一种通过核苷酸定点替换获得抗草甘膦水稻的方法与流程
本发明属于生物技术育种领域,涉及一种通过核苷酸定点替换获得抗草甘膦水稻的方法,还涉及一种可以产生核苷酸定点替换以及片段替换的方法。
背景技术:
水稻是我国乃至世界上的主要粮食作物,在我国,其总面积、总产量和单位面积产量均居首位,种植面积约5亿亩左右。我国稻田杂草发生面积约占水稻种植面积的45%左右,未进行杂草防治的稻田一般减产5-15%,每年损失稻谷约1000万吨,严重的减产15-30%。为了防除稻田杂草,不仅投入大量的人力物力和财力,还喷洒了大量除草剂。因此,培育抗除草剂的水稻十分必要。传统的育种方式周期长,可利用的种质资源匮乏。随着转基因技术以及基因组编辑技术的发展,为水稻抗草甘膦育种提供了一条有效的途径。
基因组编辑技术是近年来兴起的新技术,主要包括三大类型序列特异性核酸酶:锌指核酸酶(Zinc finger nuclease,ZFN)、类转录激活因子效应物核酸酶(Transcription activator-like effector nuclease,TALEN)和成簇的规律间隔的短回文重复序列及其相关系统(Clustered regularly interspaced short palindromic repeats/CRISPR associated,CRISPR/Cas9)。这些人工核酸酶都可以在DNA靶位点产生DNA双链断裂(Double strand breaks,DSBs),DNA损伤后产生的DSBs激活细胞内固有的非同源末端连接(Non-homologous ending-joining,NHEJ)或同源重组(Homologous recombination,HR)两种不同的修复机制对损伤的DNA进行修复:通过HR方式修复的概率很低;机体主要是通过NHEJ方式进行修复,断裂的染色体会重新连接,但往往是不精确的,断裂位置会产生少量碱基的插入或删除,造成移码突变或者使蛋白翻译提前终止,从而实现对目的基因的定点敲除。
草甘膦是一种非选择性除草剂,具有广谱、高效、低毒、低残留以及对大多数植物具有灭生性的优点,是目前世界上使用量最大的除草剂,其作用机理主要是竞争性抑制莽草酸途径中5-烯醇丙酮酰莽草酸-3磷酸合成酶(简称为EPSPS)的活性,导致芳香族氨基酸合成受阻,最终造成植物死亡。
技术实现要素:
本发明公开了一种获得抗草甘膦植物的方法。
本发明所提供的获得抗草甘膦植物的方法,包括如下步骤:仅将目的植物内源EPSPS蛋白的保守区的氨基酸序列的第8位的苏氨酸(T)替换为异亮氨酸(I),第12位的脯氨酸(P)替换为丝氨酸(S),所得植物即为抗草甘膦植物;所述目的植物内源EPSPS蛋白的保守区的氨基酸序列为序列表中SEQ ID NO:2。
发生所述替换以后的所述保守区的氨基酸序列为序列表中SEQ ID NO:7。
在所述方法中,所述“仅将目的植物内源EPSPS蛋白的保守区的氨基酸序列的第8位的苏氨酸(T)替换为异亮氨酸(I),第12位的脯氨酸(P)替换为丝氨酸(S)”是通过如下方法实现的:向所述目的植物的细胞或组织中导入如下a)或b)或c)或d)或e)或f),再将导入后的细胞或组织培养成完整植株;
a)遗传物质1、遗传物质2和donor载体;所述遗传物质1为能够表达序列特异性核酸酶1的DNA环状质粒或DNA线性片段或体外转录的RNA;所述遗传物质2为能够表达序列特异性核酸酶2的DNA环状质粒或DNA线性片段或体外转录的RNA;
b)遗传物质12和donor载体;所述遗传物质12为既能够表达所述序列特异性核酸酶1也能够表达所述序列特异性核酸酶2的DNA环状质粒或DNA线性片段或体外转录的RNA;
c)非遗传物质1、非遗传物质2和donor载体;所述非遗传物质1为表达所述序列特异性核酸酶1的mRNA;所述非遗传物质2为表达所述序列特异性核酸酶2的mRNA;
d)非遗传物质1、非遗传物质2和donor载体;所述非遗传物质1为体外表达的序列特异性核酸酶1的蛋白质;所述非遗传物质2为体外表达的序列特异性核酸酶2的蛋白质;
e)所述donor载体;
f)既能够表达所述序列特异性核酸酶1也能够表达所述序列特异性核酸酶2的donor载体;
所述donor载体为携带有突变目的靶序列的载体;所述突变目的靶序列含有将所述目的植物的基因组中包含待突变核苷酸的自靶标片段1的5’末端起到靶标片段2的3’末端止的这段DNA片段进行核苷酸突变后得到的DNA片段序列;所述靶标片段1位于所述目的植物的基因组中编码所述内源EPSPS蛋白的保守区氨基酸的核苷酸序列上游的内含子区(记为内含子区1)或启动子区;所述靶标片段2位于所述目的植物的基因组中编码所述内源EPSPS蛋白的保守区氨基酸序列的核苷酸序列下游的内含子区(记为内含子区2)或3’-UTR区;所述核苷酸突变为能够实现仅将所述目的植物的内源EPSPS蛋白的保守区氨基酸序列的第8位的苏氨酸替换为异亮氨酸,第12位的脯氨酸替换为丝氨酸的突变。
其中,所述突变目的靶序列既可以为将所述目的植物的基因组中包含待突变核苷酸的自所述靶标片段1的5’末端起到所述靶标片段2的3’末端止的这段DNA片段进行所述核苷酸突变后得到的DNA片段序列;也可以在此基础上在上游和/或下游增加同源序列后得到的DNA片段,其中上游同源序列为位于所述目的植物基因组中所述靶标片段1上游的一段序列;下游同源序列为位于所述目的植物基因组中所述靶标片段2下游的一段序列。
所述序列特异性核酸酶1能够特异性切割所述目的植物基因组中和所述donor载体中的所述靶标片段1;所述序列特异性核酸酶2能够特异性切割所述目的植物基因组中和所述donor载体中的所述靶标片段2;当所述序列特异性核酸酶1和所述序列特异性核酸酶2同时切割所述目的植物基因组中和所述donor载体中的所述靶标片段1和所述靶标片段2时,所述donor载体的两个靶标位点之间的含有待替换核苷酸的片段则有机会连接到所述目的生物基因组的两个靶标位点之间,从而获得核苷酸定点替换的基因组序列。
所述序列特异性核酸酶(如所述序列特异性核酸酶1或所述序列特异性核酸酶2)同时对所述目的生物基因组中和所述donor载体中的所述靶标片段进行特异性切割,虽然可能会造成少量碱基的插入突变、和/或缺失突变,但该突变位于启动子区,和/或内含子区,和/或UTR区(非编码区),一般不会影响蛋白的功能;所述的两个序列特异性核酸酶(即所述序列特异性核酸酶1或所述序列特异性核酸酶2)同时切割所述目的植物基因组中和所述donor载体中的所述靶标片段1和所述靶标片段2时,可以引起两个靶标位点之间序列的核苷酸替换突变。
其中,所述序列特异性核酸酶1和所述序列特异性核酸酶2均可为CRISPR/Cas9核酸酶、TALEN核酸酶、锌指核酸酶或所有能实现基因组编辑的序列特异性核酸酶。所述序列特异性核酸酶1和所述序列特异性核酸酶2既可以为同一种核酸酶也可以为不同种核酸酶。
在所述方法中,所述植物可为单子叶植物或双子叶植物;所述单子叶植物可为禾本科植物;所述禾本科植物具体可为水稻。
在本发明的一个实施例中,所述植物具体为水稻品种日本晴。
相应的,所述目的植物内源EPSPS蛋白的保守区氨基酸序列的突变位点(第8位和第12位)在基因组DNA水平上位于所述目的植物内源EPSPS基因的第二外显子;所述内含子区1为第一内含子,其核苷酸序列为序列表中SEQ ID NO:3的第1-704位;所述内含子区2为第二内含子,其核苷酸序列为序列表中SEQ ID NO:3的第950-1030位。所述序列特异性核酸酶1和所述序列特异性核酸酶2均为CRISPR/Cas9核酸酶,所述靶标片段1为序列表中SEQ ID NO:3的第1-704所示核苷酸序列中符合5’-NX-NGG-3’或5’-CCN-NX-3’序列排列规则的片段;N表示A、G、C和T中的任一种,14≤X≤30,且X为整数(如X为20),NX表示X个连续的核苷酸。所述靶标片段2为序列表中SEQ ID NO:3的第950-1030位所示核苷酸序列中符合5’-NX-NGG-3’或5’-CCN-NX-3’序列排列规则的片段;N表示A、G、C和T中的任一种,14≤X≤30,且X为整数(如X为20),NX表示X个连续的核苷酸。
进一步,所述靶标片段1的核苷酸序列为序列表中SEQ ID NO:4;所述靶标片段2的核苷酸序列为序列表中SEQ ID NO:5。
相应的,在本发明中,所述遗传物质1具体为将pHUN411载体的两个限制性内切酶BsaI之间的小片段替换为序列表中SEQ ID NO:4的第1-20位所示DNA片段后得到的重组质粒(pHUN411-C3);所述遗传物质2具体为将pHUN411载体的两个限制性内切酶BsaI之间的小片段替换为序列表中SEQ ID NO:5的第1-20位所示DNA片段后得到的重组质粒(pHUN411-C4)。
所述donor载体上携带的突变目的靶序列的核苷酸序列具体为序列表中SEQ ID NO:6。所述donor载体具体为将序列表中SEQ ID NO:6所示DNA片段与pEASY-Blunt载体(如全式金生物技术有限公司产品,目录号CB101)相连后得到的重组质粒(pEPSPS-donor)。
在本发明中,将所述pHUN411-C3、所述pHUN411-C4和所述pEPSPS-donor导入水稻品种日本晴的愈伤组织时,三者的摩尔比为1:1:2。
在所述方法中,所述细胞可为任何能作为导入受体并能经过组织培养再生为完整植株的细胞;所述组织可为任何能作为导入受体并能经过组织培养再生为完整植株的组织。具体的,所述细胞如可为原生质体细胞或悬浮细胞;所述组织如可为愈伤组织、幼胚或成熟胚。
在所述方法中,向所述目的植物的细胞或组织中导入所述a)或所述b)或所述c)或所述d)或所述e)的方法可为基因枪法、农杆菌侵染法、PEG介导的原生质体转化法或其他任何导入方法。
如下生物材料中的任一种也属于本发明的保护范围:
(1)蛋白质,为仅将水稻内源EPSPS蛋白的保守区氨基酸序列的第8位的苏氨酸替换为异亮氨酸,第12位的脯氨酸替换为丝氨酸后形成的蛋白;所述水稻内源EPSPS蛋白的保守区氨基酸序列为序列表中SEQ ID NO:2;
(2)所述蛋白质的编码基因;
(3)含有所述编码基因的表达盒、重组载体、重组菌或转基因细胞系。
其中,所述转基因细胞系为非繁殖材料。
本发明还提供了一种对目的基因中的目的核苷酸进行替换的方法。其中,所述目的核苷酸既可为1个核苷酸或者多个不连续的核苷酸,也可以为由多个连续的核苷酸组成的片段。
本发明所提供的对目的基因中的目的核苷酸进行替换的方法,是通过向目的生物的细胞或组织中导入如下a)或b)或c)或d)或e),实现对所述目的生物中所述目的基因的目的核苷酸的替换:
a)遗传物质1、遗传物质2和donor载体;所述遗传物质1为能够表达序列特异性核酸酶1的DNA环状质粒或DNA线性片段或体外转录的RNA;所述遗传物质2为能够表达序列特异性核酸酶2的DNA环状质粒或DNA线性片段或体外转录的RNA;
b)遗传物质12和所述donor载体;所述遗传物质12为既能够表达所述序列特异性核酸酶1也能表达所述序列特异性核酸酶2的DNA环状质粒或DNA线性片段或体外转录的RNA;
c)非遗传物质1、非遗传物质2和所述donor载体;所述非遗传物质1为表达所述序列特异性核酸酶1的mRNA;所述非遗传物质2为表达所述序列特异性核酸酶2的mRNA;
d)非遗传物质1、非遗传物质2和所述donor载体;所述非遗传物质1为体外表达序列特异性核酸酶1的蛋白质;所述非遗传物质2为体外表达序列特异性核酸酶2的蛋白质;
e)既能够表达序列特异性核酸酶1也能表达序列特异性核酸酶2的donor载体;
所述donor载体为携带有突变目的靶序列的载体;所述突变目的靶序列含有将所述目的生物的基因组中包含待替换核苷酸的自靶标片段1的5’末端起到靶标片段2的3’末端止的这段DNA片段进行所述目的核苷酸替换后得到的DNA片段序列;所述靶标片段1位于所述目的生物的基因组中所述目的基因的目的核苷酸上游的内含子区或启动子区;所述靶标片段2位于所述目的生物的基因组中所述目的基因的目的核苷酸下游的内含子区或3’-UTR区。两个序列特异性核酸酶的靶标位置位于启动子区,和/或内含子区,和/或UTR区(非编码区),在该区产生少量碱基的插入缺失一般不会影响目的基因的表达。
其中,所述突变目的靶序列既可以为将所述目的生物的基因组中包含待替换核苷酸的自所述靶标片段1的5’末端起到所述靶标片段2的3’末端止的这段DNA片段进行所述核苷酸突变后得到的DNA片段序列;也可以在此基础上在上游和/或下游增加同源序列后得到的DNA片段,其中上游同源序列为位于所述目的生物的基因组中所述靶标片段1上游的一段序列;下游同源序列为位于所述目的生物的基因组中所述靶标片段2下游的一段序列。
所述序列特异性核酸酶1能够特异性切割所述目的生物基因组中和所述donor载体中的靶标片段1;所述序列特异性核酸酶2能够特异性切割所述目的生物基因组中和所述donor载体中的靶标片段2。所述donor载体中的上下游同源序列引导所述donor载体到达所述目的生物的基因组中所述目的基因处,当所述序列特异性核酸酶1和所述序列特异性核酸酶2同时切割所述目的生物基因组中和所述donor载体中的所述靶标片段1和所述靶标片段2时,所述donor载体的两个靶标位点之间的含有待替换核苷酸的片段则有机会连接到所述目的生物基因组的两个靶标位点之间,从而获得核苷酸定点替换的基因组序列。
其中,所述目的生物可为目的植物、目的动物或目的微生物。
所述序列特异性核酸酶1和所述序列特异性核酸酶2均可为CRISPR/Cas9核酸酶、TALEN核酸酶、锌指核酸酶或所有能实现基因组编辑的序列特异性核酸酶。所述序列特异性核酸酶1和所述序列特异性核酸酶2既可以为同一种核酸酶也可以为不同种核酸酶。
在一些实施方案中,所述目的生物是植物,包括单子叶植物或双子叶植物,例如水稻或拟南芥。所述细胞为任何能作为导入受体并能经过组织培养再生为完整植株的细胞;所述组织为任何能作为导入受体并能经过组织培养再生为完整植株的组织。具体的,所述细胞为原生质体细胞或悬浮细胞;所述组织为愈伤组织、幼胚或成熟胚。向所述目的植物的细胞或组织中导入所述a)或所述b)或所述c)或所述d)或所述e)的方法包括基因枪法、农杆菌侵染法、PEG介导的原生质体转化法或其他任何导入方法。
在一具体实施方案中,所述植物是拟南芥,所述目的基因是SEQ ID NO:9所示的Atsnc1(At4g16890),其中所述序列特异性核酸酶1包含对应SEQ ID NO:12的sgRNA,所述序列特异性核酸酶2包含对应SEQ ID NO:13的sgRNA,所述突变目的靶序列示于SEQ ID NO:14,所述目的核苷酸的替换使得SEQ ID NO:10的Atsnc1外显子3被替换为SEQ ID NO:11,由此赋予植物对Pseudomonas syringae pv maculicola ES4326和Peronospora parasitica Noco2的抗性。
本发明利用CRISPR介导的NHEJ途径通过设计两个gRNA的位点,两个gRNA的位点分别位于EPSPS基因的第一内含子和第二内含子中,通过CRISPR/Cas9技术两个gRNA同时切割基因组中和donor载体中的靶标片段1和2,靶标位点之间的碱基替换的片段有机会连接到基因组的靶标位点之间,对水稻的第二外显子进行替换,从而实现保守区两个氨基酸的定点突变,从而产生具有草甘膦抗性的水稻,对于培育抗除草剂植物新品种具有重要意义。
本发明还提供了一种对目的基因中的目的核苷酸进行定点替换的方法。
附图说明
图1为OsEPSPS基因结构示意图及利用CRISPR/Cas9技术的两个靶位点(C3和C4)的序列。
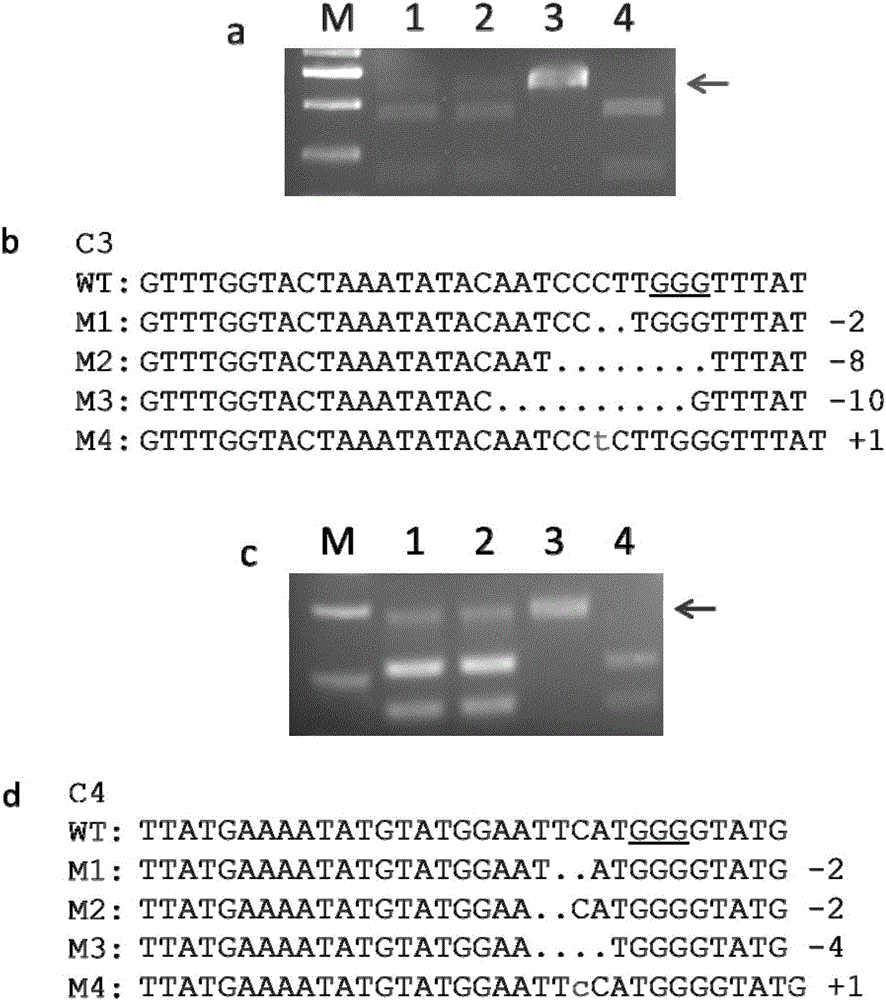
图2为OsEPSPS基因靶位点C3和C4在水稻原生质体中的活性检测及对应的测序结果。a为C3靶位点在原生质体中PCR/RE检测结果,b为C3靶位点对应的测序结果;c为C4靶位点在原生质体中PCR/RE检测结果;d为C4靶位点对应的测序结果。其中,WT表示野生型水稻日本晴的基因组序列,“-”表示发生了删除突变的序列,“+”表示发生了插入突变的序列,“-/+”后边的数字表示删除或插入的核苷酸的数量(小写字母为插入的核苷酸),M1-M4表示4种突变类型。
图3为含有替换碱基的donor载体(pEPSPS-donor)的结构示意图。a为水稻基因组中EPSPS保守区的DNA序列以及保守区编码的氨基酸序列,b为donor载体中EPSPS保守区的DNA序列及编码的氨基酸序列,加粗标记的氨基酸是替换氨基酸。为了便于后期的检测,同义突变出一个酶切位点PvuI。
图4为用PCR/RE检测CRISPR介导的水稻OsEPSPS基因定点突变的T0代突变体以及测序结果。a为T0代突变体的PCR/RE检测结果,其中1-16为不同的再生line,PCR产物利用PvuI限制性内切酶酶切,ck为野生型水稻日本晴对照;b为突变体1,5,7的测序结果,其中,G表示野生型水稻日本晴EPSPS基因的部分序列:左边序列为C3位点序列,中间为EPSPS保守区DNA序列,右边为C4位点序列;D表示donor载体上的EPSPS基因的部分序列:左边序列为C3位点序列,中间为点突变的EPSPS保守区DNA序列,右边为C4位点序列。“-”表示发生了删除突变的序列,“+”表示发生了插入突变的序列,“-/+”后边的数字表示删除或插入的核苷酸的数量(小写字母为插入的核苷酸)。
图5为水稻植株在含1mg/L草甘膦的N6培养基中的生长情况。培养10天后,a为OsEPSPS基因TIPS定点突变的突变体T0-1;b为野生型水稻日本晴对照。
具体实施方式
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
pHUN411载体:在文献“Hui-Li Xing,Li Dong,Zhi-Ping Wang,Hai-Yan Zhang,Chun-Yan Han,Bing Liu,Xue-Chen Wang,Qi-Jun Chen.A CRISPR/Cas9toolkit for multiplex genome editing in plants.BMC plant biology.14:327-338(2014)”中公开过,公众可从中国科学院遗传与发育生物学研究所获得。该质粒可同时用于转录向导RNA和表达Cas9蛋白。
实施例1、水稻OsEPSPS靶位点的选择并构建CRISPR载体
一、OsEPSPS靶位点的选择
OsEPSPS基因座位号是06g04280,位于水稻第6号染色体,包含8个外显子,7个内含子,共编码515个氨基酸。OsEPSPS基因的保守区位于第2外显子中,构建敲除载体选择的靶位点分别位于第1内含子和第2内含子中(图1)。OsEPSPS基因在水稻基因组中第1内含子、包含保守区的第2外显子及第2内含子的序列如序列表中序列3所示。其中,序列3的第1-704位为第1内含子,第705-949位为第2外显子,第950-1030位为第2内含子。
利用CRISPR技术敲除的靶标双链中的一条链具有如下结构:5-Nx-NGG-3,PAM(NGG)中的N表示A、T、C和G中的任一种,Nx中的N表示A、T、C和G中的任一种,x=20。OsEPSPS基因的靶序列如下,带下划线的碱基为PAM(原型间隔序列毗邻基序)。其中,靶标C3位于第1内含子,靶标C4位于第2内含子。
C3:5’-TACTAAATATACAATCCCTTGGG-3’(SEQ ID NO:4);
C4:5’-AAAATATGTATGGAATTCATGGG-3’(SEQ ID NO:5)。
该敲除载体转化水稻后,在gRNA的介导下,Cas9蛋白在靶序列区域切割,形成DNA双链断裂,触发机体内的自我损伤修复机制,细胞自发修复该缺口的过程中会引入突变(本处“突变”指的是广义突变,包括插入、缺失、狭义突变等形式,这些突变中绝大多数为基因功能失活突变)。
上述靶序列C3含有BsaJI酶切识别序列(斜体加粗部分的序列),可以被BsaJI限制性内切酶切割;上述靶序列C4含有EcoRI酶切识别序列(斜体加粗部分的序列),可以被EcoRI限制性内切酶切割。C3靶序列区域被切割后,如果发生了突变,则BsaJI酶切识别序列被破坏,将不能被限制性内切酶BsaJI切割;如果没有发生突变,将可以被限制性内切酶BsaJI切割。同理,C4靶序列区域被切割后,如果发生了突变,则EcoRI酶切识别序列被破坏,将不能被限制性内切酶EcoRI切割;如果没有发生突变,将可以被限制性内切酶EcoRI切割。
二、重组载体的构建
1、用限制性内切酶BsaI酶切pHUN411质粒(该质粒上含有两个BsaI限制性内切酶识别位点),回收约12.5kb的载体骨架,命名为HUN411。
2、根据步骤一设计的靶位点C3和C4序列,合成如下带有粘性末端(下划线部分)的引物:
C3-F:5’-GGCGTACTAAATATACAATCCCTT-3’;
C3-R:5’-AAACAAGGGATTGTATATTTAGTA-3’。
C4-F:5’-GGCGAAAATATGTATGGAATTCAT-3’;
C4-R:5’-AAACATGAATTCCATACATATTTT-3’。
3、将C3-F和C3-R,C4-F和C4-R分别进行退火,形成有粘性末端的双链DNA命名为C3和C4,将其和步骤1中的胶回收产物HUN411连接,得到重组质粒pHUN411-C3、pHUN411-C4。
重组质粒pHUN411-C3的结构描述为:将pHUN411质粒的两个限制性内切酶BsaI的识别序列之间的小片段(约1.2kb)替换为序列表中SEQ ID NO:4的第1-20位所示DNA片段后所得的重组质粒,该质粒可同时用于转录包含SEQ ID NO:4的向导RNA和表达Cas9蛋白。
重组质粒pHUN411-C4的结构描述为:将pHUN411质粒的两个限制性内切酶BsaI的识别序列之间的小片段(约1.2kb)替换为序列表中SEQ ID NO:5的第1-20位所示DNA片段后所得的重组质粒,该质粒可同时用于转录包含SEQ ID NO:5的向导RNA和表达Cas9蛋白。
实施例2、转化水稻原生质体及检测重组载体在原生质体中的活性
将实施例1构建的重组质粒pHUN411-C3、pHUN411-C4分别通过PEG介导的方式转入水稻品种日本晴的原生质体,然后提取原生质体的基因组DNA,用特异引物通过PCR扩增包含靶位点C3、C4的OsEPSPS基因,然后分别将含有靶位点C3、C4的PCR扩增产物用限制性内切酶BsaJI、EcoRI酶切(如果PCR扩增产物有部分条带不能被切开,说明实施例1中设计的靶位点有活性),将不能被限制性内切酶切开的PCR扩增产物进行凝胶回收,连pEASY-Blunt载体(如全式金生物技术有限公司产品,目录号CB101),挑单克隆进行测序。
分别用于扩增含有靶位点C3、C4的引物序列如下:
上游引物OsEC3-F:5’-CTAGGAATTATCTCTCAAGTCAATC-3’;
下游引物OsEC3-R:5’-CTCACTGTTCAGCAAGTTGTCC-3’。
上游引物OsEC4-F:5’-TTCTTAATAGCTTTGATCGCG-3’;
下游引物OsEC4-R:5’-TAACCTTGCCACCAGGAAGTC-3’。
实验同时设置未经酶切的野生型水稻日本晴的PCR产物对照,以及经过BsaJI或EcoRI酶切的野生型对照PCR产物对照。实验重复三次。
原生质体中C3检测重组载体活性的酶切结果见图2中a,泳道1、2为转化后的原生质体,其中含有不能被BsaJI切开的PCR条带(大小约为640bp,与预期一致),说明靶位点C3有活性;泳道3为未经酶切的野生型PCR产物对照;泳道4为经过酶切的野生型对照PCR产物,可以被BsaJI完全切开。测序(图2中b)结果表明靶位点处产生了少量碱基的插入与缺失,证实重组载体pHUN411-C3在靶位点C3处进行了基因定点编辑。
原生质体中C4检测重组载体活性的酶切结果见图2中c,泳道1、2为转化后的原生质体,其中含有不能被EcoRI切开的PCR条带(大小约为730bp,与预期一致),说明靶位点C4有活性;泳道3为未经酶切的野生型PCR产物对照;泳道4为经过酶切的野生型对照PCR产物,可以被EcoRI完全切开。测序(图2中d)结果表明靶位点处产生了少量碱基的插入与缺失,证实重组载体pHUN411-C4在靶位点处进行了基因定点编辑。
实施例3、Donor载体的构建
本实施例旨在构建含有突变目的靶序列的donor载体,以期能够与CRISPR/Cas9核酸酶一同实现将水稻内源EPSPS蛋白中SEQ ID NO:2所示的保守区多肽(保守区多肽的编码基因为序列表中SEQ ID NO:1)的第8位的苏氨酸(T)替换为异亮氨酸(I),第12位的脯氨酸(P)替换为丝氨酸(S),即使得突变后的所述保守区多肽的序列为序列表中SEQ ID NO:7,下文将该突变简称为TIPS突变。Donor载体的具体构建方法如下:
以野生型水稻日本晴的基因组DNA为模板,用引物OsEPSPS-DF/OsEPSPS-DR进行PCR扩增,电泳检测,出现1.2Kb的目的条带,PCR产物纯化后连接pEASY-Blunt载体(如全式金生物技术有限公司产品,目录号CB101)。经PCR验证获得连接EPSPS片段的克隆,摇菌提取质粒。将经测序表明在结构组成上为将序列表中SEQ ID NO:8所示DNA片段与T-Blunt载体相连后所得质粒命名为TB-EPSPS-D。其中,SEQ ID NO:8的第12-1041位与SEQ ID NO:3完全一致。
OsEPSPS-DF:5’-CCCTCTCCGAGGTGAGACG-3’(SEQ ID NO:8的第1-19位);
OsEPSPS-DR:5’-TAACCTTGCCACCAGGAAGTC-3’(SEQ ID NO:8的第1179-1199位的反向互补序列)。
以质粒TB-EPSPS-D为模板,用引物OsEPSPS-TIPSF/OsEPSPS-TIPSR扩增(为了方便后期检测,该对引物中也同义突变出PvuI限制性内切酶酶切位点),PCR产物经DpnI处理后,转化大肠杆菌,挑克隆送测序,将经测序表明在结构组成上为将序列表中SEQ ID NO:6所示DNA片段与T-Blunt载体相连后得到的重组质粒命名为pEPSPS-donor。其中,SEQ ID NO:6与SEQ ID NO:8相比,差别仅在于OsEPSPS-TIPSF/OsEPSPS-TIPSR引物中引入的突变位点,突变后的保守区氨基酸序列如序列表中SEQ ID NO:7所示。
pEPSPS-donor的结构示意图如图3所示。
OsEPSPS-TIPSF:5’-GAACGCTGGAATGCAATGCGACTTGACAGCAGCCGTGAC-3’;
OsEPSPS-TIPSR:5’-TGCTGTCAAcGATCGCATTGCAATTCCAGCGTTCCCCAAG-3’。
其中,加方框的为突变碱基,加粗字体为突变出的酶切位点PvuI。
实施例4、转化水稻及TIPS EPSPS位点突变体检测
将实施例1构建的重组质粒pHUN411-C3、pHUN411-C4和实施例3中构建的重组质粒pEPSPS-donor通过基因枪转化的方法同时导入水稻品种日本晴。以日本晴的愈伤组织为转化受体,转化时重组质粒pHUN411-C3、pHUN411-C4和pEPSPS-donor的摩尔配比为1:1:2,转化后经过组织培养获得完整再生植株(即T0代)。
提取T0代转基因植株基因组DNA,用含有靶位点C3、C4和TIPS突变位点的特异引物OsEF1和OsER对基因组DNA进行PCR扩增。
OsEF1:5’-CAACAGGATCCTCCTCCTCTC-3’(位于水稻基因组中序列8/6的上游30bp处);
OsER:5’-TAACCTTGCCACCAGGAAGTC-3’(SEQ ID NO:6的第1179-1199位的反向互补序列)。
PCR产物用PvuI单酶切,酶切结果见图4a。经PCR、酶切检测后,共获得3个line在TIPS处突变的植株,如T0-1,T0-5,T0-7(分别对应图4a的泳道1、5和7,除了大小约为1.2kb的PCR产物条带外,还含有经由PvuI酶切开的大小约为950bp和270bp的目的条带),其余编号植株(分别对应图4中的泳道2、3、4、6、8、9、10、11、12、13、14、15、16)为野生型。
进一步,对突变体的测序结果见图4b,3个line(T0-1,T0-5,T0-7)在设计的靶位点处产生了目的碱基的突变,同时在C3和C4靶位点处产生少量碱基的插入和缺失,由于C3和C4位于内含子中,少量碱基的缺失插入并不影响OsEPSPS基因的阅读框,仅发生目的碱基突变的碱基影响了翻译产生的蛋白,证明成功获得了TIPS EPSPS位点突变的突变体。
实施例5、TIPS EPSPS位点突变体的草甘膦抗性鉴定
将实施例4获得的3个TIPS EPSPS位点突变line(T0-1,T0-5,T0-7)和野生型水稻日本晴植株放置于含有1mg/L草甘膦的N6培养基中,常规培养条件下培养,10天后观察抗性结果。每个TIPS EPSPS位点突变line均进行3株以上重复。
结果显示,野生型水稻日本晴植株已经变黄萎蔫死亡,而所有的TIPS EPSPS位点突变植株均正常存活(叶片为绿色)。其中,图5a为TIPS EPSPS位点突变的T0-1,图5b为野生型水稻日本晴对照植株。
实施例6、拟南芥中内含子介导的定点基因替换
对于双子叶模式植物拟南芥的功能基因组研究,产生精确的基因组修饰(例如点突变和基因替换)具有巨大价值。本文报道在拟南芥中使用CRISPR/Cas9系统通过非同源末端连接(NHEJ)途径实现的内含子介导的位点特异性基因替换方法。
先前已经鉴别到Atsnc1(At4g16890)中的点突变(G1654-A)导致Glu552Lys置换,并导致组成型表达病程相关(PR)基因,赋予植物对Pseudomonas syringae pv maculicola ES4326和Peronospora parasitica Noco2的抗性。本实施例目的是获得内源Atsnc1基因的氨基酸替换。Atsnc1的基因组序列示于SEQ ID NO:9。
上述点突变出现于Atsnc1的外显子3,而外显子3编码的序列示于SEQ ID NO:10。为了用含有点所述突变(G1654-A)的新外显子(SEQ ID NO:11)替换内源外显子3,我们设计了分别靶向Atsnc1的内含子2和3的sgRNA。靶向Atsnc1的内含子2的sgRNA(S1)的序列示于SEQ ID NO:12,而靶向Atsnc1的内含子3的sgRNA(S2)的序列示于SEQ ID NO:13。
S1sgRNA(由AtU626启动子驱动)、S2sgRNA(由AtU629启动子驱动)以及含有单核苷酸置换(G1654-A)的供体序列被整合进携带潮霉素B磷酸转移酶基因(hpt)和Cas9表达盒的pHEE401载体,获得构建体pHEE411-S1S2Donor。含有所述核苷酸置换的供体序列示于SEQ ID NO:14。
终载体pHEE411-S1S2Donor通过冻融法转化进农杆菌菌株GV3101。通过浸花法转化拟南芥Col-0野生型植物。收集的种子在含有25mg/L潮霉素的MS平板上进行筛选。从土壤生长的T1代转基因植物提取基因组DNA。使用基因特异性引物PCR扩增包含靶位点的片段并进行测序。结果确认获得了具有所需基因替换的植物。
- 还没有人留言评论。精彩留言会获得点赞!
- 5-烯醇丙酮莽草酸-3-磷酸合酶抗体的免疫原及其制备方法与应用的制作方法
- 一种便于使用的抗草甘膦转基因大豆pcr-elisa检测试剂盒的制作方法
- 一种快速检测抗草甘膦g2-epsps蛋白的金标试剂盒的制作方法
- 一种以基因优化方法获得的抗草甘膦基因及其表达载体的制作方法
- 一种双甘膦废水套用制备草甘膦中间体双甘膦的工艺的制作方法
- 草甘膦降解酶基因和含该基因的载体的制作方法
- 草甘膦抗性基因及含该基因的载体和转化子的制作方法
- 分离自土壤的高抗草甘膦epsp合酶及其编码序列的制备和用途的制作方法
- 一种抗草甘膦EPSP合成酶GmEPSPS-2及其编码基因与应用的制作方法
- 一种抗草甘膦EPSP合成酶GmEPSPS及其编码基因与应用的制作方法