1/5水头孢孟多酯钠化合物的制作方法
本发明属于化学工程医药结晶
技术领域:
,具体涉及一种1/5水头孢孟多酯钠化合物及其制法。
背景技术:
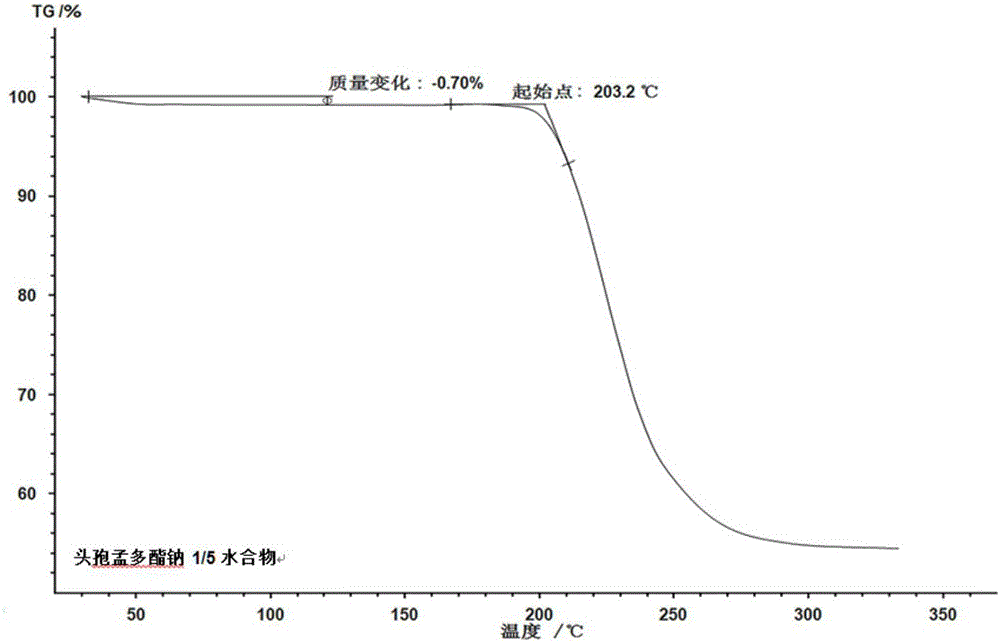
:头孢孟多酯钠是由美国Lilly公司于1972年创制成功,1978年首先在英国上市,头孢孟多酯钠别名为甲酸(酰)苄四唑头孢菌素钠、羟苄四唑头孢菌素、头孢羟唑、羟唑头孢菌素、先锋羟苄唑、头孢孟多甲酸酯钠、先锋羟苄唑等,是一种杀菌作用强的第2代半合成头孢菌素,兼有第1代和第3代头孢菌素的某些优点,对β-内酰胺酶较稳定,抗菌谱广。文献报道,头孢孟多酯钠的合成主要有以下几条路线:路线1,以3-乙酰氧甲基-5-硫-7-氨基-8-氧-1-氮杂二环辛-2-烯-2羧酸为起始原料,3-乙酰氧甲基-5-硫-7-氨基-8-氧-1-氮杂二环辛-2-烯-2羧酸与1-甲基-5-巯基-1,2,3,4-四氮唑反应生成中间体(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸,(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸与甲酰基扁桃酰氯反应生成头孢孟多酯酸,与异辛酸钠成盐得到头孢孟多酯钠,该工艺采用水相法引入甲巯四氮唑,需在较高温度下进行,反应物3-乙酰氧甲基-5-硫-7-氨基-8-氧-1-氮杂二环辛-2-烯-2羧酸和生成物(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸均容易降解,产品纯度较低。同时草酰氯价格高、毒性大,且使用草酰氯使得手性碳原子产生外消旋体,产物中含有异构体。路线2,甲酰基扁桃酸与1-甲基-5-巯基-1,2,3,4-四氮唑反应生成活性酯,再与(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸反应生成头孢孟多酸,与异辛酸钠成盐得到头孢孟多酯钠。该工艺使用了DCC等毒性较大和过敏性较大的物质,生产上需要进行严格的劳动保护。路线3,以(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸为起始原料,经硅烷化保护的(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸与甲酰基扁桃酰氯反应生成头孢孟多酸,与异辛酸钠成盐得到头孢孟多酯钠。该工艺使用了六甲基二硅胺烷保护7位氨基和2位羧基,反应中产生的副产物NH3影响酰化反应,需要较长时间才能将其清除干净,反应时间长,成本高,同时加入傅酸剂N,N-二甲基苯胺中和反应中产生的HCl,由于N,N-二甲基苯胺有致癌毒性,因此生产安全性难以保证。路线4,以(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸为起始原料,步骤与合成路线2基本相同,只是硅烷化试剂不同,采用氯硅烷和胺烷混合硅烷化试剂,硅胺烷产生的副产物NH3可以被氯硅烷产生的HCl中和,不需要加入N,N-二甲基苯胺,增加了操作的安全性,减少了环境污染。此外,上述方法制备的头孢孟多酯钠化合物为无水物,存在着储存稳定性差、热分解温度低、流动性差、易吸湿等等严重问题。从而影响产品质量,造成制剂产品不澄清,浊度不合格,并且降低了制剂的稳定性。本发明在路线3和路线4的基础上对头孢孟多酯钠化合物的合成工艺进行了改进和优化,从而获得一种性能更为优良的头孢孟多酯钠化合物。技术实现要素:本发明公开了一种头孢孟多酯钠新的溶剂化物,更具体的为1/5水头孢孟多酯钠化合物,即每摩尔头孢孟多酯钠化合物含1/5摩尔水,分子式为:分子量为:516.09,结构式为:本发明所述的1/5水头孢孟多酯钠化合物,具体制备方法包括:(1)将乙腈、3-乙酰氧甲基-5-硫-7-氨基-8-氧-1-氮杂二环辛-2-烯-2羧酸、甲巯四氮唑、三氟化硼/乙腈络合物混合,升温,快速搅拌反应,反应结束后降温,将反应液与预冷的纯化水进行混合,缓慢搅拌至有晶体析出后,再搅拌30min,加入氨水调节pH值,控制温度,养晶,过滤,丙酮洗涤,真空干燥得到白色结晶性粉末(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸;(2)将(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸与乙酸乙酯混合,常温搅拌,加入N,O-双三甲硅基乙酰胺,升温,快速搅拌反应至体系澄清,降温,缓慢加入甲酰基扁桃酸酰氯,加毕升温反应,加入纯化水,搅拌,静置分层,弃去水相,有机相加入活性碳和无水MgSO4搅拌脱色,过滤,控制温度减压浓缩至得到白色粘稠固体,加入有机溶剂,搅拌溶解,加入活性炭搅拌脱色,过滤,得到头孢孟多酯酸溶液;(3)将头孢孟多酯酸溶液降温,快速加入用有机溶剂溶解的异辛酸钠,搅拌升温,养晶,过滤,洗涤,真空干燥,得到头孢孟多酯钠化合物。上述制备方法中,步骤(1)中所述的升温至30~40℃,所述的反应时间为2~3h,所述的反应结束后降温至0~5℃,所述的纯化水冷却温度为0~5℃,所述的滴加氨水调节pH值至3.0~3.4。上述制备方法中,步骤(2)中所述的加入N,O-双三甲硅基乙酰胺后升温至30~35℃,所述的反应结束后降温至-16~-10℃,加入甲酰基扁桃酸酰氯后升温至-5~0℃,所述的加入甲酰基扁桃酸酰氯后反应1~2h,所述的减压浓缩的温度控制为≤30℃,所述的有机溶剂为体积比为2:1的丙酮和无水乙醇的混合溶液。上述制备方法中,步骤(3)中所述的头孢孟多酯酸溶液降温至8~10℃,所述的有机溶剂为体积比为2:1的丙酮和无水乙醇的混合溶液,所述的升温至28~32℃。卡尔费休法是各种测定物质中含有水分方法中最为专一、准确的方法之一,已被列为许多物质中水分测定的基本方法,尤其是对有机化合物,结果准确可靠。本发明公开的头孢孟多酯钠化合物用卡尔费休法测定水分含量在0.68%-0.72%之间,理论水分含量为0.70%,可确定本发明头孢孟多酯钠化合物含有1/5摩尔水。本发明所述的1/5水头孢孟多酯钠化合物,其红外光谱在波数为3345.2±2cm-1,3319.5±2cm-1,1760.9±2cm-1,1682.7±2cm-1,1637.8±2cm-1,1533.7±2cm-1,1394.4±2cm-1,1238.5±2cm-1,1166.9±2cm-1,1065.9±2cm-1处有特征吸收峰,如附图1所示。红外光谱测试条件为:安捷伦Cary630,溴化钾压片。本发明所述的1/5水头孢孟多酯钠化合物,其TG分析结果显示,1/5水头孢孟多酯钠化合物的失重百分率计算结果可知,失重约为0.70%,头孢孟多酯钠分子中水的理论百分含量是0.70%,参照卡尔费休法测得头孢孟多酯钠水分含量为0.68~0.72%,而实验测得TG失重为0.70%,与理论水基本相符。因此可推断头孢孟多酯钠TG失重是脱除水所致,且每摩尔头孢孟多酯钠含1/5摩尔水。如附图2所示。数据由热分析-质谱联用仪(NETZSCHSTA449C)分析得到。分析条件为:样品2~10mg,氧化铝坩埚,高纯氮气做反应气和保护气,流量分别为40ml/min和30ml/min,升温速率10K/min,温度测试范围为25~450℃。样品分解温度约为203.2℃。本发明所述的1/5水头孢孟多酯钠化合物,其DSC分析结果显示,在约200.8℃有吸热峰,在约206.9℃有放热峰。如附图3所示。DSC数据由热分析-质谱联用仪(NETZSCHSTA449C)分析得到,分析条件为:样品2~10mg,氧化铝坩埚,高纯氮气做反应气和保护气,流量分别为40ml/min和30ml/min。升温速率10℃/min,温度范围25~450℃。本发明提供的1/5水头孢孟多酯钠化合物的制备方法优点为:收率高、成本低、产品品质稳定。所制备的1/5水头孢孟多酯钠化合物含量和纯度优于药典规定。本发明提供的1/5水头孢孟多酯钠化合物热稳定性好,DSC分析表明其在约200.8℃有吸热峰,在约206.9℃有放热峰。加速稳定性试验表明本发明1/5水头孢孟多酯钠化合物的稳定性优于头孢孟多酯钠无水物。本发明提供的1/5水头孢孟多酯钠化合物与头孢孟多酯钠无水物相比更不易吸湿。因此本发明提供的1/5水头孢孟多酯钠化合物在稳定性、抗湿性方面优于头孢孟多酯钠无水物,具有更广泛的应用前景。本发明的进一步的目的,提供了一种含1/5水头孢孟多酯钠化合物的药物组合物。优选地,所述药物组合物包含1/5水头孢孟多酯钠化合物和药学上接受的赋形剂。更有选地,药物组合物选自药学上可接受的剂型。附图说明图1为1/5水头孢孟多酯钠化合物的傅里叶红外光谱图。图2为1/5水头孢孟多酯钠化合物的TG分析图谱。图3为1/5水头孢孟多酯钠化合物的DSC分析图谱。具体实施方式下面将通过具体实施方式对本发明做进一步说明,但并不因此将本发明限制在所述的实施例范围中,本领域的技术人员应理解,对本
发明内容所做的等同替换,或相应的改进,仍属于本发明的保护范围之内。实施例11/5水头孢孟多酯钠化合物的制备(1)将1L乙腈、200g3-乙酰氧甲基-5-硫-7-氨基-8-氧-1-氮杂二环辛-2-烯-2羧酸、1kg甲巯四氮唑、900ml三氟化硼/乙腈络合物混合,升温至30℃,快速搅拌反应2h,反应结束后降温至0℃,将反应液与1.5L预冷至0℃的纯化水进行混合,缓慢搅拌至有晶体析出后,再搅拌30min,加入10%的氨水调节pH值至3.0,控制温度0~5℃,养晶2h,过滤,1L的丙酮洗涤2次,真空干燥,得到白色结晶性粉末(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸221g;(2)将(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸与2L乙酸乙酯混合,常温搅拌20min,加入300gN,O-双三甲硅基乙酰胺,升温至30℃,快速搅拌反应至体系澄清,降温至-16℃,缓慢加入150ml甲酰基扁桃酸酰氯,加毕升温至0℃反应1h,加入1L纯化水,搅拌30min,静置分层,弃去水相,有机相加入20g活性碳和60g无水MgSO4搅拌脱色,过滤,控制温度≤30℃,减压浓缩至得到白色粘稠固体,加入2.25L体积比为2:1的丙酮和无水乙醇的混合溶剂,搅拌溶解,加入20g活性炭搅拌脱色,过滤,得到头孢孟多酯酸溶液;(3)将头孢孟多酯酸溶液降温至8℃,快速加入用600ml丙酮和无水乙醇(2:1)的混合溶液溶解的异辛酸钠(100g),搅拌升温至30℃,养晶1.5h,过滤,分别用1L无水乙醇和1L丙酮洗涤,真空干燥,得到头孢孟多酯钠化合物299g。实施例21/5水头孢孟多酯钠化合物的制备(1)将1.5L乙腈、300g3-乙酰氧甲基-5-硫-7-氨基-8-氧-1-氮杂二环辛-2-烯-2羧酸、1.5kg甲巯四氮唑、1.4L三氟化硼/乙腈络合物混合,升温至40℃,快速搅拌反应1h,反应结束后降温至5℃,将反应液与2.25L预冷至5℃的纯化水进行混合,缓慢搅拌至有晶体析出后,再搅拌30min,加入10%的氨水调节pH值至3.4,控制温度0~5℃,养晶3h,过滤,1.5L的丙酮洗涤2次,真空干燥,得到白色结晶性粉末(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸324g;(2)将(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸与3L乙酸乙酯混合,常温搅拌30min,加入450gN,O-双三甲硅基乙酰胺,升温至35℃,快速搅拌反应至体系澄清,降温至-10℃,缓慢加入225ml甲酰基扁桃酸酰氯,加毕升温至-2℃反应1.5h,加入1.5L纯化水,搅拌30min,静置分层,弃去水相,有机相加入30g活性碳和90g无水MgSO4搅拌脱色,过滤,控制温度≤30℃,减压浓缩至得到白色粘稠固体,加入3.38L体积比为2:1的丙酮和无水乙醇的混合溶剂,搅拌溶解,加入30g活性炭搅拌脱色,过滤,得到头孢孟多酯酸溶液;(3)将头孢孟多酯酸溶液降温至10℃,快速加入用900ml丙酮和无水乙醇(2:1)的混合溶液溶解的异辛酸钠(150g),搅拌升温至32℃,养晶2h,过滤,分别用1.5L无水乙醇和1.5L丙酮洗涤,真空干燥,得到头孢孟多酯钠化合物451g。实施例31/5水头孢孟多酯钠化合物的制备(1)将1.25L乙腈、250g3-乙酰氧甲基-5-硫-7-氨基-8-氧-1-氮杂二环辛-2-烯-2羧酸、1.25kg甲巯四氮唑、1.17L三氟化硼/乙腈络合物混合,升温至35℃,快速搅拌反应2h,反应结束后降温至3℃,将反应液与1.88L预冷至2℃的纯化水进行混合,缓慢搅拌至有晶体析出后,再搅拌30min,加入10%的氨水调节pH值至3.2,控制温度0~5℃,养晶2h,过滤,1.3L的丙酮洗涤2次,真空干燥,得到白色结晶性粉末(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸262g;(2)将(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸与2.5L乙酸乙酯混合,常温搅拌30min,加入375gN,O-双三甲硅基乙酰胺,升温至32℃,快速搅拌反应至体系澄清,降温至-12℃,缓慢加入188ml甲酰基扁桃酸酰氯,加毕升温至-5℃反应1.5h,加入1.25L纯化水,搅拌30min,静置分层,弃去水相,有机相加入25g活性碳和75g无水MgSO4搅拌脱色,过滤,控制温度≤30℃,减压浓缩至得到白色粘稠固体,加入2.82L体积比为2:1的丙酮和无水乙醇的混合溶剂,搅拌溶解,加入25g活性炭搅拌脱色,过滤,得到头孢孟多酯酸溶液;(3)将头孢孟多酯酸溶液降温至10℃,快速加入用750ml丙酮和无水乙醇(2:1)的混合溶液溶解的异辛酸钠(125g),搅拌升温至28℃,养晶2h,过滤,分别用1.25L无水乙醇和1.25L丙酮洗涤,真空干燥,得到头孢孟多酯钠化合物371g。对比例1头孢孟多酯钠化合物的制备(无水物)根据文献报道线路1的方法制备头孢孟多酯钠化合物(1)272g3-乙酰氧甲基-5-硫-7-氨基-8-氧-1-氮杂二环辛-2-烯-2羧酸溶于2L水和1L丙酮的混合溶剂中,边搅拌边加入碳酸氢钠饱和溶液,控制温度45℃,加入2L10%的1-甲基1H-四唑-5-硫醇丙酮溶液,搅拌混合,80℃反应3h,冷却至10℃,6mol/L的盐酸调节pH值至3.9,结晶,过滤,洗涤,干燥,得(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸269g;(2)将15.2gD-扁桃酸加入2.5L甲酸中,室温放置2天,加压干燥,将干燥固体溶于苯中,加入180ml草酰氯和4ml甲基甲酰胺,搅拌2h,真空浓缩,得D-扁桃酸酰氯草酸酯;(3)将D-扁桃酸酰氯草酸酯、(6R,7R)-7氨基-3[(Z)-2-(4-甲基噻唑)-5-基]乙烯基-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸和360g碳酸氢钠溶于1L水和5L丙酮混合溶液(2℃)中,搅拌1h,然后室温搅拌2h,减压除掉丙酮,加入1L水和2L乙酸乙酯的混合溶液,盐酸溶液调节pH值至2.0,过滤,分层,收集有机层,真空浓缩得油状物,加入10%碳酸氢钠溶液2.5L,混合搅拌2h,加入乙酸乙酯5L,冰浴冷却,盐酸调节pH值至2.0,收集有机层,减压浓缩,得头孢孟多酯酸,将头孢孟多酯酸溶于500ml乙醇中,加入260ml1mol/L的乙酸钠乙醇溶液,搅拌,冷却,结晶,得头孢孟多酯钠化合物(无水物)115g。对比例2根据现有技术文献CN101829118A制备得到2.5个结晶水的头孢孟多酯钠水合物采用该专利文献实施例1相同的制备方法,在室温条件下,将2.6g头孢孟多酯钠固体溶解于5.2mL蒸馏水中,然后加入47mL的异丙醇和乙醚的有机混合溶液(异丙醇与乙醚的体积比为1∶2.5),在5℃静置5小时,得到白色晶体,过滤,滤饼用乙醇洗涤二次,真空干燥后2小时,得到含2.5个结晶水的头孢孟多酯钠水合物晶体。实验1测定1/5水头孢孟多酯钠化合物的傅里叶红外光谱图1、仪器试剂:安捷伦Cary630红外光谱仪、手动粉末压片机、红外灯、研钵、镊子、药勺、样品支架、溴化钾(光谱纯)。2、样品制备:(1)打开电脑、安装连接红外光谱仪,预热待用;(2)取500mg溴化钾放入研钵中,研磨20分钟,至溴化钾为均匀白色粉末;(3)取大约200mg适量溴化钾粉末放入压片模具中,将压片模具置于压片机上,调整至20MPa,保持1min;(4)将压好的溴化钾空白片置于红外灯下烘烤2小时;(5)另取约2mg样品和300mg溴化钾放入研钵中,研磨20分钟,至溴化钾与样品混合均匀;(6)取大约200mg适量溴化钾与样品的混合粉末放入压片模具中,将压片模具置于压片机上,调整至20MPa,保持1min;(7)将压好的溴化钾样品片置于红外灯下烘烤2小时。3、测试方法:(1)首先将空白片用样品支架固定好,放入红外光谱仪的透射仓中,按照程序设定进行背景信号采集;(2)背景信号采集完成后,将空白片取下,用样品支架把样品片固定好,按同样的方法进行信号采集;(3)样品信号采集完成后,将数据导出,保存待处理。4、数据处理:(1)打开安捷伦数据处理软件,导入数据文件;(2)对数据文件进行基线校正、标峰等操作,处理完成后,将文件保存。5、结果实验2测定1/5水头孢孟多酯钠化合物的TG分析图谱检测仪器:热分析-质谱联用仪(NETZSCHSTA449C)。检测条件:样品2~10mg,氧化铝坩埚,高纯氮气做反应气和保护气,流量分别为40ml/min和30ml/min,升温速率10K/min,温度测试范围为25~450℃。实验操作:1、开机,基线测试(浮力效应修正——修正曲线)。放坩埚准备一对干净的坩埚,分别作为参比坩埚与样品坩埚放到支架上。坩埚是否加盖视后面的样品测试而定。关闭炉体。(若已由原先做好的基线,可跳过此步骤,直接放样品)。2、设置参数,开始测试。输入样品名称、编号、所使用的气体及流量等参数,开始测量。3、测量完成,进行数据分析,拿出样品,关闭仪器。实验3测定1/5水头孢孟多酯钠化合物的DSC分析图谱检测仪器:热分析-质谱联用仪(NETZSCHSTA449C)检测条件:样品2~10mg,氧化铝坩埚,高纯氮气做反应气和保护气,流量分别为40ml/min和30ml/min。升温速率10℃/min,温度范围25~450℃。实验操作:1、开机,基线测试(浮力效应修正——修正曲线)。放坩埚准备一对干净的坩埚,分别作为参比坩埚与样品坩埚放到支架上。坩埚是否加盖视后面的样品测试而定。关闭炉体。(若已由原先做好的基线,可跳过此步骤,直接放样品)。2、设置参数,开始测试。输入样品名称、编号、所使用的气体及流量等参数,开始测量。3、测量完成,进行数据分析,拿出样品,关闭仪器。试验例1稳定性考察本发明人对本发明实施例1制备的1/5水头孢孟多酯钠化合物和对比例1制备的头孢孟多酯钠化合物,以及对比例2制备的2.5结晶水的头孢孟多酯钠水合物进行了加速稳定性考察试验。考察条件为温度40℃±2℃、相对湿度75%±5%。放置6个月,分别于0、1、2、3、6月末取样。考察指标为性状、澄清度、水分、酸度、头孢孟多、2-乙基己酸和有关物质。试验例2引湿性考察将本发明实施例1制备的1/5水头孢孟多酯钠化合物与对比例1制备的头孢孟多酯钠化合物,以及对比例2制备的2.5结晶水的头孢孟多酯钠水合物置于动态蒸汽吸附仪中40℃条件下,记录三小时内重量变化小于0.01g时的重量,试验结果如下:相对湿度RH/%实施例1重量变化率对比例1重量变化率对比例2重量变化率00001000020000300.010.030.01400.040.070.06500.420.890.75600.560.940.84700.911.521.27结论:本发明实施例1提供的1/5水头孢孟多酯钠化合物更不易吸湿。试验例3流动性考察本发明人对本发明实施例3和对比例1所制备头孢孟多酯钠,以及对比例2制备的2.5结晶水的头孢孟多酯钠水合物采取漏斗法测定休止角,对头孢孟多酯钠的流动性进行了研究。休止角检测结果:休止角检测结果:实施例休止角θ实施例329.1°对比例135.4°对比例232.4°结论:本发明制备的1/5水头孢孟多酯钠化合物的流动性明显高于对比例1头孢孟多酯钠化合物和对比例2的2.5结晶水的头孢孟多酯钠水合物,可以满足不同制剂制备的需要。试验例4溶解性考察将实施例1和对比例1所制备的头孢孟多酯钠,以及对比例2制备的2.5结晶水的头孢孟多酯钠水合物分别溶于水溶液中,震摇20min,通过检测含量计算实施例1、对比例1和对比例2所制备化合物在水中的溶解度。溶解度检测结果实施例溶解度实施例10.75g/ml对比例10.43g/ml对比例20.52g/ml结论:本发明实施例1制备的1/5水头孢孟多酯钠化合物的溶解性明显好于对比例1头孢孟多酯钠化合物和对比例2的2.5结晶水的头孢孟多酯钠水合物。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1