,所述新crispr系统与crispr/cas系统不同,并且最近已报道cpf1作为可编程核酸酶的作用(cell,2015,163(3):759-71)。cpf1是由单一rna驱动的可编程核酸酶,不需要tracrrna,并且尺寸与cas9相比相对较小。另外,cpf1使用富含胸腺嘧啶的前间区序列邻近基序(pam)序列并且裂解dna双链以形成粘端。cpf1可以但不限于来源于候选帕西菌(candidatuspaceibacter)、毛螺菌属(lachnospiragenus)、丁酸弧菌属(butyrivibriogenus)、peregrinibacteria、氨基酸球菌属(acidaminococcusgenus)、卟啉单胞菌属(porphyromonasgenus)、普雷沃氏菌属(prevotellagenus)、弗朗西斯菌属、候选甲烷支原体(candidatusmethanoplasma)或真细菌属(eubacteriumgenus)。
在本披露的具体实施方案中,中靶位点和一些预测脱靶位点由于用在体外分离的基因组dna处理靶向hbb基因的rgen而被裂解。在体内,在该位点处诱导插缺(插入和缺失)(图1)。然而,并非所有预测脱靶位置都被裂解。
步骤(b)是使用通过步骤(a)裂解的dna进行全基因组测序(wgs)的步骤。与寻找和中靶位点处的序列具有同源性的序列并预测其为脱靶位点的间接方法不同,进行步骤(b)以检测在整个基因组规模上基本上由可编程核酸酶裂解的脱靶位点。
在本披露中,术语“全基因组测序”意指通过下一代测序用于全基因组测序的以10x、20x和40x格式以多个倍数(multiple)读取基因组的方法。“下一代测序”意指以基于芯片的和基于pcr的末端配对格式对全基因组或基因组的靶向区域造型(sculpt)并以超高速度基于片段的化学反应(杂交)进行测序的技术。
步骤(c)是在通过全基因组测序获得的序列读取中测定dna被裂解的位点的步骤,并且可编程核酸酶的中靶位点和脱靶位点可通过分析测序数据而容易地检测。从序列读取测定dna被裂解的特定位点可以多种方法进行,并且本披露提供了多种合理方法来测定该位点。然而,这只是被包括在本披露的技术构思中的一个例子,并且本披露的范围不受限于这些方法。
例如,作为测定裂解位点的例子,在使用分析程序(例如,bwa/gatk或isaac)根据基因组中的位点比对通过全基因组测序获得的序列读取时,5'端经垂直比对的位点可意指dna被裂解的位点。换句话说,在本披露中,术语“垂直比对”意指如下布置,其中对于邻近的沃森链(watsonstrand)和克里克链(crickstrand)中的每一者,在用程序(例如bwa/gatk或isaac)分析全基因组测序结果时,两个或更多个序列读取的5'端在基因组的相同位点(核苷酸位置)处开始。因为对由可编程核酸酶裂解并且因此具有相同5'端的每一dna片段进行测序,所以显示此垂直比对。
也就是说,在可编程核酸酶在中靶位点和脱靶位点处具有核酸酶活性并裂解所述位点时,如果比对序列读取,那么垂直比对共同的经裂解位点,因为其每一位点都在该5'端开始。然而,该5'端在未裂解位点中并不存在,使得该5'端可在比对中以交错方式布置。因此,垂直比对位点可视为由可编程核酸酶裂解的位点,其意指该可编程核酸酶的中靶位点或脱靶位点。
比对意指将序列读取映射至参考基因组,并且随后比对在基因组中具有相同位点的碱基以配合(fit)每一位点。因此,可使用任何计算机程序,只要序列读取可以与上述相同的方式布置即可,所述计算机程序可为相关领域中已知的已知程序,或适应该目的的程序。在本披露的一个实施方案中,比对是使用isaac进行的,但并不限于此。
由于比对,dna被可编程核酸酶裂解的位点可通过例如寻找5'端如上文所述经垂直比对的位点的方法来确定,并且如果该经裂解位点不是中靶位点,其可被确定为脱靶位点。换句话说,与经设计具有可编程核酸酶的中靶位点的碱基序列一致的序列是中靶位点,并且与该碱基序列不一致的序列被视为脱靶位点。这根据上文所述的脱靶位点的定义显而易见。具体地,脱靶位点可由与中靶位点序列具有同源性的序列组成,具体地包括具有中靶位点和一个或多个核苷酸错配的序列,且更具体地,具有中靶位点和1至6个核苷酸错配,但并不特别受限于此。如果该脱靶位点为可编程核酸酶可裂解的位点,那么其可被包括于本披露的范围内。这时,中靶位点可为与指导rna互补的15-30个核苷酸的序列,并且可进一步包括由核酸酶识别的序列(例如,在cas9情形中由cas9识别的pam序列)。
除了寻找5'端经垂直比对的位点的方法以外,如果当在5'端图中观察到双峰图案时,脱靶位点并非中靶位点,可将该位点确定为脱靶位点。在通过计数构成基因组中每一位点处相同碱基的5'端的核苷酸数目绘图时,在特定位点处出现双峰图案。这是因为由可编程核酸酶裂解的每一双链都指示双峰。
在本披露的具体实施方案中,基因组dna被裂解为rgen,并且在全基因组分析后,将其与isaac比对,并且鉴别在裂解位点处垂直比对的模式和在未裂解位点处的交错模式。经鉴别,在通过5'端图表示时,在裂解位点处出现唯一双峰图案(图2至图4)。
此外,其并不限于此,但作为具体例子,两个或更多个对应于沃森链和克里克链的序列读取经垂直比对的位点可被确定为脱靶位点。另外,20%或更多的序列读取经垂直比对并且在沃森链和克里克链的每一者中具有相同5'端的序列读取数目为10或更大的位点被确定为脱靶位点位置,也就是裂解位点。
在本披露的具体实施方案中,搜索在两条链处具有相同5'端的序列读取数目为10或更大,并且至少19%的序列读取经垂直比对的位点。结果,经鉴别,消化基因组测序通过检测125个先前已验证的位点(包括中靶位点和脱靶位点)而具有高再现性(图5至图7)。
在本披露的另一具体实施方案中,经鉴别,脱靶位点可用另一靶基因vegf-a的消化基因组测序来检测(图8至图10)。在另一具体实施方案中,经鉴别,消化基因组测序还可检测除了rgen以外的zfn的脱靶位点(图24)。总之,从这些结果可以看出,本披露的消化基因组测序是用于在不限于中靶位点和可编程核酸酶的类型的情况下检测可编程核酸酶的脱靶位点的方法。
脱靶位点是在体外通过在基因组dna中处理可编程核酸酶来进行的。因此,可鉴别脱靶效应是否也在体内在通过这种方法检测的脱靶位点中实际产生。然而,这只是另外的验证过程,并且因此不是本披露的范围必须伴随的步骤,并且只是可根据需求另外进行的步骤。在本披露中,术语“脱靶效应”是与脱靶位点不同的概念。也就是说,如上文所述,在本披露中,脱靶位点的概念意指在可编程核酸酶可发挥作用的位点中,除了中靶位点以外的,并且是作为核酸酶裂解的位点来提及的位点。脱靶效应是指通过可编程核酸酶在细胞中的脱靶位点处显示插缺(插入和缺失)的效应。在本披露中,术语“插缺”是在dna碱基序列中间插入或缺失一些碱基的突变的通用术语。另外,由可编程核酸酶引起插缺的脱靶位点还称为脱靶插缺位点。总之,本披露的脱靶位点被认为是包括脱靶插缺位点的概念,并且如果该脱靶位点是可编程核酸酶在其中可能具有活性,并且插缺不一定必须通过可编程核酸酶来鉴别的位点,那么这个概念是足够的。同时,本披露中的脱靶位点被称为候选脱靶位点,并且脱靶插缺位点还称为经验证脱靶位点。
具体地,验证过程可包括但不限于从表达针对脱靶位点的可编程核酸酶的细胞中分离基因组dna,在dna脱靶位点处鉴别插缺,和在脱靶位点处鉴别脱靶效应。脱靶效应可通过使用t7e1分析和cel-i酶分析突变体检测以及相关领域中已知的鉴别插缺的方法(例如靶向深度测序)来鉴别。鉴别脱靶效应的步骤可为直接确认在脱靶位点处是否出现插缺。然而,即使在体内验证过程期间不出现插缺,也应将其视为辅助手段,因为其无法鉴别插缺以低于可检测水平的频率出现的情形。
通过鉴别如上文所述的垂直比对位点,或通过鉴别5'端图中的双峰,可充分检测脱靶位点,这种方式可高度再现。然而,存在可能遗失一些具有不均匀裂解图案或低测序深度的位点的问题。基于序列读取的比对模式,诸位发明人研发了用于计算每一核苷酸位点处的dna裂解分数的公式(图11),如下:
fi:在i位点处开始的正向序列读取的数目
ri:在i位点处开始的反向序列读取的数目
di:在i位点处的测序深度
c:任意常数。
通过这个公式,可检测出多个在现有消化基因组测序中未检测到的另外的位点,从而容许容易地过滤假阳性位点。这个公式中的c值不受限于本发明的例子,因为本领域技术人员可应用任意常数。具体地,其不限于此,但是例如,在c值为100并且所计算分数为25,000或更高时,可将位点确定为脱靶位点。然而,评分标准可由本领域技术人员根据目的进行适当调整或改变。
在本披露的具体实施方案中,脱靶位点是通过将dna裂解分数引入现有消化基因组测序方法中来检测的。结果,与只寻找垂直比对位点的方法相比较,可检测另外的位置,并且其具有高再现性(图12和图13)。在本披露的另一具体实施方案中,在rgen的sgrna中,在使用从质粒模板转录的sgrna与从质粒模板转录的sgrna和从寡核苷酸双链转录的sgrna相比较时,所检测的脱靶位点与中靶位点相比具有高同源性(图14,表1和表2)。
另外,本披露的消化基因组测序可使用多种可编程核酸酶来进行,并且诸位发明人已将这种方法命名为“多重消化基因组测序”。在这种情形中,该可编程核酸酶可为2个或更多个、具体地2至100个靶的可编程核酸酶的混合物,但不限于此。
在多重消化基因组测序的情形中,重要的是检查裂解位点是否由可编程核酸酶裂解,因为基因组dna是由每一种可编程核酸酶裂解的。这可通过根据到中靶位点的编辑距离归类脱靶位点来实现,并且是基于以下假设:脱靶位点处的碱基序列与中靶位点同源。这使得针对每一可编程核酸酶的中靶位点与脱靶位点之间有明显区别。
在本披露的具体实施方案中,进行在消化基因组测序中使用11个不同中靶位点的sgrna的多重消化基因组测序,并且根据与中靶位点的编辑距离将所鉴别的964个位置归类以鉴别每一中靶位点的脱靶位点(图15-图19)。
在另一具体实施方案中,使用100个不同中靶位点的sgrna进行多重消化基因组测序,并且同样在这种情形中,可在没有特别限制的情况下鉴别脱靶位点(图23)。经鉴别,本披露的消化基因组测序可没有限制地应用于任何数目的中靶位点。
在本披露的具体实施方案中,对于靶向特定位点的rna指导的工程化核酸酶(rgen),在全基因组中通过消化基因组测序检测的脱靶位点之间,在与中靶位点具有6个或更少的核苷酸错配的同源性位点为13,000个或更少并且其不具有含有2个或更少的核苷酸错配的同源性位点时,经鉴别,脱靶效应可通过选择该特定位点作为rgen的中靶位点而降到最低。这是个显示使用本披露的消化基因组测序建立用于选择中靶位点的优选标准的方法的例子,并且预期可编程核酸酶的脱靶效应可通过消化基因组测序降至最低。
在本披露的另一具体实施方案中,经鉴别,随着核苷酸错配水平增加,通过消化基因组测序以小比率检测出与中靶位点处的序列具有同源性的位点数目(图16)。
这是因为在rgen的中靶位点的选择中,在靶序列和基因组中具有同源性的核苷酸序列越小,具有高同源性的核苷酸序列的特异性越高。通过这种方法选择的rgen的中靶位点可使脱靶效应降至最低。
在另一方面中,本披露提供了用于在基因组编辑中降低脱靶效应的方法,其包括将在体外转录的指导rna引入具有质粒作为模板的细胞中。
这种脱靶效应降低归因于在使用质粒作为模板时,在凸起型脱靶位点处防止插缺。也就是说,在通过体外转录方法制备指导rna时,在使用寡核苷酸双链作为模板时检测出大量凸起型脱靶位点,但大多数凸起型脱靶位点在使用质粒模板时消失。除了消化基因组测序以外,rgen也可用于裂解基因组dna和诱导插缺,其可使用质粒作为模板代替寡核苷酸双链来降低脱靶效应。这是因为寡核苷酸含有失效序列,其被称为(n-1)聚体。
实施例
在下文中,将参照实施例详细描述本披露。然而,本披露的这些实施例在本文中仅出于说明目的来描述,并且本披露的权利范围不受限于这些实施例。
实施例1:cas9和体外sgrna
重组cas9蛋白是从大肠杆菌(e.coli)中纯化的或购自toolgen(南韩)。sgrna是使用t7rna聚合酶通过体外转录合成的。具体地,将sgrna模板与t7rna聚合酶在反应缓冲液(40mmtris-hcl、6mmmgcl2、10mmdtt、10mmnacl、2mm亚精胺、ntp和rna酶抑制剂)中在37℃下混合8小时。在与dnasei一起孵育以移除模板dna后,使用pcr纯化试剂盒(macrogen)纯化所转录sgrna。
实施例2:细胞培养和转化条件
在含有10%fbs的dmem培养基中培养hela细胞。使用lipofectamine2000(lifetechnologies)将cas9表达质粒(500ng)和编码sgrna的质粒(500ng)引入8×104个hela细胞中。在48小时后,根据制造商说明书用dneasy组织试剂盒(qiagen)分离基因组dna。
实施例3:基因组dna的体外裂解
使用dneasy组织试剂盒(qiagen)从hap1细胞中纯化基因组dna。进行该基因组dna的体外裂解用于消化基因组测序。具体地,将cas9蛋白和sgrna在室温下孵育10分钟以形成rnp(核糖核蛋白)。之后,使rnp复合物与该基因组dna在反应缓冲液(100mmnacl、50mmtris-hcl、10mmmgcl2和100μg/mlbsa)中在37℃下反应8小时。将在这个过程期间裂解以分解sgrna的基因组dna用rna酶a(50ug/ml)处理,并再次用dneasy组织试剂盒(qiagen)纯化。
实施例4:全基因组测序和消化基因组测序(裂解基因组测序)
对于全基因组测序(wgs),将裂解的dna用超声波仪破碎并用衔接子连接以制备文库。wgs是在来自macrogen(南韩)的illuminahiseqxten测序仪上使用这个文库来进行的。之后,使用isaac比对人类参考基因组hg19的序列文件。使用裂解评分系统来鉴别dna裂解位点。
对于多重消化基因组测序,根据编辑距离将检测位点结果归类为11个组。分开生成用于检测体外rgen裂解位点的计算机程序和用于消化基因组(digenome)检测位点归类的计算机程序。
实施例5:靶向深度测序
使用phusion聚合酶(newenglandbiolabs)扩增中靶位点和潜在脱靶位点。用naoh使pcr扩增产物变性,使用illuminamiseq进行末端配对测序,随后计算插入和缺失(插缺)的频率。
实验实施例1:在体外使用rgen的基因组dna裂解
为了研发用于检测可编程核酸酶的脱靶位点的方法,诸位发明人已使用rgen(rna指导的工程化核酸酶)作为代表来进行实验。然而,这只是用于解释本披露技术的例子,并且可应用的可编程核酸酶的种类不限于rgen。本披露的用于检测基因组中可编程核酸酶的脱靶位点的方法的特征在于,在体外将基因组裂解为针对特定靶的可编程核酸酶,随后通过进行并分析全基因组测序(wgs)来检测可编程核酸酶的脱靶位点。诸位发明人将这种方法命名为消化基因组测序(核酸酶裂解的基因组dna测序)。
诸位发明人推断,他们可通过消化基因组测序在大细胞群中鉴别由可编程核酸酶诱导的脱靶突变。
应该可能在体外在高rgen浓度下有效裂解脱靶dna序列,产生许多具有相同5'端的dna片段。这些rgen裂解的dna片段会产生在核酸酶裂解位点垂直比对的序列读取。相反,未通过rgen裂解的序列读取会以交错方式比对。开发计算机程序以搜索对应于脱靶位点的具有垂直比对的序列读取。
首先,诸位发明人测试了rgen是否可在体外在基因组中有效裂解潜在脱靶dna序列。为此,选择了已显示可在rgen的中靶位点和高同源位点(称为ot1位点)处诱导脱靶突变的hbb基因特异性rgen。除了这个位点以外,还分析了另外三个与rgen的该中靶位点相差3个核苷酸的潜在脱靶位点(称为ot3、ot7和ot12位点)。
使用与在从0.03nm至300nm浓度范围下的hbb特异性sgrna一起预孵育的cas9蛋白裂解从野生型hap1细胞中分离的基因组dna(图1a)。随后,使用定量pcr测量这些位点的dna裂解。即使在极低rgen浓度下也几乎完全裂解hbb中靶位点和ot1位点二者(图1b)。相比之下,ot3位点只在高rgen浓度下被完全裂解。另两个位点ot7和ot12即使在最高浓度下也裂解较差。
之后,将此rgen转化到hap1细胞中并使用t7内切核酸酶i(t7e1)和靶向深度测序检测在这些位点处诱导的插缺(插入和缺失)。
对于t7e1分析,根据制造商说明书使用dneasy组织试剂盒(qiagen)分离基因组dna。通过pcr扩增中靶位点。之后,通过加热使扩增的pcr产物变性并使用热循环仪缓慢冷却。将冷却的产物与t7内切核酸酶i(toolgen)一起在37℃下孵育20分钟,并通过琼脂糖凝胶电泳根据大小进行分离。
对于靶向深度测序,使用phusion聚合酶(newenglandbiolabs)扩增跨越中靶位点和脱靶位点的基因组dna区段。使用illuminamiseq使pcr扩增子经历末端配对测序。
在解释这些结果时,位于pam(前间区序列邻近基序)上游3-bp的插缺被视为由rgen诱导的突变。如所预期,hbbrgen在hbb中靶位点和ot1脱靶位点二者处具有高活性,分别以71%和55%(t7e1)的频率产生插缺(图1c)。在ot3位点处也以3.2%(t7e1)或4.3%(深度测序)的频率诱导脱靶插缺(图1c、图1d)。同时,在另两个在体外裂解较差的潜在脱靶位点处,使用t7e1(检出限,约1%)和深度测序(检出限,约0.1%)未检测到插缺。应注意,ot7位点在种子区域(在pam上游的10-nt至12-nt序列)中无核苷酸错配,但在体外或在细胞中未被裂解,从而鉴别pam远端区域的重要性。
这些结果与我们先前的如下发现一致:rgen可在体外裂解脱靶dna序列,但通常在细胞中在相同位点处无法诱导插缺。因此,rgen在体外比在细胞中在靶特异性方面似乎杂乱得多。也许,通过rgen产生的大多数dna双链断裂(dsb)在细胞中通过非同源末端连接(nhej)或同源重组(hr)得到修复。
实验实施例2:序列读取分析
使4个不同的基因组dna组经历全基因组测序(wgs)以研究使用rgen在体外裂解基因组dna是否可产生在裂解位点处具有垂直比对的序列读取。
将从经rgen转化的hap1细胞和未转化hap1细胞中分离的基因组dna在体外用靶向hbb基因的300nmcas9和900nmsgrna完全裂解。平行地,在体外没有rgen裂解的情况下通过使用从这些细胞中分离的基因组dna进行wsg(图2a)。在将序列读取映射到参考基因组中之后,使用igv(整合基因组学查看器)观察在中靶位点和4个同源位点处的序列比对模式。
首先,检查从对照组hap1细胞中分离的消化基因组(裂解的基因组)。在中靶位点、ot1位点和ot3位点处,观察到不常见的垂直比对模式(图2b和图3a、图3b)。跨越裂解位点的序列读取极其罕见。相比之下,在分析尚未经rgen处理的完整基因组时,在这些位点处未观察到这样的垂直比对。在ot7位点和ot12位点处,大多数序列读取跨越潜在裂解位点(在pam上游3-bp),产生交错比对(图3c、图3d)。
其次,将从经rgen转化细胞中分离的消化基因组与相应的完整基因组相比较。在所有5个位点处,完整基因组产生典型的交错比对模式(图2b和图3)。相比之下,消化基因组在中靶位点和ot1位点处显示垂直和交错比对。在这两个位点处,几乎所有对应于交错比对的序列读取都含有插缺(图2b及图3a和图3b)。也就是说,应注意,rgen无法裂解由其自身诱导的插缺序列。同时,对于跨越ot7和ot12裂解位点的序列读取,未发现插缺,与t7e1和深度测序结果一致。在ot3位点处,对于少数跨越该裂解位点的序列读取,消化基因组显示出直线比对模式。具体地,一个序列读取含有由rgen诱导的插缺(图3b)。
这些结果表明,消化基因组测序足够灵敏,容许鉴别后脱靶突变,并且序列读取的垂直比对是体外rgen裂解的独特标志。
实验实施例3:在信号核苷酸规模下的5'端图
为了在基因组规模上鉴别潜在的rgen脱靶位点,开发搜索序列读取的直线比对的计算机程序。首先,对在单核苷酸规模下5'端在hbb中靶位点和两个经验证脱靶位点(ot1和ot3)附近的核苷酸位置处开始的序列读取绘图(图4a)。因为对沃森链和克里克链二者进行测序,假设应该在裂解位点处就在彼此旁边观察到几乎相等数目的对应于每条链的序列读取,产生双峰。如所预期,消化基因组在三个裂解位点(中靶位点、ot1和ot3)处产生双峰(图2c和图4b、图4c)。已经历体外rgen处理的完整基因组在这些位点处未产生所述双峰图案。
之后,将这种方法应用于整个经rgen转化的消化基因组、未转化消化基因组、完整的经rgen转化的基因组和完整的未转化基因组。另外,将未转化基因组dna在体外用cas9蛋白在sgrna不存在下或用浓度低100倍的rgen(3nmcas9)处理,并且经历wgs和消化基因组分析。对具有相同5'端的序列读取的计数在两条链中都大于10并且至少19%的序列读取经垂直比对的位点实施搜索。在经3nm和300nmrgen处理的未转化消化基因组中鉴别总共17个和78个位点(包括中靶位点和两个经验证脱靶位点)(图5a),其显示出5'端图中的双峰图案和igv图像中的直线比对。在这些位点之间,在经3nm和300nmrgen处理的消化基因组中的一个和两个位点是源自天然存在的插缺的假阳性。另外,在经rgen转化的消化基因组中在总共125个位点(包括三个经验证中靶位点和脱靶位点)处观察到这样的图案。同时,未验证ot7和ot12位点在这三个消化基因组中未显示出双峰图案。此外,在这三个消化基因组中共同鉴别出大多数位点,证实了消化基因组测序的高再现性。具体地,发现于未转化消化基因组(3nmrgen)中的16个候选位点中的15个(94%)(不包括一个假阳性位点)也在另两个消化基因组中被鉴别出。发现于未转化消化基因组(300nm)中的76个候选位点中的74个(97%)也在经rgen转化的消化基因组中被鉴别出(图5a)。除了这三个经验证裂解位点外,在经rgen转化的消化基因组中其他122个位点都不伴随插缺,表明这些候选位点处很少发生突变。同时,只在完整基因组中的两个位置、完整的经rgen转化的基因组中的三个位置和仅用cas9(300nm)处理的未转化基因组中的一个位置处观察到这样的双峰图案。在这三个完整基因组中鉴别的所有这些位置都是假阳性,其源自hap1基因组中相对于参考基因组的天然存在的插缺(图6a至图6c)。因此,双峰图案或序列读取的垂直比对是在消化基因组中发现的独特特征。
之后,将在经rgen转化和未转化消化基因组中鉴别的74个共有位点的dna序列与20bp中靶位点相比较,并且发现该20个核苷酸除了在5'端的一个以外全部都是保守的(图5b)。另外,通过将74个位点的dna序列彼此比较而不是与中靶序列比较获得的序列标志或从头基序明确显示出在除了最初两个核苷酸外的所有位置处与中靶序列匹配(图5c)。另外,这些双峰位置中的70个(95%)伴随确切地在预期裂解位置下游3个核苷酸的5'-nag-3'pam。预期仅6.25%(=1/16)的位点偶然伴随pam。两个位点含有5'-nag-3'pam。一些位点通过容许dna或rna凸起或假设5'-nga-3'作为非标准pam而与中靶位点匹配。可疑的是,5'-nga-3'是否可在细胞中用作pam,但是在本披露的极端体外裂解条件下,rgen可裂解这些位点。其他位点与中靶序列不具有序列同源性,表明其可为假阳性。
另外,在同源位点中的核苷酸错配越少,其越可能被消化基因组测序检测到。也就是说,检测到15个中的7个(47%)和142中的14个(10%)同源位点与中靶位点相差3和4个核苷酸,但仅检测到1,191个位点中的15个(1.2%)和7,896个位点中的1个(0.013%)相差5和6个核苷酸(图5d)。
总而言之,这些结果指示,大多数双峰图案是由体外rgen裂解引起的,并且消化基因组测序可在基因组规模上发现核酸酶裂解位点。
实验实施例4:用于鉴别候选位点处的脱靶效应的深度测序
进行深度测序以验证在两个消化基因组中鉴别的74个共有位点处的脱靶效应(图5e)。此外,还测试了另外8个与中靶位点相差3个核苷酸但未被消化基因组测序检测到的位点。在这8个位点处以至少0.1%并且大于阴性对照组的频率未检测到脱靶插缺(费雪精确检验,p<0.01)(图5d)。在74个位点中,总共5个位点(包括已验证的中靶位点、ot1位点和ot3位点)处以在0.11%至87%范围内的频率观察到插缺(图5e和图7a、图7b)。在另外两个刚验证的脱靶位点(称为hbb_48和hbb_75)处,以0.11%和2.2%的频率检测到插缺。这两个位点与中靶位点相差3个核苷酸。相对于在5'端与中靶位点相差1个核苷酸的20-ntsgrna序列,在该hbb_48位点处存在3个核苷酸错配并且在该hbb_75位点处存在2个错配。这些经验证脱靶位点与该20-ntsgrna序列相比都不具有dna或rna凸起,其也都不伴随非标准pam,例如5'-nga-3'或5'-nag-3'。应注意,这两个新脱靶位点和另外三个位点是在三个消化基因组的每一者中被独立地鉴别。这些结果显示,消化基因组测序是灵敏并且可再现的在基因组规模上鉴别核酸酶脱靶效应的方法。
实验实施例5:用于vegf-a特异性rgen的消化基因组测序
之后,诸位发明人设法鉴别消化基因组测序是否适用于除了hbb基因以外的其他基因。用已显示在vegf-a基因座处诱导中靶突变并且另外在4个同源位点处诱导脱靶突变的另一rgen进行消化基因组测序。鉴别到总共81个位点(包括中靶位点和4个已验证的脱靶位点)显示双峰图案(图8a和图9)。这81个位点处的所有dna序列都含有标准5'-ngg-3'pam序列。将这些序列与中靶序列相比较显示在每个核苷酸位点处都匹配。此外,还将这些序列彼此比较以获得从头基序:所得序列标志还显示在几乎每个核苷酸位置处都与靶序列匹配,表明在该20-ntsgrna序列中的每个核苷酸都有助于rgen的特异性(图8b和图8c)。
之后,使用靶向深度测序在由消化基因组测序鉴别的81个位点和与中靶位点相差3个或更少核苷酸但未被消化基因组测序鉴别出的28个位点处鉴别中靶效应和脱靶效应。这种rgen在hap1细胞中具有高活性,在中靶位点除以87%的频率产生插缺并且在4个先前经验证的脱靶位点出以0.32%至79%范围内的频率产生插缺。此外,另外鉴别出4个脱靶位点,在这些位点处以0.065±0.021%至6.4±1.2%范围内的频率诱导插缺(图8e和图10)。使用该rgen获得的在这些位点处的插缺频率显著大于使用空载体对照组获得的频率(费雪精确检验,p<0.01)。这些脱靶位点含有与该20-nt靶序列的1至6个核苷酸错配和在pam近端种子区域中的至少一个错配。在人类基因组中存在13,892个具有6-nt错配的位点,但通过消化基因组测序仅鉴别到6个位点(0.043%),并且在所述位点中,仅1个位点通过深度测序得到了验证(图8d和图8e)。迄今,与中靶位点具有6-nt核苷酸错配的rgen脱靶位点先前从未被鉴别过。这些脱靶位点都不含dna或rna凸起,但通过消化基因组测序鉴别的81个位点中的40个与该20-nt靶序列相比含有丢失或额外的核苷酸。在所有这些其他位点(包括那些未被消化基因组测序鉴别出的位点)处,使用rgen获得的插缺频率为0.05%或更低,或小于使用空载体对照组获得的频率或与其无显著不同。
从这些实验实施例1至5可以看出,本频率的消化基因组测序是用于检测可编程核酸酶的脱靶位点的极高可再现的方法。
实验实施例6:改良的消化基因组测序
首先,诸位发明人开发了能使用人类基因组的全基因组测序(wgs)数据鉴别体外裂解位点的评分系统。在这些实验实施例1至5中鉴别的消化基因组测序分析具有高再现性,但存在一些具有不均匀裂解图案或低测序深度的位点可能丢失的问题。诸位发明人已发现,这些位点可通过估计cas9蛋白在钝端制造一个或两个核苷酸悬突的情形来鉴别。基于序列读取的比对模式,将dna裂解分数分配给每一核苷酸位点(图11)。通过这个程序,检测到多个在现有消化基因组测序中未检测到的另外的位点。裂解分数的基因组规模图显示,在未裂解基因组dna中几乎未发现假阳性位点(图12a):
在全基因组中鉴别的少量假阳性位点包括插缺(插入和缺失),其在基因组dna中天然存在,可易于筛选。如在两个独立消化基因组测序分析中可以看出的,人类基因组的裂解分数具有高再现性(r2=0.89)(图13)。
诸位发明人还发现,在消化基因组测序分析中通过质粒模板转录的sgrna甚至不裂解在中靶位点处的任何核苷酸缺陷型假阳性的凸起型脱靶位点,其中该脱靶位点(it)是以使用寡核苷酸双链转录的sgrna来检测的(图12b和图14)。
这是因为从寡核苷酸双链转录的sgrna不是均匀组分,包括从合成失败的寡核苷酸转录的不完整分子。结果,使用从质粒模板转录的sgrna鉴别到的裂解位点比使用从寡核苷酸模板转录的sgrna鉴别到的裂解位点具有更高的与中靶位点的同源性(表1和表2)。裂解位点周围的dna序列可从通过比较所述序列获得的序列标志加以鉴别(图12c)。
[表1]
[表2]
因此,假阴性位点数目可使用本披露的裂解评分系统显著减少,并且假阳性位点数目可使用在质粒模板中转录的sgrna显著减少。
实验实施例7:多重消化基因组测序
与其他方法不同,消化基因组测序可组合使用而无需增加与核酸酶数目成比例的测序深度。诸位发明人选择了10个sgrna,使用guide-seq对其单独分析,所述guide-seq比idlv检测和其他方法更灵敏。诸位发明人用另一种靶向sgrna的cas9蛋白、10个sgrna和hbb基因的混合物裂解人类基因组dna,并且进行两个独立wgs分析(图15a)。之后,使用该评分系统在基因组规模上研究体外裂解位点。结果,在人类基因组中鉴别了总共964个位点(表3至12)。之后,根据与中靶位点的编辑距离将该位点归类(图15a和表3至表12)。
[表3]
[表4]
[表5]
[表6]
[表7]
[表8]
[表9]
[表10]
[表11]
[表12]
guide-seq和其他方法需要过滤步骤,所述步骤移除约90%的与中靶位点缺少同源性的检测位点,但多重消化基因组测序不过滤位点,而是基于编辑距离进行比对。将该964个位点明确分为11个组。另外,体外裂解位点的11个组中的每组与11个靶序列中的一个序列具有高同源性。因此,通过比较每组内的序列获得的从头基序或序列标志在几乎所有核苷酸位点处匹配靶序列(图15a)。
结果显示,尽管其小于前间区序列邻近基序(pam)序列和由cas9识别的pam近端10-nt“种子”位点,但在23-nt靶序列5'端的10-nt位点有助于rgen的特异性。另外,经鉴别,除了由该11个rgen裂解的964个位点中的一个位点外,所有位点都具有5'-ngg-3'的pam序列或类似于5'-nng-3'/5'-ngn-3'的pam的序列。因此,多重消化基因组测序可用于在无需同源序列的程序搜索的情况下精确寻找体外裂解位点并且简单,可应用于多种可编程核酸酶,并且与其他已知方法(例如guide-seq和htgts)相比具有许多优点。
之后,鉴别是否每一sgrna都能裂解中靶位点和脱靶位点。通过在高浓度(900nm)的hbb特异性sgrna下用cas9(300nm)处理而裂解的30个位点中的17个位点(=57%)在使用低浓度(82nm)的相同sgrna进行多重消化基因组测序时被检测到(图15b和图16c)。这些结果表明,11个sgrna中的每一者都可彼此独立地将cas9引导至其中靶位点和脱靶位点,并且可理解,消化基因组测序具有复杂性。
实验实施例8:体外裂解位点
该11个sgrna在基因组规模上显示宽特异性范围;人类基因组中每个sgrna的裂解位点数目都在13至302范围内(图16a和表3至表12)。如所预期,在进行多重消化基因组测序时检测到在人类基因组中使用cas-offinder鉴别出的所有中靶位点,以及每个中靶位点和大多数具有一个或两个核苷酸的位点(图16b)。然而,检测到极少数具有3个或更多个核苷酸错配的位点。也就是说,通过消化基因组测序检测出的位点的比率随着核苷酸错配数目从3增加到6,以指数方式减少(图16b)。另外,与具有0或1个错配的位置相比,种子区域中具有两个或更多个核苷酸错配的位点在体外不被裂解(p<0.01,学生t检验)。
另一方面,经鉴别,用消化基因组测序检测出的位点数目和人类基因组中具有6个或更少核苷酸错配的同源位点数目(定义为“正交性”)具有显著相关性(r2=0.93)(图16c)。也就是说,人类基因组中5个具有16,000个或更多同源位点的sgrna在体外裂解63个或更多(每个sgrna平均161个),而6个具有13,000个或更少同源位点的sgrna在体外裂解46个或更少(每个sgrna平均28个),并且因此特异性相对更高(p<0.01,学生t检验)(图16c)。该结果与在guide-seq阳性位点数目与人类基因组的中靶位点的正交性之间观察到的缺乏相关性(r2=0.29)不同(图17)。然而,如guide-seq所鉴别的5个特异性最高的sgrna在细胞中裂解10个或更少位点,与通过消化基因组测序鉴别的特异性最高的sgrna一致。
结果表明,可需要人类基因组中的某些位点以使脱靶效应降至最低,在所述位点中存在少于13,000个具有6个或更少核苷酸错配的同源位点且不存在具有2个或更少核苷酸错配的同源位点。就这一点而言,在1715个可靶向位点(包括5'-ngg-3'pam序列)中的368个位点(=21.5%)对应于上文关于本披露中测试的4个基因的概念(表13)。
[表13]
实验实施例9:消化基因组测序相对于其他方法
平均来说,多重消化基因组测序成功地鉴别到80±8%的通过常规guide-seq检测出的位点(图16a)。例如,用guide-seq使用3个对vegfa1、rnf2和hek293-3具有特异性的sgrna检测到的所有位点也被消化基因组测序所鉴别。另外,多重消化基因组测序检测出703个未被guide-seq检测到的新位点(平均每个sgrna70个)(图16a)。结果,guide-seq检测出25±6%的通过多重消化基因组测序检测到的位点。rnf2特异性sgrna是显示出消化基因组测序的优点的很好的例子。先前研究已进行了两个独立的guide-seq分析,但无法检测到这个sgrna的脱靶位点。然而,除了中靶位点外,消化基因组测序还鉴别到12个裂解位点。另外,在消化基因组测序阳性位点数目与guide阳性位点数目之间观察到缺乏相关性(r2=0.20)(图16d)。
对于该10个sgrna中的9个,消化基因组测序可比guide-seq获得更多的候选脱靶位点,但这不是综合结果。也就是说,hbbsgrna未通过guide-seq进行分析。总之,guide-seq检测出总共168个在消化基因组测序中未被检测到的位点。
另一方面,还针对两个靶向vegfa1和emxl位点的sgrna进行htgts(图16a)。大多数通过另外两种方法(guide-seq和htgts)中的至少一种检测到的位点(vegfa1中40个位点中的31个和emx1中19个位点中的17个)也如消化基因组测序所研究,但vegfa中的9个和emx1中的2个未被检测到。这是因为一些位点是pcr引物所致的假结果或因天然存在的dsb产生的假阳性,这是guide-seq和htgts的固有限制。然而,常见地在这个位置中发现、最常见地在另外两种方法中发现的两个emx1脱靶位点是在该特定位点处具有低测序深度(图18)或低浓度(82nm)的sgrna,并且因此在多重消化基因组测序中不被鉴别出。这个问题可以通过多次进行wgs以增加平均测序深度和与通过使用在单一分析中具有高浓度的sgrna获得的序列读取合并来克服。
vegfa2特异性sgrna是消化基因组测序可检测出比guide-seq更多的候选位点的规则的唯一例外。也就是说,guide-seq鉴别出122个未在消化基因组测序中检测到的位点。靶序列是由胞嘧啶伸长段(stretch)组成的不常见序列。可从映射程序中移除用wgs在均聚物位点处获得的多个序列读取。另一方面,guide-seq将能够使用pcr扩增所检测的寡核苷酸位点来检测这些位置。
之后,将在本披露中鉴别的裂解位点与使用chip-seq(染色质免疫沉淀测序)检测的裂解位点相比较。首先,对本披露中所用的4个sgrna进行chip-seq。dcas9未结合至如消化基因组测序鉴别的大多数cas9裂解位点(288个,98%)(图19)。结果显示,cas9的dna结合是与dna裂解分开的概念,并且使用dcas9的chip-seq可用于检查基于dcas9的转录因子和表观基因组调节因子的特异性,但其不适于分析cas9rgen的基因组规模特异性。
实验实施例10:细胞内脱靶位点的鉴别
之后,使用下一代测序(ngs)平台,鉴别在消化基因组测序和guide-seq中鉴别的位点(表14至表23)中的一些位点的每一sgrna和cas9蛋白是否在人类细胞中诱导脱靶插缺。
[表14]
[表15]
[表16]
[表17]
[表18]
[表19]
[表20]
[表21]
[表22]
[表23]
在消化基因组测序和guide-seq中共同检测到的132个位点中的116个位点(=88%)处检测到高于由测序误差造成的背景噪音水平的插缺。另一方面,在消化基因组测序中以及仅在guide-seq中检测到的大多数位置未通过靶向深度测序进行鉴别。另一方面,仅在消化基因组测序中以及在guide-seq中检测到的大多数位点未通过靶向深度测序来鉴别插缺。也就是说,仅在消化基因组测序中检测到的127个位点中的21个(=17%)和仅在guide-seq中检测的45个位点中的23个(=51%)诱导高于噪音水平的插缺。经鉴别,两种方法都不是通用方法。在大多数经验证位点中,插缺频率低于1%,远低于在相应中靶位点处鉴别的插缺频率。例如,靶向rnf2的sgrna在本披露中验证的中靶位点和两个脱靶位点处诱导插缺,其显示频率分别为68%、0.25%和0.09%(图20)。可以看出,可在未在ngs中鉴别的位点处以低于噪音水平的频率(0.001%至4%,取决于位点)诱导插缺。
为了降低脱靶效应,另外使用在5'端包括两个鸟嘌呤的sgrna(称为ggx20sgrna)(图21a)。经修饰sgrna的特异性是相应gx19sgrna的598倍(图21b-图22g)。rnf2特异性ggx20sgrna未检测到高于噪音水平的脱靶插缺(图21d)。
实验实施例11:脱靶位点处的插缺频率
通过ngs验证的脱靶位点(=160)和未验证脱靶位点(=144)处的插缺频率特定地用于鉴别脱靶效应。经鉴别,发现中靶位点和脱靶位点的插缺频率图中的错配核苷酸数目和具有2个或更少核苷酸错配的脱靶位点可在细胞内被有效裂解(平均插缺频率=5.38%),并且所述位点在具有3个或更多个核苷酸错配的情形中未被有效裂解(平均插缺频率=0.14%或更低)(图22a)。中靶位点处的插缺频率为60±7%。在经验证或未验证位点处,核苷酸错配在pam远端区域和pam近端区域中几乎均匀分布。具有3个或更多个核苷酸错配的经验证或未验证位点与pam远端位点同样重要(图22b和图23c)。也就是说,在种子位点具有0或1个核苷酸错配的位点处,插缺频率与具有2个或更多个错配的位点同样低。
结果显示,计算基因组中的潜在脱靶位点数目、通过消化基因组测序鉴别的位点比率(图16a)和从该位点的平均可插缺频率(图20a)计算的脱靶分数(表24)。
[表24]关于人类基因组中的emx1靶序列
(5'-gagtccgagcagaagaagaangg-3')的脱靶分数的计算
a通过使用cas-offinder获得
b如图16b中所示来鉴别
c通过靶向深度测序鉴别(图22a)。
为了汇总上述结果,诸位发明人已开发了能检测可编程核酸酶的脱靶位点的消化基因组测序方法,所述方法与其他常规方法相比高度可再现,并且经配置以易于检测脱靶位点。另外,诸位发明人开发了体外dna裂解评分系统并开发了强化消化基因组测序,其可使用从质粒模板而不是合成寡核苷酸双链转录的sgrna减少假阳性和假阴性位点数目。另外,多重消化基因组测序是通过用11个sgrna的混合物裂解基因组dna来进行的,并且平均每个sgrna鉴别70个在guide-seq中未检测到的另外的裂解位点。在经rgen转化的人类细胞中在这些位点中的多个中诱导脱靶插缺。因此,通过检查插缺频率、核苷酸错配数目和数百个脱靶位点中的错配位点,经鉴别,rgen特异性中的pam远端区域与种子区域同样重要。另外,已鉴别,与总错配核苷酸数目为0或1的情形相比,在种子位点具有两个或更多个核苷酸错配的位点不在体外被裂解。
实验实施例12:大规模多重消化基因组测序
诸位发明人尝试鉴别即使在大规模扩展多重消化基因组测序的靶的情形中是否可有效地检测脱靶位点。
具体地,对各自不同的100个中靶位点进行多重消化基因组测序。即使将中靶位点扩展至100个,该100个靶的脱靶位点也可通过消化基因组测序有效检测。
就这一点而言,在通过计算机程序关于中靶位点细化具有6个或更少核苷酸错配的位点后,将这个部分归类为rgen的裂解位点和非裂解位点。之后,通过基于神经网络的机器学习来分析裂解位点序列与非裂解位点序列之间的差异,并产生能关于中靶位点预测脱靶位点的程序。发现与已开发的其他程序(crop-it)相比,通过该程序可检测大量脱靶位点(图23)。
实验实施例13:用于zfn的消化基因组测序
另外,诸位发明人还尝试通过相同方法检测zfn而不是rgen的脱靶位点。
与rgen一样,用在体外分离的无细胞基因组dna处理zfn蛋白,然后进行wgs。在zfn情形中,经鉴别,在通过igv观察中靶位点时出现垂直比对(图24a),并且在整个基因组规模上给出裂解分数(图24b)。经鉴别,通过比较在体外裂解位点周围的dna序列获得的序列标志与大多数位点处的靶序列一致(图24c和图24d)。
在针对一部分从消化基因组测序获得的具有4个或更少核苷酸错配区域的候选中靶位点和候选脱靶位点(表25)通过zfn转化后,进行靶向深度测序。
[表25]
结果,经鉴别,插缺存在于62个候选脱靶位点中的35个中靶位点和脱靶位点中。具体地,经鉴别,0.028%至5.9%被诱导(表25)。这显示消化基因组测序方法也预测zfn的脱靶位点。在通过在foki位点处修饰(kk或el)制得的zfn的情形中,特异性增加(图24)。因此,在通过foki修饰的zfn进行消化基因组测序时发现总共16个候选脱靶位点。另外经鉴别,在通过使用foki修饰的zfn转化的细胞中,插缺在16个候选脱靶位点中的15个位点处出现,并且这指示与其他常规方法(ildv、体外选择)相比,可发现大量脱靶位点(图25)。
总之,上述结果表明,本披露的消化基因组测序可应用于任何可具有rgen、zfn以及中靶位点和脱靶位点的可编程核酸酶。
如上所述,本披露所属技术领域的普通技术人员将理解,在不背离本披露的技术精神或本质特征的情况下,本披露可以其他具体形式来体现。就这一点而言,应理解,上述实施方案旨在在每个方面中进行说明,但并不旨在是限制性的。本发明的范围应视为涵盖在如通过所附权利要求书而不是前述说明书定义的含义和范围内的所有修改和改变及其等效概念。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种新型的15种人乳头瘤病毒亚型分型试剂盒和方法与流程
- 一种与水稻稻瘟病抗性基因紧密连锁的分子标记、引物及其应用的制造方法与工艺
- 一种无创高通量甲基化结肠癌诊断、研究和治疗方法与流程
- 用于检测CDKN2B基因变异的试剂盒及CDKN2B基因变异率的定量检测方法与流程
- 用于基因甲基化检测的引物和探针、采样方法、试剂盒与流程
- 一种癌症相关基因突变高灵敏度检测方法和试剂盒与流程
- 基于高通量测序法的人类多基因突变检测试剂盒的制造方法与工艺
- 一种用于检测德尔卑沙门氏菌的靶基因、特异性引物对及检测方法和试剂盒与流程
- 一种检测测序平台索引序列污染的方法及引物与流程
- 无物种限制的真核生物同时进行基因敲除和基因过表达的制造方法与工艺