一种3‑氨基‑5‑甲硫基‑1,2,4‑三氮唑的制备方法与流程
本发明属于抗病毒药物化学合成技术领域,特别涉及一种3-氨基-5-甲硫基-1,2,4-三氮唑的制备方法。
背景技术:
3-氨基-5-甲硫基-1,2,4-三氮唑是一种新型抗病毒药物的重要原料,可用于合成具有广谱抗病毒活性的三唑-三嗪类药物。这类药物能有效治疗克里米亚-刚果出血热、裂谷热、西尼罗河病毒感染等疾病。可以保护动物(60%-90%)免受流感病毒、蜱传播的脑炎、呼吸道感染以及其它多种社会影响大、危害高的病毒性传染病。在抑制高致病人类流感病毒的再生方面疗效显著,包括H1N1、H3N5、H5N1和非流感病毒如西部马脑脊髓炎病毒、呼吸道合胞病毒、鸡传染性喉气管炎病毒、禽流感病毒等,能够用于预防和治疗人类和动物病毒感染性疾病。该化合物具有抗病毒活性高,作用持续时间长和毒性低等特点。
到目前为止,仅有CN201310116564-一种5-氨基-3-巯基-1,2,4-三氮唑晶体及其制备方法公开了一种利用5-氨基-3-巯基-1,2,4三氮唑和碘甲烷为原料,进行甲基化反应得到产品的制备方法,尽管该方法相对其他现有制备方法已经具有一定环境污染减少,成本降低,纯度提高等效果,但其仍然存在诸多弊端。
目前的3-氨基-5-甲硫基-1,2,4-三氮唑多是以5-氨基-3-巯基-1,2,4三氮唑和碘甲烷为原料,进行甲基化反应得到产品,次工艺所用原料比较昂贵,成本高,且会产生大量含碘废水,很难处理,工业化放大受到限制。
三唑杂环化合物具有广泛的生物活性,如抗菌,镇静,抗焦虑等作用,因此,一直是杂环化合物的中药研究领域,其中3-氨基-5-甲硫基-1,2,4-三氮唑是合成三唑杂环衍生物的重要中间体。
因此提供一种新的3-氨基-5-甲硫基-1,2,4-三氮唑的合成方法,节省成本的同时减少环境污染,便于工业化大范围加工就显得尤为必要。
技术实现要素:
为解决上述现有技术存在的问题,本发明的目的在于提供一种3-氨基-5-甲硫基-1,2,4-三氮唑的制备方法,以氰氨基二硫化碳酸二甲酯、水合肼为原料,以醇或水作为溶剂,通过一步反应制备合成目标产物,原料成本低,不产生污染环境的含碘废水,操作步骤简单,且产物纯度能达到98%以上,具有很好的商业应用前景,值得大范围推广。
为达到上述目的,本发明的技术方案为:
一种3-氨基-5-甲硫基-1,2,4-三氮唑的制备方法,反应器进行氮气置换后,以氰氨基二硫化碳酸二甲酯、水合肼为原料,溶剂为醇类有机溶剂,将氰氨基二硫化碳酸二甲酯加入溶剂中搅拌溶解后,在25-28℃条件下,缓慢滴加水合肼,氰氨基二硫化碳酸二甲酯和水合肼摩尔比为1:1.1-1:15,滴加1.5-2小时,之后升温至65-70℃,在回流条件下,反应2-3小时,通过一步反应制备合成类白色固体,经核磁确认为目标产物3-氨基-5-甲硫基-1,2,4-三氮唑,反应方程式为:
进一步的,所述有机溶剂为甲醇或乙醇。
进一步的,所述方法3-氨基-5-甲硫基-1,2,4-三氮唑的纯度≥98%。
进一步的,所述溶剂的体积为氰氨基二硫化碳酸二甲酯重量的5-7倍。
进一步的,所述方法反应合成收率≥90%。
相对于现有技术,本发明的有益效果为:
本发明一种3-氨基-5-甲硫基-1,2,4-三氮唑的制备方法,以氰氨基二硫化碳酸二甲酯、水合肼为原料,以醇或水作为溶剂,通过一步反应制备合成目标产物,原料成本低,不产生污染环境的含碘废水,操作步骤简单,且产物纯度能达到98%以上,具有很好的商业应用前景,值得大范围推广。
附图说明
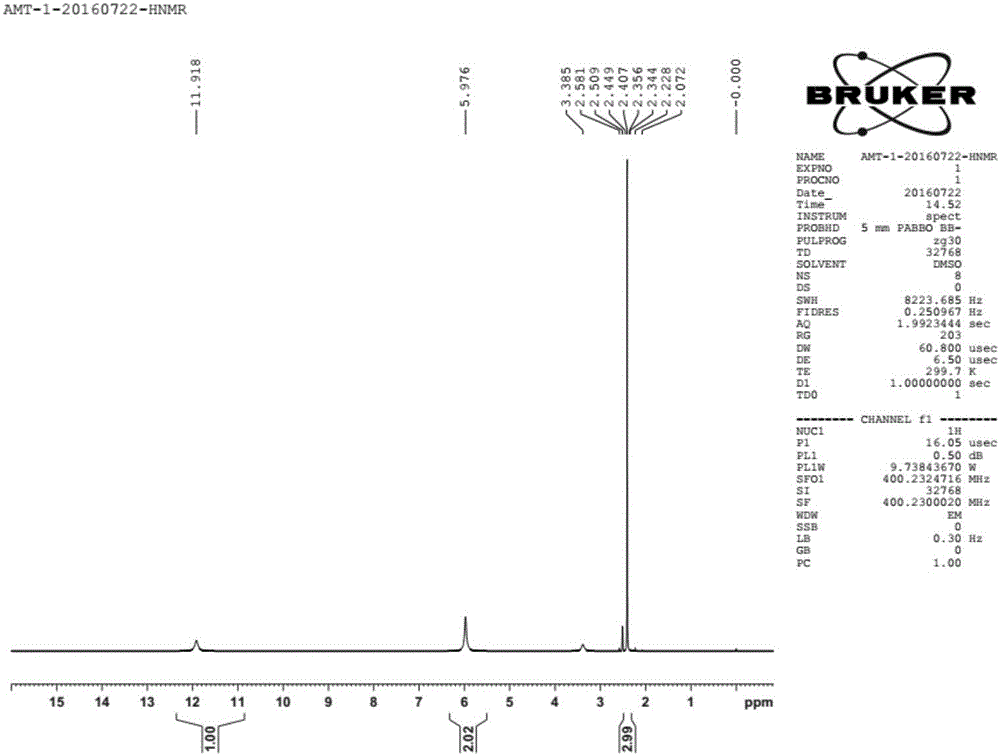
图1为目标产物的氢核磁共振HNMR图谱。
图2为目标产物的碳核磁共振CNMR图谱。
图3为目标产物的液质联用LC-MS图谱。
具体实施方式
下面结合实施例对本发明做进一步的说明,以下所述,仅是对本发明的较佳实施例而已,并非对本发明做其他形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更为同等变化的等效实施例。凡是未脱离本发明方案内容,依据本发明的技术实质对以下实施例所做的任何简单修改或等同变化,均落在本发明的保护范围内。
如图1-3所示,一种3-氨基-5-甲硫基-1,2,4-三氮唑的制备方法,反应器进行氮气置换后,以氰氨基二硫化碳酸二甲酯、水合肼为原料,溶剂为醇类有机溶剂,将氰氨基二硫化碳酸二甲酯加入溶剂中搅拌溶解后,在25-28℃条件下,缓慢滴加水合肼,氰氨基二硫化碳酸二甲酯和水合肼摩尔比为1:1.1-1:15,滴加1.5-2小时,之后升温至65-70℃,在回流条件下,反应2-3小时,通过一步反应制备合成类白色固体,经核磁确认为目标产物3-氨基-5-甲硫基-1,2,4-三氮唑,反应方程式为:
进一步的,所述有机溶剂为甲醇或乙醇。
进一步的,所述方法3-氨基-5-甲硫基-1,2,4-三氮唑的纯度≥98%。
进一步的,所述溶剂的体积为氰氨基二硫化碳酸二甲酯重量的5-7倍。
进一步的,所述方法反应合成收率≥90%。
以下结合数个较佳实施例对本发明技术方案作进一步非限制性的详细说明。
实施例1
反应器进行氮气置换后,加入250ml甲醇,50g氰氨基二硫化碳酸二甲酯,搅拌溶解后,自然升至室温,在25℃搅拌下,然后缓慢滴加32g水合肼,滴加时间保持在1.5小时,然后升温至65℃至回流反应2小时,然后反应液降温至10℃,过滤,滤饼用水洗涤,干燥后得到类白色固体40g,反应方程式为:
,液相含量为99.2%。
实施例2
反应器进行氮气置换后,加入1323ml乙醇,189g氰氨基二硫化碳酸二甲酯,搅拌溶解后,自然升至室温,在28℃搅拌下,然后缓慢滴加96g水合肼,滴加时间保持在1.8小时,然后升温至67℃至回流2.5小时,冷却至10℃,过滤,滤饼用水洗涤,干燥后得到类白色固体134g,反应方程式为:
,液相含量为99.5%,所得产物的氢核磁共振HNMR图谱如图1所示。碳核磁共振CNMR图谱如图2所示。液质联用LC-MS图谱如图3所示。
实施例3
反应器进行氮气置换后,加入350ml甲醇,50g氰氨基二硫化碳酸二甲酯,搅拌溶解后,自然升至室温,在27℃搅拌下,然后缓慢滴加40g水合肼,滴加时间保持在2小时,然后升温至70℃至回流3小时,冷却至10℃,过滤,滤饼用水洗涤,干燥后得到类白色固体32g,反应方程式为:
,液相含量为98.5%。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何不经过创造性劳动想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应该以权利要求书所限定的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!