制造聚烯烃反应性远螯预聚物的方法与流程
本发明涉及一种制造聚烯烃反应性远螯预聚物的方法。
背景技术:
聚烯烃作为高摩尔质量聚合物为有用的材料。饱和聚烯烃材料的高耐化学品性和抗氧化性伴随着有竞争力的价格使得聚烯烃对于塑料产业高度合乎需要。已证实聚烯烃上官能团的受控制的包括可引起特性显著增强。然而,尽管从聚烯烃衍生大量材料和应用,但是其预聚物型式的制造仍是尚未充分探索的领域。形成快速固化的和高分子量聚合物所需要的聚烯烃的精确可控官能化一直充满挑战。大多数将反应性基团并入聚烯烃中的方法涉及后聚合反应,这通常对官能化位置和数量控制不良并导致机械特性减弱。形成固化和/或高分子量聚合物的可模制、可注入和另外可处理反应性聚烯烃预聚物的合成将合乎期望,因为这类方法将开辟目前由如硅酮和聚氨酯弹性体的材料主导的市场的应用空间。
技术实现要素:
在一个实施例中,本发明提供一种制造聚烯烃反应性远螯预聚物的方法,包含在具有两个或更多个氨基并且所述两个或更多个氨基受一个或多个保护基保护的多官能链转移剂存在下,使烷基-顺-环辛烯与任选地顺-环辛烯在开环复分解聚合条件下接触以形成二氨基甲酸酯远螯不饱和聚烯烃预聚物。
附图说明
图1是说明根据astmd1708在127mmmin-1下所测量的交联聚合物xt-a-ph(3ecoe)-2的应力应变曲线的曲线。
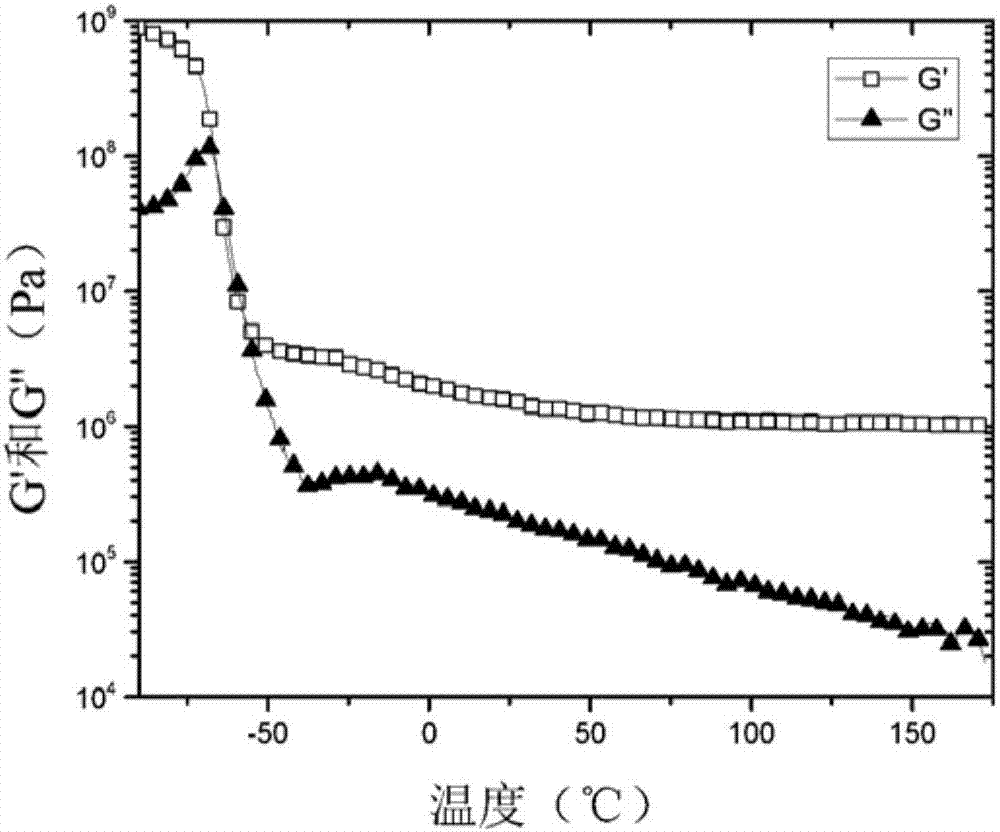
图2是说明交联聚合物xt-a-ph(3ecoe)-2在5℃min-1下在-90℃到200℃进行扭转试验的动态机械热分析的曲线。ω=6.28rads-1并且γ=0.05%。
具体实施方式
在制造聚烯烃反应性远螯预聚物的本发明方法中,使用烷基-顺-环辛烯和/或芳基-顺-环辛烯。此项技术中已知可用于本发明的实施例的烷基-顺-环辛烯。示例性烷基-顺-环辛烯包括3-经取代-顺-环辛烯、这类3-甲基-顺-环辛烯、3-乙基-顺-环辛烯和3-己基-顺-环辛烯,示例性芳基-顺-环辛烯包括3-苯基-顺-环辛烯。
在具有两个或更多个氨基的多官能链转移剂存在下使烷基-顺-环辛烯与任选地顺-环辛烯接触,其中所述两个或更多个氨基受一个或多个保护基保护。示例性保护基包括以下化合物类别∶氨基甲酸酯(氨基-酯)、酰胺(氨基-酮)、苯甲基-胺和磺酸酯。可用于本发明的示例性特异性保护基包括氨基甲酸叔丁酯(“boc胺”);氨基甲酸9-芴基甲酯(“fmoc胺”);氨基甲酸苯甲酯;三氟乙酰胺;邻苯二甲酰亚胺;苯甲胺;和对甲苯磺酰胺(“甲苯磺酰胺”)。示例性保护链转移剂包括丁-2-烯-1,4-二基(e)-二氨基甲酸二-叔丁酯;n,n'-(丁-2-烯-1,4-二基)双(2,2,2-三氟乙酰胺);和2,2'-(丁-2-烯-1,4-二基)双(异吲哚啉-1,3-二酮)。
接触在开环复分解聚合(romp)条件下发生,这为此项技术中所熟知并描述在例如“3-取代环辛烯的区域选择性和立体选择性开环复分解聚合(regio-andstereoselectivering-openingmetathesispolymerizationof3-substitutedcyclooctenes),”shingokobayashi等人,《美国化学会志(j.am.chem.soc.)》2011,133,5794-5797和“使用顺丁烯二酸作为链转移剂的通过romp的羧基-远螯聚烯烃(carboxy-telechelicpolyolefinsbyrompusingmaleicacidasachaintransferagent),”pitet和hillmyer,《大分子(macromolecules)》2011,44,2378-2381中。已知多种催化剂可用于romp,包括简单金属类化合物,如rucl3/醇混合物和更多复合grubbs'催化剂,所述grubbs'催化剂包括第一代grubbs'催化剂和第二代grubbs'催化剂以及hoveyda-grubbs催化剂。第一代grubbs'催化剂是具有以下通式的过渡金属碳烯复合物:
第二代grubbs'催化剂具有通式:
hoyveda-grubbs催化剂具有通式:
熟练技术人员将理解可以使用适合于romp的任何催化剂。本发明并不受前述催化剂结构限制或受使用钌作为用于此类催化剂的金属限制。
在开环复分解聚合条件下,在具有两个或更多个保护氨基的多官能链转移剂存在下使烷基-顺-环辛烯与任选地顺-环辛烯接触之后,形成二氨基甲酸酯远螯不饱和聚烯烃预聚物。所得预聚物的分子量和身份取决于烷基-顺-环辛烯的烷基官能团。
本发明进一步公开本文所述的方法,其进一步包含部分氢化二氨基甲酸酯远螯不饱和聚烯烃预聚物以制造饱和聚烯烃二氨基甲酸酯远螯预聚物。在一特定实施例中,部分氢化通过在对甲苯磺酰基酰肼存在下使二氨基甲酸酯远螯不饱和聚烯烃预聚物回流实现。下文反应方案总体上描绘二氨基远螯饱和聚烯烃预聚物的形成:
在一特定实施例中,氢化提供至少90%的饱和度并产生具有至少1.7官能度/预聚物链的饱和聚烯烃二氨基甲酸酯远螯预聚物。本文中包括并且本文中公开1.7官能度/预聚物链的下限的所有个别值和子范围。举例来说,官能度的下限可以是1.7、1.8、1.9、或2.0官能度/预聚物链。在一替代实施例中,氢化聚烯烃反应性远螯预聚物等于或小于10官能度/预聚物链,或在替代方案中,等于或小于7官能度/预聚物链,或在替代方案中,等于或小于4官能度/预聚物链。
在一替代实施例中,本发明提供一种根据本文中公开的任何实施例制造饱和聚烯烃二氨基甲酸酯远螯预聚物的方法,不同之处在于在氢化之后保持至少60%的官能度。本文中包括并且本文中公开至少60%的所有个别值和子范围。举例来说,在氢化之后保持的官能度的百分比可以在60、70、80、90或95的下限范围内。
在一替代实施例中,本发明提供一种根据本文中公开的任何实施例制造饱和聚烯烃二氨基甲酸酯远螯预聚物的方法,不同之处在于氢化引起预聚物中存在的至少90%的不饱和度被氢化。本文中包括并且本文中公开至少90%的所有个别值及子范围;举例来说,氢化水平下限可以是90%、92.5%、95%或97%。
本发明进一步提供本文中公开的方法,其进一步包含将一个或多个保护基从饱和二氨基甲酸酯远螯聚烯烃预聚物中移除以制造饱和聚烯烃二氨基远螯预聚物。可以使用用于使与保护基反应且将其移除的任何合适方法(例如使其与酸接触)。在一特定实施例中,保护基通过在室温下使饱和二氨基甲酸酯远螯聚烯烃预聚物与三氟乙酸接触移除。在一替代实施例中,保护基通过在吡啶或三甲胺溶剂中在100℃下使饱和二氨基甲酸酯远螯聚烯烃预聚物与ph<1的此类酸接触持续数分钟到数小时的反应时间来移除。
在又一实施例中,本发明提供一种根据本文所述的方法制造的二氨基甲酸酯远螯聚烯烃预聚物。
在另一实施例中,本发明提供一种制造交联聚合物的方法,其包含任选地在不存在催化剂的情况下使二氨基远螯聚烯烃预聚物与一种或多种与预聚物反应的多官能化合物接触以形成交联和/或扩链聚合物。如本文所用,术语多官能化合物是指具有与预聚物的氨基反应的超过一种官能团的化合物。取决于多官能化合物中的官能团,预聚物可以充当双官能预聚物或四官能预聚物。举例来说,每个胺基可以与两个环氧基反应,这意味着预聚物是四官能的。可以使用的示例性多官能化合物包括多官能环氧化物,如双官能环氧化物、多异氰酸酯、多羧酸、多酰基氯和聚环氧化物。
本发明进一步提供一种制造高分子量聚合物的方法,包含任选地在不存在催化剂的情况下使二氨基远螯聚烯烃预聚物与一种或多种与远螯预聚物反应的双官能化合物接触以形成高分子量聚合物。如本文所用,高分子量聚合物意味分子量是聚烯烃反应性远螯预聚物分子量至少两倍的的聚合物。本文中包括并且本文中公开至少两倍的所有个别值和子范围。举例来说,高分子量聚合物的分子量下限可以是聚烯烃反应性远螯预聚物分子量的2倍,或在替代方案中高分子量聚合物的分子量下限可以是聚烯烃反应性远螯预聚物分子量的5倍,或在替代方案中高分子量聚合物的分子量下限可以是聚烯烃反应性远螯预聚物分子量的10倍,或在替代方案中高分子量聚合物的分子量下限可以是聚烯烃反应性远螯预聚物分子量的15倍。
在一替代实施例中,本发明提供一种根据本文中公开的任何实施例的方法,不同之处在于所述方法进一步包含任选地在不存在催化剂的情况下同时扩链氢化聚烯烃反应性远螯预聚物和双官能化合物的混合物并使扩链氢化聚烯烃反应性远螯预聚物与多官能化合物热交联,这两者都与远螯预聚物反应,以形成扩链交联聚合物。示例性扩链多官能化合物包括双-异氰酸酯(如4,4′-亚甲基双(异氰酸苯酯)和甲代亚苯基-2,4-二异氰酸酯)、二-酰基氯(如癸二酰基氯)和双-环氧化物(如1,4-丁二醇二缩水甘油醚和双酚a二缩水甘油醚)。示例性交联多官能化合物包括三-环氧化物(如三(2,3-环氧丙基)异氰尿酸酯和三羟甲基丙烷三缩水甘油基醚)和双-环氧化物(如1,4-丁二醇二缩水甘油醚和双酚a二缩水甘油醚)。
扩链反应总体上展示于以下反应方案中∶
交联反应总体上描绘于以下反应方案中∶
在又一实施例中,本发明提供一种按本文所述制造的交联和/或扩链聚合物。
在一替代实施例中,本发明提供一种根据本文中公开的任何实施例制造聚烯烃反应性远螯预聚物、不饱和聚烯烃反应性远螯预聚物、氢化聚烯烃反应性远螯预聚物、交联聚合物和高分子量聚合物的方法,不同之处在于不饱和和/或氢化聚烯烃反应性远螯预聚物具有1kg/mol到20kg/mol的摩尔质量。本文中包括并且本文中公开1kg/mol到20kg/mol摩尔质量的所有个别值和子范围;例如,不饱和聚烯烃反应性远螯预聚物的摩尔质量可以在1kg/mol、3kg/mol、6kg/mol、9kg/mol、12kg/mol、15kg/mol或18kg/mol的下限到2kg/mol、5kg/mol、8kg/mol、11kg/mol、14kg/mol、17kg/mol或20kg/mol的上限。
在一替代实施例中,本发明提供一种根据本文中公开的任何实施例制造聚烯烃反应性远螯预聚物、不饱和聚烯烃反应性远螯预聚物、氢化聚烯烃反应性远螯预聚物、交联聚合物和高分子量聚合物的方法,不同之处在于多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比是1:2到2:1。本文中包括并且本文中公开1:2到2:1的所有个别值和子范围;例如,多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比可以是1:2,或在替代方案中,多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比可以是2:1,或在替代方案中,多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比可以是1.5:2,或在替代方案中,多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比可以是2:1.5,或在替代方案中,多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比可以是1:1.05,或在替代方案中,多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比可以是1:0.95。在一特定实施例中,多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比是1:0.94到1:1.06。
在一替代实施例中,本发明提供一种根据本文中公开的任何实施例制造聚烯烃反应性远螯预聚物、不饱和聚烯烃反应性远螯预聚物、氢化聚烯烃反应性远螯预聚物、交联聚合物和高分子量聚合物的方法,不同之处在于双官能和多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比是1:2到2:1。本文中包括并且本文中公开1:2到2:1的所有个别值和子范围;例如,双官能和多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比可以是1:2,或在替代方案中,双官能和多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比可以是2:1,或在替代方案中,双官能和多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比可以是1.5:2,或在替代方案中,双官能和多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比可以是2:1.5,或在替代方案中,双官能和多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比可以是1:1.05,或在替代方案中,双官能和多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比可以是1:0.95。在一特定实施例中,双官能和多官能化合物上的官能团与聚烯烃反应性远螯预聚物的官能团的摩尔比是1:0.94到1:1.06。
在一替代实施例中,本发明提供一种根据本文中公开的任何实施例的方法的反应产物,不同之处在于所述方法进一步包含将填充剂添加到反应产物中。填充剂可以是强化或非强化填充剂。合适填充剂的非限制性实例包括滑石、碳酸钙、白垩、硫酸钙、粘土、高岭土、二氧化硅、玻璃、热解法二氧化硅、云母、硅灰石、长石、硅酸铝、硅酸钙、氧化铝、水合氧化铝(如氧化铝三水合物)、玻璃微球体、陶瓷微球体、热塑性微球体、重晶石、木粉、玻璃纤维、碳纤维、大理石粉、水泥粉、氧化镁、氢氧化镁、氧化锑、氧化锌、硫酸钡、二氧化钛和钛酸盐。在另一实施例中,方法进一步包含将两种或更多种前述填充剂添加到反应产物中。添加一种或多种填充剂可用于提高反应产物的机械特性,例如拉伸和撕裂特性、模量和耐热性。
实例
以下实例说明本发明但是并不意欲限制本发明的范围。
链转移剂(cta)合成
使用《生物化学与生物物理学报(biochimicaetbiophysicaacta)》(bba);he等人,1995,第1253卷,第117页和《大分子》nagarkar,等人2012,第45卷,第4447页中描述的方法制造叔丁氧基羰基保护的氨基链转移剂;并且如下文反应方案1中所示∶
方案1
确切地说,1,4-二溴-2-丁烯中的溴在亲核攻击中被邻苯二甲酰亚胺取代以产生化合物1;在酸性条件下移除此基团得到化合物2,二氨基化合物的二-盐酸盐。此二-氨基化合物的衍生物随后用叔-丁氧基羰基保护以获得化合物3,丁-2-烯-1,4-二基(e)-二氨基甲酸二-叔丁酯。
如下文方案2中所示,随后二-叔丁基丁-2-烯-1,4-二基(e)-二氨基甲酸酯作为链转移剂(cta)与3-乙基环辛烯接触以制造二氨基甲酸酯远螯不饱和聚烯烃预聚物,p(3ecoe):
方案2
其中g2是第二代grubbs催化剂,确切地说是(1,3-双(2,4,6-三甲基苯基)-2-咪唑烷亚基)二氯(苯基亚甲基)(三环己基膦)钌(“(imesh2)-(cy3p)rucl2(chph)”)。
用特氟龙(teflon)涂布的磁力搅拌棒将二-叔丁基丁-2-烯-1,4-二基-二氨基甲酸酯(cta)放置在单颈20ml烧瓶中。烧瓶用橡胶隔片封闭。使用针,将烧瓶和其内容物放置在高度真空下持续10分钟并随后用氩气回填;此抽空-填充循环进行3次。借助注射器将无水氯仿和3-乙基-顺-环辛烯添加到烧瓶中。用氩气净化系统20分钟,并且随后浸没在40℃下的油浴中。借助注射器添加g2催化剂作为含1ml无水脱气氯仿的溶液。在20h之后用0.1ml乙基乙烯基醚淬灭溶液并搅拌另外10分钟。预聚物通过沉淀到室温甲醇中分离。溶液搅拌1小时,并随后倾析甲醇以留下高粘性液体预聚物。将预聚物溶解于8mlch2cl2中并随后添加2mg丁基化羟基甲苯(bht)。移除溶剂并使预聚物在高真空下在40℃下干燥。
1hnmr(500mhz,cdcl3):δ=5.32(m,1h,=c(1)h),5.07(m,1h,=c(2)h),4.45(b,nh),3.65(m,c(11)h),1.98(m,2h,-c(8)h2),1.76(m,1h,-c(3)h),1.45(s,c(12)h)1.40-1.07(m,10h,-ch2-),0.83(t,3h,-c(10)h3).13cnmr(125mhz,cdcl3):δ=134.78(c2),130.39(c1),44.67(c3),35.33(c4),32.77(c8),29.90(c6),29.40(c5),28.31(c9),27.26(c7),11.89(c10)。
随后使用对甲苯磺酰基酰肼作为氢化催化剂氢化二氨基甲酸酯远螯不饱和聚烯烃预聚物p(3ecoe),如下文方案3中所示∶
方案3
将p(3ecoe)(1.5g,10mmol链烯烃)、对-甲苯磺酰肼(3.74g,20mmol)、三丁胺(4.75mlg,20mmol)、少量bht(约5mg)与二甲苯(80ml)的混合物回流9h,并随后使其冷却到室温。将反应混合物倒入甲醇中并使预聚物沉淀。沉淀的预聚物通过倾析分离并通过使用甲醇系统反复再沉淀来纯化。随后在50℃下在高度真空下干燥预聚物以获得呈粘性液体的ph(3ecoe)。
1hnmr(500mhz,cdcl3):δ=δ4.5(b,nh),3.10(m,c(11)h),1.45(s,c(12)h),1.43-1.07(b,17h,-ch2-,-c(3)h-),0.85(t,3h,-c(10)h3).13cnmr(125mhz,cdcl3):δ=38.98(c3),33.34(c2,c4),30.32(c6,c8),29.91(c7),26.88(c1,c5),26.00(c9),10.99(c10)。
在氢化反应9小时之后,获得饱和二氨基甲酸酯远螯聚烯烃预聚物ph(3ecoe)的95摩尔%转化率。
根据下文方案4进行饱和二氨基甲酸酯远螯聚烯烃预聚物ph(3ecoe)的酸性脱保护,从而得到官能/反应性聚烯烃预聚物a-ph(3ecoe):
方案4
将ph(3ecoe)与二氯甲烷(0.5m)混合并在室温下剧烈搅拌。一次性将三氟乙酸(5当量/叔-丁氧基羰基保护基)添加到溶液中并在室温下将溶液搅拌5小时。此后一次性添加三乙基胺(5当量/boc基团,即1:1三乙基胺:三氟乙酸)并将反应物搅拌超过5分钟。在真空下将溶液浓缩到1/3原始体积并随后在甲醇中沉淀。随后在50℃下在高度真空下干燥预聚物以获得呈粘性液体的a-ph(3ecoe)。
1hnmr(500mhz,cdcl3):δ=δ2.73(m,c(11)h),1.45(s,c(12)h),1.43-1.07(b,17h,-ch2-,-c(3)h-),0.85(t,3h,-c(10)h3).13cnmr(125mhz,cdcl3):δ=38.98(c3),33.34(c2,c4),30.32(c6,c8),29.91(c7),26.88(c1,c5),26.00(c9),10.99(c10)。
表1提供不饱和及饱和预聚物和反应性聚烯烃的分子特征和玻璃转移温度。
表1
b通过假设每条链恰好两个cta端基使用1hnmr光谱仪进行端基分析来测定。c通过thf相对于聚苯乙烯标准物的sec来测定。d通过10℃min-1的dsc(第2次加热循环)来测定。e在9h中获得的95%氢化。
使用反应性聚烯烃(a-ph(3ecoe)),进行三种不同的交联反应。首先,使用1,4-丁二醇二缩水甘油醚使反应性聚烯烃交联以制造交联聚合物xd-a-ph(3ecoe),如下文方案5中所示∶
方案5
表2提供交联聚合物xd-a-ph(3ecoe)的分子特征。
表2
cch2cl2,72h,每24h替换溶剂。使样品在高度真空下在50℃下干燥直至获得恒重。d在凝胶分数实验之后获得的可溶性部分的sec(dri、thf相对于聚苯乙烯标准物)。e在使用己烷作为溶剂交联之前经由矽胶过滤的聚合物。
在第二和第三交联反应中,三甲基丙烷三缩水甘油基醚和三(2,3-环氧丙基)异氰尿酸酯分别用作交联剂,如下文方案6中所示∶
方案6
热交联,一般过程
在1800rpm下使交联剂与氨基-远螯聚烯烃预聚物在速度混合器(dac150.1fvz,flacktekinc.)中混合,每45秒20段。随后将混合物缓慢转移到特氟龙模具中。随后将模具放置在在100℃下预加热的烘箱中并使材料固化16小时。获得浅黄色透明热固性弹性体。以3:2聚合物与交联剂的摩尔比混合三官能交联剂。在实例中,使用三(2,3-环氧丙基)异氰尿酸酯,将交联剂溶解于最少量ch2cl2中,随后在速度混合器中与聚合物混合。随后在室温下将混合物置于高度真空下持续72h直至溶剂移除。此后应用一般交联过程。
三甲基丙烷三缩水甘油基醚可购自西格玛-奥德里奇公司(sigma-aldrich)作为试剂级,具有如通过1hnmr所测量的92%的纯度。三(2,3-环氧丙基)异氰尿酸酯可购自西格玛-奥德里奇公司,具有98%的纯度。将三(2,3-环氧丙基)异氰尿酸酯溶解于二氯甲烷中,随后用于交联反应。图1提供交联聚合物xt-a-ph(3ecoe)-2的应力应变试验曲线。图2说明交联聚合物xt-a-ph(3ecoe)-2的动态机械热分析。
试验方法
试验方法包括以下∶
通过1hnmr端基分析测定数均分子量(mn)。
在1ml/min的流速下使用thf作为流动相使用尺寸排阻色谱(sec)仪器在25℃测定分散度
在用铟标准物校准的tainstrumentsdiscoverydsc上进行差示扫描量热法(differentialscanningcalorimetry,dsc)。在气密密封式铝罐中制备最低质量4mg的样品,并在n2下以10℃/min的加热速率分析。自第二次加热测定热转变温度以擦除热历程。
使用ares-g2流变仪(ta仪器)(ω=6.28rad/s,γ=0.05%)在8mm或25mm平行板几何形状中进行动态机械温度分析(dynamicmechanicaltemperatureanalysis,dmta)。在实验期间,以5℃/min的速率升高温度。
在shimadzuags-x仪器上进行固化弹性体的拉伸应变试验。以127mm/min的应变速率测试astmd1708微拉伸棒的拉伸特性;所有值以至少四个样品的平均值和标准偏差形式报告。
在不背离本发明精神和基本特质的情况下可以其它形式实施本发明,并且因此,应参考所附权利要求书而非前文说明书来指定本发明的范围。
- 还没有人留言评论。精彩留言会获得点赞!