一种聚呋喃类化合物及其制备方法与流程
本发明属于有机合成技术领域,具体涉及一种聚呋喃类化合物及其制备方法。
背景技术:
发展新的聚合反应对于高分子材料科学来说是非常重要的。炔烃是易得或易合成的化学原料之一,利用炔烃构建功能性高分子具有重要的学术意义和技术意义,已经吸引了科学家们的广泛关注。
氧化反应在有机反应里面是一类非常重要的反应,在官能团转换方面扮演着重要的角色。虽然近些年,很多科研工作者发展了一些新的氧化方法,但是化学领域还是在“如何使用绿色氧化试剂”以及“怎样才能达到最大原子经济性”方面面临着很大的挑战。传统氧化剂,如:高锰酸钾,双氧水等等,存在很多弊端:价格昂贵,对环境有危害,难以回收利用等等。然而氧气作为自然界取之不尽,用之不竭的一种资源,是一个非常好的氧化剂。拿氧气作为氧化剂参与有机化学反应有诸多优点:环境友好,成本低,反应温和等等。虽然很多科研工作者已经发展了很多用氧气氧化的有机反应体系,但是还是存在着诸多问题,如:反应条件苛刻(需要高温高压的环境),反应的适用性有限,反应底物较难合成等等,这就限制了氧气氧化体系的发展。目前,还没有氧气参与氧化并参与形成产物的聚合反应的报道。
技术实现要素:
为了解决以上现有技术的缺点和不足之处,本发明的首要目的在于提供一种聚呋喃类化合物的制备方法。该制备方法可以在常压氧气环境下进行,反应绿色、高效、容易操作。
本发明的另一目的在于提供一种通过上述方法制备得到的聚呋喃类化合物。
本发明目的通过以下技术方案实现:
一种聚呋喃类化合物的制备方法,包括如下制备步骤:
(1)在常压氧气环境下,双官能团芳基内炔单体和氧气在有机溶剂中通过催化剂和助催化剂共同作用进行聚合反应;
(2)反应完毕后,将产物溶解在有机溶剂中,然后加入到甲醇中进行沉淀,收集沉淀物,干燥至恒重,得到聚呋喃类化合物;
所述的双官能团芳基内炔单体的结构通式如式(Ⅱ)所示:
作为优选,所述的有机溶剂包含两种成分,一种选自全氟萘烷,另一种选自四氢呋喃、二氯甲烷、氯仿、甲苯、1,4-二氧六环、二甲基亚砜、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、乙腈、乙醇中的至少一种。从溶剂的对聚合反应的影响程度考虑,进一步优选,所述的有机溶剂选自全氟萘烷和N,N-二甲基乙酰胺,此时,得到的聚呋喃类化合物溶解度较好,产率和分子量也较高,便于下一步应用。
反应单体的浓度会对反应的产率产生影响,作为优选,步骤(1)所述聚合反应中双官能团芳基内炔单体在有机溶剂中的物质的量浓度为0.05~5mol/L,进一步优选,所述双官能团芳基内炔单体在有机溶剂中的物质的量浓度为0.20mol/L。
反应催化剂的种类和用量会对反应的时间和产物的产率和分子量产生影响,作为优选,所述的催化剂为醋酸钯、氯化钯、四(三苯基膦)钯、双三苯基磷二氯化钯、二(二苯膦基)二茂铁二氯化钯和三(二亚苄基丙酮)二钯中的至少一种,所述的催化剂的用量为双官能团芳基内炔单体的1%~40mol%;进一步优选,所述的催化剂为醋酸钯,所述的催化剂的用量为双官能团芳基内炔单体的20mol%。
反应助催化剂的种类和用量会对反应的时间和产物的产率和分子量产生影响,作为优选,所述的助催化剂为氯化锌、一水合氯化锌、氯化铜、三氟甲磺酸锌中的至少一种。所述的助催化剂的用量为双官能团芳基内炔单体的2%~100mol%;进一步优选,所述的助催化剂为氯化锌,所述的助催化剂的用量为双官能团芳基内炔单体的60~100mol%。
作为优选,所述聚合反应的温度为0~200℃,时间为0.25~72小时。
考虑到聚合反应的产率和聚合所得产物的分子量及其分布,同时从节省能源和高速高效的角度出发,将所述的聚合反应的温度进一步优选为70℃,时间优选为8小时。
一种聚呋喃类化合物,所述聚呋喃类化合物具有式(I)所示的结构通式:
式中,x,y,z为0~200的整数,R1,R2为氢原子或者有机基团。
优选地,所述R1为以下1~24中的任意一种有机基团;R2为氢原子或以下25~28中任意一种有机基团;
其中,m、h、k为1~20的整数;X选自N、P、O、S或Si元素;*表示取代位置。
本发明的制备方法及所得到的产物具有如下优点及有益效果:
(1)本发明的制备方法可以在常压氧气环境下进行,并且氧气参与聚合物的形成,无需控制单体的投料比例;据我们所知,该聚合反应此前未见报道,因此,具有创新性和极其重要的意义;
(2)本发明的制备方法操作简单,反应原料和催化剂易得,可直接购买或通过简单的反应制备;聚合反应条件温和,节约能源;聚合反应使用的氧化剂为氧气,成本低,绿色环保;
(3)本发明的制备方法具有良好的官能团耐受性,可以引入多种功能性基团;制得的功能化聚呋喃类化合物具有较好的热稳定性和优异的可加工性,由于特殊的光物理性质,在爆炸物检测和双光子吸收等领域具有巨大的应用前景。
附图说明
图1为本发明实施例1制备的聚呋喃类化合物(C)与其相应单体(A)和模型化合物(B)在CD2Cl2中核磁共振氢谱对比图(*代表溶剂峰);
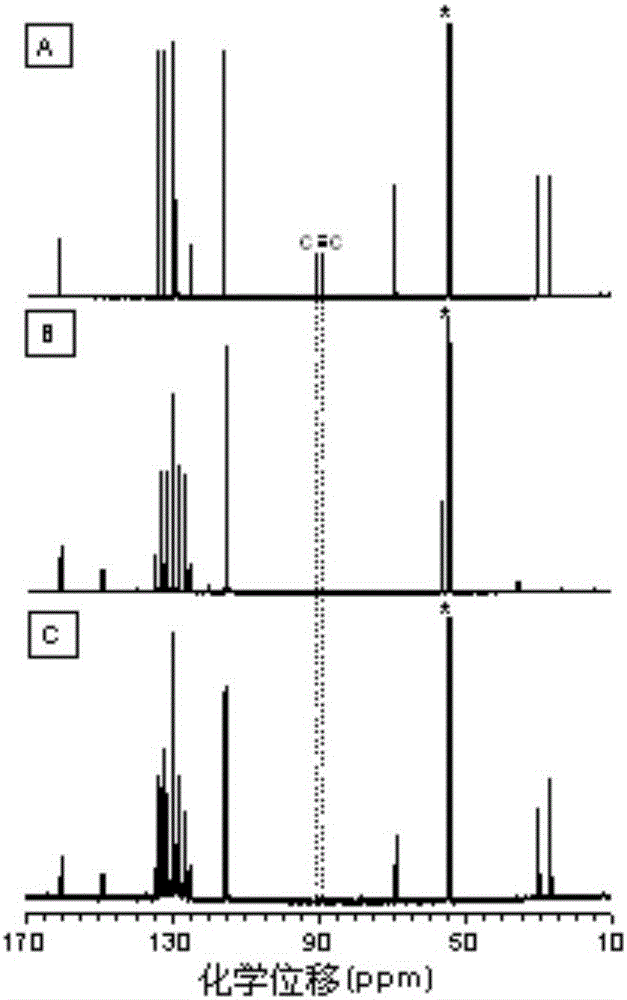
图2为本发明实施例1制备的聚呋喃类化合物(C)与其相应单体(A)和模型化合物(B)在CD2Cl2中核磁共振碳谱对比图(*代表溶剂峰);
图3为本发明实施例1制备的聚呋喃类化合物(C)与其相应单体(A)和模型化合物(B)的红外吸收光谱对比图;
图4为本发明实施例2制备的聚呋喃类化合物(C)与其相应单体(A)和模型化合物(B)在CD2Cl2中核磁共振氢谱对比图(*代表溶剂峰);
图5为本发明实施例2制备的聚呋喃类化合物(C)与其相应单体(A)和模型化合物(B)在CD2Cl2中核磁共振碳谱对比图(*代表溶剂峰);
图6为本发明实施例2备的聚呋喃类类化合物(C)与其相应单体(A)和模型化合物(B)的红外吸收光谱对比图;
图7为本发明实施例3制备的聚呋喃类化合物(C)与其相应单体(A)和模型化合物(B)在CD2Cl2中核磁共振氢谱对比图(*代表溶剂峰);
图8为本发明实施例3制备的聚呋喃类化合物(C)与其相应单体(A)和模型化合物(B)在CD2Cl2中核磁共振碳谱对比图(*代表溶剂峰);
图9为本发明实施例3备的聚呋喃类类化合物(C)与其相应单体(A)和模型化合物(B)的红外吸收光谱对比图;
图10为本发明实施例4制备的聚呋喃类化合物(B)及其相应单体(A)在CD2Cl2中核磁共振氢谱对比图(*代表溶剂峰);
图11为本发明实施例4制备的聚呋喃类化合物(B)及其相应单体(A)在CD2Cl2中核磁共振碳谱对比图(*代表溶剂峰);
图12为本发明实施例4制备的聚呋喃类化合物(B)及其相应单体(A)的红外吸收光谱对比图;
图13为本发明实施例1制备的聚呋喃类化合物在四氢呋喃溶液中检测三硝基苯酚的光谱图;
图14为本发明实施例4制备的聚呋喃类化合物在四氢呋喃溶液中检测三硝基苯酚的光谱图;
图15为本发明实施例4制备的聚呋喃类化合物在二氯甲烷溶液中不同激发波长下的双光子吸收截面曲线图;
图16为本发明实施例1~4制备的聚呋喃类化合物的热失重曲线图,测试条件:氮气气氛下,升温速率为10℃/min。
具体实施方式
下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例1
本实施例的一种聚呋喃类化合物,其结构式如P1(6)所示:
上述聚呋喃类化合物通过双官能团芳基内炔单体和氧气聚合反应进行制备,反应方程式如式(一):
其中,单体M1的合成方法可按照申请人在已公开文献中(Polym.Chem.,2013,4,2841–2849.)的合成方法合成。
本实施例所述的聚呋喃类化合物的具体制备步骤如下:
在一个干燥的施伦克管中加入M1(94mg,0.2mmol),Pd(OAc)2(20mol%),ZnCl2(60mol%),抽真空0.5小时,扎入一个充满氧气的气球,加N,N-二甲基乙酰胺(DMAc)和全氟萘烷(C10F18)各0.5mL,在70℃反应8小时,然后冷却至室温,反应完之后的溶液通过一个棉花过滤装置,逐滴滴加到装有200mL甲醇溶液的锥形瓶中,并伴随有强烈的搅拌。沉淀出来的聚合物放置过夜,然后通过过滤获得,得到的聚合物用甲醇溶液冲洗,然后在真空干燥箱干燥到重量不会再发生变化,得到产物。
表征数据:黄色固体,产率80%。凝胶渗透色谱(GPC)结果显示:重均分子量(Mw)为14900,分子量分布(PDI)为2.6。
为了表征聚呋喃类化合物结构,我们做了模型反应,拿到了模型化合物2~4,反应方程式如式(二):
本实施例所得聚呋喃类化合物与其相应单体以及模型化合物的核磁共振谱对比图(*代表溶剂峰)见图1、图2,红外吸收光谱图见图3,从图1中可以看出聚合物的谱图和模型产物的谱图非常相似,从图2中可以看出单体中碳碳三键在化学位移91ppm和89ppm处的核磁共振峰在聚合物中已经消失,从图3也可以看到单体中碳碳三键在波数2217cm-1处的红外吸收峰在聚合物中已经消失,从而可以确定该聚合物为聚呋喃类化合物。此外,该聚呋喃类化合物在室温下易溶于二氯甲烷、氯仿、四氢呋喃、N,N-二甲基甲酰胺等常见有机溶剂,表明具有优异的可加工性。从图16可以看出,该聚呋喃类化合物有良好的热稳定性。图13为该聚呋喃类化合物在四氢呋喃溶液中检测三硝基苯酚光谱图,图中可以看出,随着三硝基苯酚含量的增加,该聚呋喃类化合物在四氢呋喃溶液中的荧光发射有很明显的减弱,说明该聚呋喃类化合物在爆炸物检测方面有潜在的应用价值。
实施例2
本实施例的一种聚呋喃类化合物,其结构式如P1(4)所示:
上述聚呋喃类化合物通过双官能团芳基内炔单体和氧气聚合反应进行制备,反应方程式如式(三):
其中,单体M2的合成方法可按照申请人在已公开文献中(Polym.Chem.,2013,4,2841–2849.)的合成方法合成。
本实施例所述的聚呋喃类化合物的具体制备步骤如下:
在一个干燥的施伦克管中加入M2(88.5mg,0.2mmol),Pd(OAc)2(20mol%),ZnCl2(60mol%),抽真空0.5小时,扎入一个充满氧气的气球,加N,N-二甲基乙酰胺(DMAc)和全氟萘烷(C10F18)各0.5mL,在70℃反应8小时,然后冷却至室温,反应完之后的溶液通过一个棉花过滤装置,逐滴滴加到装有200mL甲醇溶液的锥形瓶中,并伴随有强烈的搅拌。沉淀出来的聚合物放置过夜,然后通过过滤获得,得到的聚合物用甲醇溶液冲洗,然后在真空干燥箱干燥到重量不会再发生变化,得到产物。
表征数据:黄色固体,产率81%。凝胶渗透色谱(GPC)结果显示:重均分子量(Mw)为12200,分子量分布(PDI)为2.0。
为了表征聚呋喃类化合物结构,我们做了模型反应,拿到了模型化合物2~4,反应方程式如式(二),该聚呋喃类化合物与其相应单体以及模型化合物的核磁共振谱对比图(*代表溶剂峰)见图4、图5,红外吸收光谱图见图6,从图4中可以看出聚合物的谱图和模型产物的谱图非常相似,从图5中可以看出单体中碳碳三键在化学位移91ppm和89ppm处的核磁共振峰在聚合物中已经消失,从图6也可以看到单体中碳碳三键在波数2217cm-1处的红外吸收峰在聚合物中已经消失,从而可以确定该聚合物为聚呋喃类化合物。此外,该聚呋喃类化合物在室温下易溶于二氯甲烷、氯仿、四氢呋喃、N,N-二甲基甲酰胺等常见有机溶剂,表明具有优异的可加工性。从图16可以看出,该聚呋喃类化合物有良好的热稳定性。
实施例3
本实施例的一种聚呋喃类化合物,其结构式如P1(8)所示:
上述聚呋喃类化合物通过双官能团芳基内炔单体和氧气聚合反应进行制备,反应方程式如式(四):
其中,单体M3的合成方法可按照申请人在已公开文献中(Polym.Chem.,2013,4,2841–2849.)的合成方法合成。
本实施例所述的聚呋喃类化合物的具体制备步骤如下:
在一个干燥的施伦克管中加入M3(99.7mg,0.2mmol),Pd(OAc)2(20mol%),ZnCl2(60mol%),抽真空0.5小时,扎入一个充满氧气的气球,加N,N-二甲基乙酰胺(DMAc)和全氟萘烷(C10F18)各0.5mL,在70℃反应8小时,然后冷却至室温,反应完之后的溶液通过一个棉花过滤装置,逐滴滴加到装有200mL甲醇溶液的锥形瓶中,并伴随有强烈的搅拌。沉淀出来的聚合物放置过夜,然后通过过滤获得,得到的聚合物用甲醇溶液冲洗,然后在真空干燥箱干燥到重量不会再发生变化,得到产物。
表征数据:黄色固体,产率76%。凝胶渗透色谱(GPC)结果显示:重均分子量(Mw)为14000,分子量分布(PDI)为2.0。
为了表征聚呋喃类化合物结构,我们做了模型反应,拿到了模型化合物2~4,反应方程式如式(二),该聚呋喃类化合物与其相应单体以及模型化合物的核磁共振谱对比图(*代表溶剂峰)见图7、图8,红外吸收光谱图见图9,从图4中可以看出聚合物的谱图和模型产物的谱图非常相似,从图5中可以看出单体中碳碳三键在化学位移91ppm和89ppm处的核磁共振峰在聚合物中已经消失,从图6也可以看到单体中碳碳三键在波数2217cm-1处的红外吸收峰在聚合物中已经消失,从而可以确定该聚合物为聚呋喃类化合物。此外,该聚呋喃类化合物在室温下易溶于二氯甲烷、氯仿、四氢呋喃、N,N-二甲基甲酰胺等常见有机溶剂,表明具有优异的可加工性。从图16可以看出,该聚呋喃类化合物有良好的热稳定性。
实施例4
本实施例的一种聚呋喃类化合物,其结构式如P2所示:
上述聚呋喃类化合物通过双官能团芳基内炔单体和氧气聚合反应进行制备,反应方程式如式(五):
其中,单体M4的合成方法可按照申请人在已公开文献中(Polym.Chem.,2013,4,2841–2849.)的合成方法合成。
所述的聚呋喃类化合物的制备步骤如下:
在一个干燥的施伦克管中加入M4(106.5mg,0.2mmol),Pd(OAc)2(20mol%),ZnCl2(60mol%),抽真空0.5小时,扎入一个充满氧气的气球,加N,N-二甲基乙酰胺(DMAc)和全氟萘烷(C10F18)各0.5mL,在70℃反应8小时,然后冷却至室温,反应完之后的溶液通过一个棉花过滤装置,逐滴滴加到装有200mL甲醇溶液的锥形瓶中,并伴随有强烈的搅拌。沉淀出来的聚合物放置过夜,然后通过过滤获得,得到的聚合物用甲醇溶液冲洗,然后在真空干燥箱干燥到重量不会再发生变化,得到产物。
表征数据:黄色固体,产率74%。凝胶渗透色谱(GPC)结果显示:重均分子量(Mw)为11600,分子量分布(PDI)为2.3。
该聚呋喃类化合物与其相应单体的核磁共振谱对比图(*代表溶剂峰)见图10、图11,红外吸收光谱图见图12,从图11中可以看出单体中碳碳三键在化学位移91ppm和90ppm处的核磁共振峰在聚合物中已经消失,从图12也可以看到单体中碳碳三键在波数2217cm-1处的红外吸收峰在聚合物中已经消失,说明碳碳三键在反应过程中已经反应完全。此外,该聚呋喃类化合物在室温下易溶于二氯甲烷、氯仿、四氢呋喃、N,N-二甲基甲酰胺等常见有机溶剂,表明具有优异的可加工性。从图16可以看出,该聚呋喃类化合物有良好的热稳定性。图14为该聚呋喃类化合物在四氢呋喃溶液中检测三硝基苯酚光谱图,图中可以看出,随着三硝基苯酚含量的增加,该聚呋喃类化合物在四氢呋喃溶液中的荧光发射有很明显的减弱,说明该聚呋喃类化合物在爆炸物检测方面有潜在的应用价值。图15是该聚呋喃类化合物在二氯甲烷中不同激发波长下的双光子截面曲线,说明该聚呋喃类化合物具有较大的双光子吸收截面,在双光子成像、光限幅等领域具有潜在的应用价值。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其它的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!