伊格列净中间体的制备方法与流程
本发明涉及一种医药中间体的制备方法。属于医药技术领域。
背景技术:
(1s)-2,3,4,6-四乙酰氧基-1,5-脱水-1-[3-(1-苯并噻吩-2-基甲基)-4-氟苯基]-d-山梨糖醇是合成伊格列净l-脯氨酸(ipragliflozinl-proline)的关键中间体,后者是sglt2抑制剂的重要代表性药物,主要用于2型糖尿病的治疗。
专利申请ep2105442a1公开了(1s)-2,3,4,6-四乙酰氧基-1,5-脱水-1-[3-(1-苯并噻吩-2-基甲基)-4-氟苯基]-d-山梨糖醇的一种制备方法,具体步骤如下:
上述制备方法存在以下问题:
(1)在制备化合物3时,会产生大量的f邻位取代的副产物;
(2)在制备化合物5时,使用了吡啶,毒性较大;
(3)在脱甲氧基制备化合物6时使用了三氟甲磺酸,有可能产生遗传毒性物质,不适用于医药领域。
而化合物6的纯度会直接影响下一步制备所得(1s)-1,5-脱水-1-c-[3-[(1-苯并噻吩-2-基)甲基]-4-氟苯基]-d-葡糖醇的纯度,进而影响终产物原料药的质量。
技术实现要素:
本发明的目的是为克服上述现有技术的不足,提供伊格列净中间体的制备方法。
为实现上述目的,本发明采用下述技术方案:
2-(3-苯并[b]噻吩-2-基甲基-4-氟-苯基)-3,4,5-三-三甲基硅氧基-6-三甲基硅氧基甲基-四氢-吡喃-2-醇(化合物3)的制备方法,是将2-(5-溴-2-氟苄基)苯并噻吩(化合物1)溶于有机溶剂ⅰ中,降温至-60~-70℃,惰性气体或氮气保护下,在控制反应液温度不高于-60℃的前提下,尽快滴加正丁基锂的正己烷溶液,并于-60~-70℃保温0.5~1小时,之后滴加2,3,4,6-四-o-三甲基硅基-d-葡萄糖酸内酯(化合物2),滴加过程中控制反应液温度不高于-60℃,-60~-70℃加成反应5~6小时,即得。反应式如下:
优选的,2-(5-溴-2-氟苄基)苯并噻吩、正丁基锂和2,3,4,6-四-o-三甲基硅基-d-葡萄糖酸内酯的配比为22g:0.07875mol:36.8g。
优选的,惰性气体为氩气。
优选的,有机溶剂ⅰ为四氢呋喃和甲苯的混合液,两者的体积比为1:10。
优选的,正丁基锂的浓度为2.5mol/l,其滴加速率为3滴/秒。
优选的,化合物2溶于甲苯后进行滴加,化合物2和甲苯的质量体积比为36.8g:20ml。
优选的,化合物2的滴加时间为10~60分钟。
(1s)-2,3,4,6-四乙酰氧基-1,5-脱水-1-[3-(1-苯并噻吩-2-基甲基)-4-氟苯基]-d-山梨糖醇的制备方法,包括步骤:
(1)按照前述方法制备2-(3-苯并[b]噻吩-2-基甲基-4-氟-苯基)-3,4,5-三-三甲基硅氧基-6-三甲基硅氧基甲基-四氢-吡喃-2-醇(化合物3);
(2)加入酸化剂,0~10℃反应16~20小时,化合物3经酸化反应,得化合物4;
(3)加入缚酸剂和乙酸酐,5~15℃反应4~5小时,化合物4经乙酰化反应,得化合物5;
(4)加入有机溶剂ⅱ和三乙基硅烷,-5~5℃下滴加三氟化硼乙醚,0~10℃反应3~5小时,脱甲氧基,得(1s)-2,3,4,6-四乙酰氧基-1,5-脱水-1-[3-(1-苯并噻吩-2-基甲基)-4-氟苯基]-d-山梨糖醇(化合物6);反应式如下:
优选的,2-(5-溴-2-氟苄基)苯并噻吩、正丁基锂、2,3,4,6-四-o-三甲基硅基-d-葡萄糖酸内酯、酸化剂、缚酸剂、乙酸酐、三乙基硅烷、三氟化硼乙醚的配比为22g:0.07875mol:36.8g:18.8g:41.6g:38.5g:39.8g:34.0g。
优选的,步骤(2)中的酸化剂为浓硫酸,具体方法是:使用玻璃反应釜,向步骤(1)所得反应体系中依次加入甲醇和浓硫酸,缓慢升温至0~10℃反应;甲醇和浓硫酸的质量比为87:18.8。
优选的,步骤(2)中的酸化剂为浓硫酸,具体方法是:使用不锈钢釜(适合低温反应),向步骤(1)所得反应体系中依次加入甲醇和第一部分浓硫酸,缓慢升温至-5~-15℃,转入搪瓷釜(不耐低温),再加入第二部分浓硫酸,0~10℃反应;甲醇、第一部分浓硫酸和第二部分浓硫酸的质量比为87:4:14.8。
进一步优选的,所述浓硫酸的质量分数为98%。
优选的,步骤(3)中的缚酸剂为三乙胺,具体方法是:向步骤(2)所得反应体系中加入三乙胺、dmap(4-二甲氨基吡啶,催化剂)和乙酸酐,三乙胺、dmap和乙酸酐的质量比为41.6:1.7:38.5。
优选的,步骤(4)中的有机溶剂ⅱ为乙腈,乙腈与三乙基硅烷的体积质量比为100ml:39.8g。
优选的,步骤(4)还包括精制步骤,是将所得化合物6粗品加入乙醇中,加热至50~60℃,搅拌1小时,再降温至0~10℃,搅拌2小时,过滤,减压干燥至少6小时,得化合物6精制品。
进一步优选的,化合物6粗品与乙醇的质量体积比为20g:100ml。
进一步优选的,减压干燥条件为50~60℃、真空度-0.09mpa以上。
本发明的有益效果:
1、步骤(1),化合物1和化合物2反应制备化合物3的过程中,会产生f邻位取代的杂质,其结构式如下:
在控制反应液温度不高于-60℃的前提下,应当尽快滴加正丁基锂,其滴加速率基本控制在3滴/秒,5分钟左右滴毕,放大生产后,滴加时间可适当延长。通常在使用正丁基锂促进羰基和卤代物的加成反应时,正丁基锂的滴加速度越慢,反应效果会越好,但是在本发明中,正丁基锂的滴加速度越快,f邻位取代的副产物反而越少,在某种程度上克服了正丁基锂使用的技术偏见。应当注意的是,化合物3的f邻位取代的副产物多必然相应的所得化合物4的f邻位取代的副产物多,由于化合物3中的tms保护基并不稳定,在hplc检测过程中易脱保护基,故本发明中检测化合物4的hplc情况,其可直接反映化合物3的纯度情况。通过对化合物4的hplc检测可以发现,提高正丁基锂的滴加速度,能将f邻位取代的副产物的比例由25%降至2%。
2、步骤(2),酸化制备化合物4时,选择了硫酸-甲醇体系,避免现有技术中盐酸-乙酸乙酯-甲醇体系容易挥发带来的投料不精确的问题。
3、步骤(3),乙酰化制备化合物5时,采用三乙胺作为缚酸剂,降低了毒性。
4、步骤(4),去甲氧基反应制备化合物6时,采用三氟化硼乙醚-三乙基硅烷体系,与现有技术相比,使用三氟化硼乙醚代替三氟甲磺酸可以有效避免遗传毒性,三乙基硅烷代替叔丁基二甲基硅烷也能降低成本。
5、在反应过程和精制步骤中,前期生成的f邻位取代的副产物逐渐被精制除去,直接影响了最后所得化合物6的收率,使得化合物6的收率较低。
6、本发明经放大无放大效应,适于工业化生产。
附图说明
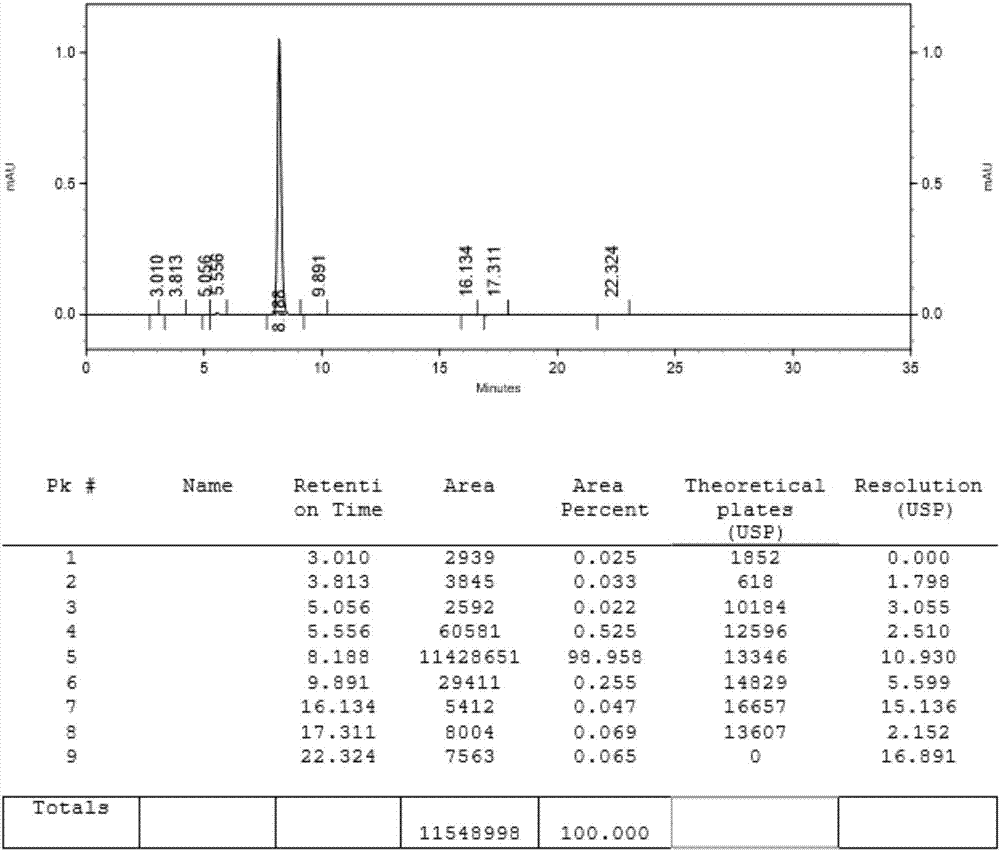
图1为实施例1步骤(2)制备化合物4的hplc图;
图2为实施例2制备所得化合物6的hplc图;
图3为对比例1步骤(2)制备化合物4的hplc图;
图4为对比例2步骤(2)制备化合物4的hplc图。
具体实施方式
下面结合实施例对本发明进行进一步的阐述,应该说明的是,下述说明仅是为了解释本发明,并不对其内容进行限定。
实施例1:
(1s)-2,3,4,6-四乙酰氧基-1,5-脱水-1-[3-(1-苯并噻吩-2-基甲基)-4-氟苯基]-d-山梨糖醇的制备方法,包括步骤:
(1)向反应瓶加入四氢呋喃22ml、甲苯220ml,搅拌下,加入2-(5-溴-2-氟苄基)苯并噻吩(化合物1)22.0g。将反应液降温至-70℃,氮气保护下滴加正丁基锂的正己烷溶液(2.5mol/l)31.5ml,控制反应液温度不高于-60℃,滴加速率3滴/秒,约5min滴毕,-70℃保温1小时。
将2,3,4,6-四-o-三甲基硅基-d-葡萄糖酸内酯(化合物2)36.8g溶于甲苯20ml,滴加到上述体系中,控制反应液温度不高于-60℃,约10min滴毕,保温-70℃下继续反应6小时,得化合物3。
(2)向反应体系中依次加入甲醇87g、浓硫酸4.0g,缓慢升温至-5℃。加入浓硫酸14.8g,保温0℃,继续反应16小时,得化合物4。
经hplc检测(图1),f邻位取代的副产物含量2%,化合物4含量67%,中间态含量30%,未知杂质1%。其中,中间态为绿色杂质,如果延长时间可以继续转化为化合物4,如果不延长时间,也会在后续反应中转化为目标产物,不影响最终收率。
向反应体系中加入乙酸乙酯200ml,从而提高萃取效果,再缓慢加入碳酸氢钠溶液(将28.8g碳酸氢钠溶于400ml水中,或者碳酸氢钠饱和溶液),搅拌约20分钟,静置,分液。有机层再依次用饱和碳酸氢钠溶液90ml和饱和食盐水90ml洗涤后,分液。减压浓缩至60~80ml(50~60℃,真空度-0.09mpa以上)。然后加入甲苯100ml,并浓缩至约60~80ml,再重复一次。残余液降温至5℃。
(3)加入甲苯200ml,然后依次加入三乙胺41.6g、dmap1.7g、乙酸酐38.5g,保温5℃反应5小时,即得化合物5。
保温15℃下加入质量百分比20%磷酸水溶液300ml(洗涤反应中加入的碱),搅拌20分钟,静置,分液。有机层用饱和碳酸氢钠溶液180ml洗涤,再用饱和食盐水90ml洗涤。有机相减压浓缩至60~80ml(50~60℃、真空度-0.09mpa以上)。
(4)加入乙腈100ml、三乙基硅烷39.8g,降温至-5~5℃,滴加三氟化硼乙醚34.0g,滴毕,控制内温0~10℃继续反应5小时。
(5)控温0~10℃下加入碳酸氢钠溶液(将57.5g碳酸氢钠溶于700ml水中,或者碳酸氢钠饱和溶液),析出固体,过滤,用55ml水洗涤,再用50ml乙醇洗涤,得到化合物6,即(1s)-2,3,4,6-四乙酰氧基-1,5-脱水-1-[3-(1-苯并噻吩-2-基甲基)-4-氟苯基]-d-山梨糖醇粗品。
加入乙醇100ml和所得的化合物6粗品,加热至50~60℃,搅拌1小时,再降温至0~10℃,搅拌2小时,过滤,减压干燥至少6小时(50~60℃、真空度-0.09mpa以上),得化合物6精制品18.0g,白色,纯度98.96%,摩尔收率为46%。
实施例2:
(1s)-2,3,4,6-四乙酰氧基-1,5-脱水-1-[3-(1-苯并噻吩-2-基甲基)-4-氟苯基]-d-山梨糖醇的制备方法,包括步骤:
(1)向反应瓶加入四氢呋喃22ml、甲苯220ml,搅拌下,加入2-(5-溴-2-氟苄基)苯并噻吩(化合物1)22.0g。将反应液降温至-60℃,氮气保护下滴加正丁基锂的正己烷溶液(2.5mol/l)31.5ml,控制反应液温度不高于-60℃,滴加速率3滴/秒,约5min滴毕,-60℃保温1小时。
将2,3,4,6-四-o-三甲基硅基-d-葡萄糖酸内酯(化合物2)36.8g溶于甲苯20ml,滴加到上述体系中,控制反应液温度不高于-60℃,约10min滴毕,保温-60℃下继续反应5小时,得化合物3。
(2)向反应体系中依次加入甲醇87g、浓硫酸4.0g,缓慢升温至-15℃。加入浓硫酸14.8g,保温10℃,继续反应约20小时,得化合物4。
向反应体系中加入乙酸乙酯200ml,再缓慢加入碳酸氢钠溶液(将28.8g碳酸氢钠溶于400ml水),搅拌约20分钟,静置,分液。有机层再依次用饱和碳酸氢钠溶液90ml和饱和食盐水90ml洗涤后,分液。
减压浓缩至60~80ml(50~60℃、真空度-0.09mpa以上)。然后加入甲苯100ml,并浓缩至约60~80ml,再重复一次。残余液降温至15℃。
(3)加入甲苯200ml,然后依次加入三乙胺41.6g、dmap1.7g、乙酸酐38.5g,保温15℃反应5小时,得化合物5。
保温35℃下加入质量百分比20%磷酸水溶液300ml,搅拌20分钟,静置,分液。有机层用饱和碳酸氢钠溶液180ml洗涤,再用饱和食盐水90ml洗涤。有机相减压浓缩至60~80ml(50~60℃、真空度-0.09mpa以上)。
(4)加入乙腈100ml、三乙基硅烷39.8g,降温至-5℃,滴加三氟化硼乙醚34.0g。滴毕,控制内温0℃继续反应4小时。
(5)控温10℃下加入碳酸氢钠溶液(将57.5g碳酸氢钠溶于700ml水),析出固体。过滤,用55ml水洗涤,再用50ml乙醇洗涤,得到化合物6,即(1s)-2,3,4,6-四乙酰氧基-1,5-脱水-1-[3-(1-苯并噻吩-2-基甲基)-4-氟苯基]-d-山梨糖醇粗品。
加入乙醇100ml和所得的化合物6粗品,加热至60℃,搅拌1小时,再降温至0℃,搅拌2小时,过滤,减压干燥至少6小时(50~60℃、真空度-0.09mpa以上),得化合物6精制品21g,性状为白色,纯度98.50%(图2),摩尔收率为53%。
实施例3:
(1s)-2,3,4,6-四乙酰氧基-1,5-脱水-1-[3-(1-苯并噻吩-2-基甲基)-4-氟苯基]-d-山梨糖醇的制备方法,包括步骤:
(1)向反应瓶加入四氢呋喃2.2l、甲苯22l,搅拌下,加入2-(5-溴-2-氟苄基)苯并噻吩(化合物1)2200g。将反应液降温至-70℃,氮气保护下滴加正丁基锂的正己烷溶液(2.5mol/l)3150ml,控制反应液温度不高于-60℃,滴加速率3滴/秒,约1小时滴毕,再保温-60℃反应1小时。
将2,3,4,6-四-o-三甲基硅基-d-葡萄糖酸内酯(化合物2)3680g溶于甲苯2l,滴加到上述体系中,控制反应液温度不高于-60℃,约1小时滴毕,保温-70℃下继续反应6小时,得化合物3。
(2)向反应体系中依次加入甲醇8.7kg、浓硫酸400g,缓慢升温至-5℃。加入浓硫酸1.5kg,保温0℃,继续反应约20小时,得化合物4。
向反应体系中加入乙酸乙酯20l,再缓慢加入碳酸氢钠溶液(将2.88kg碳酸氢钠溶于40l水),搅拌约20分钟,静置,分液。有机层再依次用饱和碳酸氢钠溶液9l和饱和食盐水9l洗涤后,分液。
减压浓缩至6~8l(50~60℃、真空度-0.09mpa以上)。然后加入甲苯10l,并浓缩至约6~8l,再重复一次。残余液降温至15℃。
(3)加入甲苯20l,然后依次加入三乙胺4.16kg、dmap170g、乙酸酐3.85kg,保温15℃反应5小时,得化合物5。
保温15℃下加入质量百分比20%磷酸水溶液30l,搅拌20分钟,静置,分液。有机层用饱和碳酸氢钠溶液18l洗涤,再用饱和食盐水9l洗涤。有机相减压浓缩至6~8l(50~60℃、真空度-0.09mpa以上)。
(4)加入乙腈10l、三乙基硅烷3.98kg,降温至-5℃,滴加三氟化硼乙醚3.40kg。滴毕,控制内温0℃继续反应5小时。
(5)控温0℃下加入碳酸氢钠溶液(将5.75kg碳酸氢钠溶于70l水),析出固体。过滤,用5.50l水洗涤,再用5l乙醇洗涤,得到化合物6,即(1s)-2,3,4,6-四乙酰氧基-1,5-脱水-1-[3-(1-苯并噻吩-2-基甲基)-4-氟苯基]-d-山梨糖醇粗品。
加入乙醇10l,化合物7粗品,加热至60℃,搅拌1小时,再降温至0℃,搅拌2小时。过滤,减压干燥至少6小时(55℃、真空度-0.09mpa以上),得化合物6精制品2000g,性状为白色,纯度99.1%,摩尔收率为51%。
对比例1
(1s)-2,3,4,6-四乙酰氧基-1,5-脱水-1-[3-(1-苯并噻吩-2-基甲基)-4-氟苯基]-d-山梨糖醇的制备方法,包括步骤:
(1)向32.5ml甲苯和25ml异丙醚中加入5.0g2-(5-溴-2-氟苄基)苯并噻吩(化合物1),-40℃滴加正丁基锂的正己烷溶液(1.6mol/l)10ml,滴加速率0.5滴/秒(滴加速度是实施例1~3的1/5),约30min滴毕,滴加完毕搅拌10min,降温至-75℃。将8.0g2,3,4,6-四-o-三甲基硅基-d-葡萄糖酸内酯(化合物2)溶于17.5ml甲苯中,滴加至上述体系,后搅拌6小时,得化合物3。
(2)升温至0℃,加入甲醇25ml和4mol/l的盐酸乙酸乙酯溶液7.8ml。搅拌17小时,得化合物4。经hplc检测(图3),化合物4含量为70%,副产物含量为25%,5%未知杂质。
加入1.29g碳酸钾35ml水的溶液,乙酸乙酯萃取,分液,水相用20ml甲苯10ml乙酸乙酯萃取。有机相合并后,浓缩至40ml,加入25ml甲苯,浓缩至40ml,重复一次。
3℃下向上述体系加入7.39g吡啶,19mgdmap,7.94g乙酸酐,搅拌12小时。
加入50ml2mol/l的盐酸溶液,分液,有机相用75ml质量浓度5%的碳酸氢钠溶液洗涤,然后用50ml25%的溶液洗涤,然后浓缩至16ml。
(3)加入10ml乙腈,溶解后,0℃下把此溶液加入到4.67g三氟甲磺酸和3.62g叔丁基二甲基硅烷溶于20ml乙腈的溶液。搅拌3小时。加入70ml四氢呋喃、25ml甲苯。2.8g碳酸钾、1.5g氯化钠溶于30ml水,加入上述体系,温度为5℃。然后升温至30℃分液。有机相用25ml25%的食盐水洗涤,常压蒸馏至55ml,冷却到0℃搅拌50小时过滤,固体用5ml*2淋洗,得到2.3g化合物6,纯度98.1%,摩尔收率25.8%。
对比例2
(1s)-2,3,4,6-四乙酰氧基-1,5-脱水-1-[3-(1-苯并噻吩-2-基甲基)-4-氟苯基]-d-山梨糖醇的制备方法,包括步骤:
(1)向反应瓶加入四氢呋喃22ml、甲苯220ml,搅拌下,加入2-(5-溴-2-氟苄基)苯并噻吩(化合物1)22.0g。将反应液降温至-70℃左右,氮气保护下滴加正丁基锂的正己烷溶液(2.5mol/l)31.5ml,控制反应液温度不高于-60℃,滴加速率0.5滴/秒(滴加速度是实施例1~3的1/5),约30min滴毕,-70℃左右保温1小时。
将2,3,4,6-四-o-三甲基硅基-d-葡萄糖酸内酯(化合物2)36.8g溶于甲苯20ml,滴加到上述体系中,控制反应液温度不高于-60℃,约10min滴毕,保温-70℃左右继续反应6小时,得化合物3。
(2)向反应体系中依次加入甲醇87g、浓硫酸4.0g,缓慢升温至-5~15℃。加入浓硫酸14.8g,保温0~10℃,继续反应约16小时,得化合物4。经hplc检测(图4),化合物4含量为86%,副产物含量为12%,2%未知杂质。
向反应体系中加入乙酸乙酯200ml,再缓慢加入碳酸氢钠溶液(将28.8g碳酸氢钠溶于400ml水),搅拌约20分钟,静置,分液。有机层再依次用饱和碳酸氢钠溶液90ml和饱和食盐水90ml洗涤后,分液。
减压浓缩至60~80ml(50~60℃、真空度-0.09mpa以上)。然后加入甲苯100ml,并浓缩至约60~80ml,再重复一次。残余液降温至15℃。
(3)加入甲苯200ml,然后依次加入三乙胺41.6g、dmap1.7g、乙酸酐38.5g,保温15℃反应5小时,得化合物5。
保温25℃下加入质量百分比20%磷酸水溶液300ml,搅拌20分钟,静置,分液。有机层用饱和碳酸氢钠溶液180ml洗涤,再用饱和食盐水90ml洗涤。有机相减压浓缩至60~80ml(50~60℃、真空度-0.09mpa以上)。
(4)加入乙腈100ml、三乙基硅烷39.8g,降温至-5℃,滴加三氟化硼乙醚34.0g。滴毕,控制内温0℃继续反应4小时。
(5)控温10℃下加入碳酸氢钠溶液(将57.5g碳酸氢钠溶于700ml水),析出固体。过滤,用55ml水洗涤,再用50ml乙醇洗涤,得到化合物6,即(1s)-2,3,4,6-四乙酰氧基-1,5-脱水-1-[3-(1-苯并噻吩-2-基甲基)-4-氟苯基]-d-山梨糖醇粗品。
加入乙醇100ml和所得的化合物6粗品,加热至60℃,搅拌1小时,再降温至0℃,搅拌2小时,过滤,减压干燥至少6小时(50~60℃、真空度-0.09mpa以上),得化合物6精制品12g,性状为白色,纯度97.9%,摩尔收率为30.6%。
上述虽然对本发明的具体实施方式进行了描述,但并非对本发明保护范围的限制,在本发明的技术方案的基础上,本领域技术人员不需要付出创造性劳动即可做出的各种修改或变形仍在本发明的保护范围以内。
- 还没有人留言评论。精彩留言会获得点赞!