一种高效重组表达限制性内切酶的方法与流程
本发明属于生物工程领域,特别是一种高效重组表达限制性内切酶的方法。
背景技术:
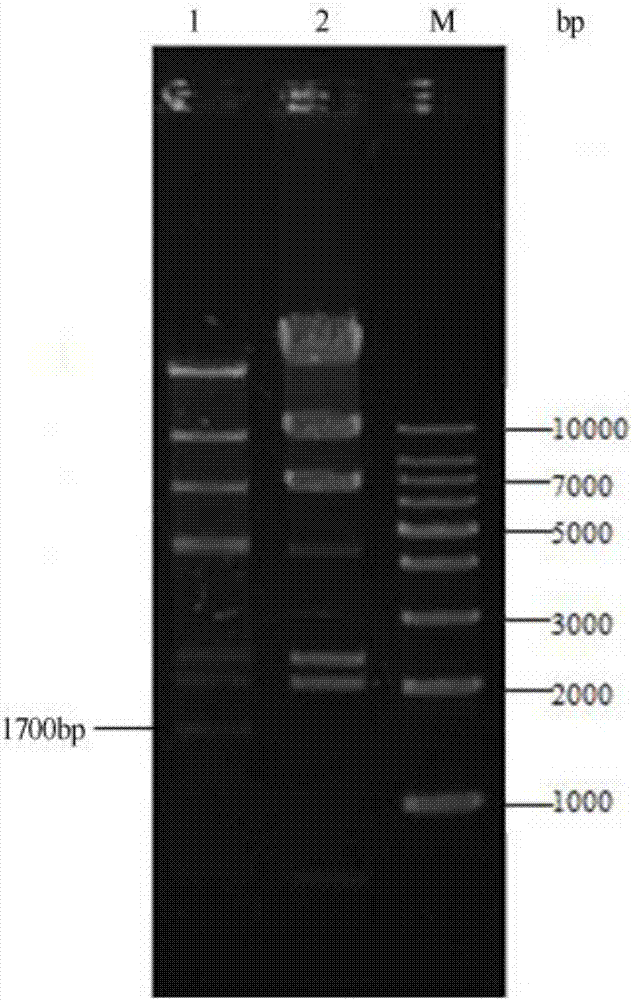
:限制性内切酶是当代基因工程研究不可缺少的一类重要工具酶,是现代分子生物学的基础。自20世纪60年代末第一个限制性内切酶被发现之后,越来越多的限制性内切酶被发现、提纯并获得应用。现有技术中生产限制性内切酶的方法主要是将限制-修饰基因克隆入质粒,使用与限制性内切酶对应的特异性甲基化酶制备限制性内切酶,靠质粒的高拷贝数实现目的蛋白高表达,这种方法的制备程序繁琐、限制性内切酶得率低、生产成本高,获得的甲基化保护的菌株只能特异性保护宿主dna免于某一种限制性内切酶的切割,应用范围狭窄。技术实现要素:本发明要解决的技术问题是针对现有技术的不足,提出了一种操作简便、成本低、酶活力好的高效重组表达限制性内切酶的方法。本发明要解决的技术问题是通过以下技术方案来实现的,一种高效重组表达限制性内切酶的方法,其特点是,包括如下步骤:(1)先将限制性内切酶基因插入,再将载体转化导入大肠杆菌,筛选培养、测序,获得限制性内切酶的重组表达质粒;所述质粒载体为利用乳糖操纵子和t7启动子调控,iptg诱导的pet系列表达载体;(2)将限制性内切酶的重组表达质粒转化至bl21(de3)plyss感受态细胞中,筛选获得限制性内切酶的重组表达菌株;(3)扩大培养限制性内切酶的重组表达菌株并诱导表达限制性内切酶。本发明所要解决的技术问题还可以通过以下的技术方案来进一步实现。以上所述的方法中,所述的限制性内切酶选自:bcli,dpni,dpnii,ecori,hindiii,kpni,mlui,msei,ncoi,ndei,nhei,noti,nsii,nspv,psti,sali,sbfi,sgei,sspi,stui,taqi,xbai,xhoi。本发明所要解决的技术问题还可以通过以下的技术方案来进一步实现。以上所述的方法的步骤(1)中,所述的质粒载体为pet-28b。本发明所要解决的技术问题还可以通过以下的技术方案来进一步实现。以上所述的方法的步骤(1)中,将限制性内切酶基因片段连接至质粒载体上后,将连接产物转化至大肠杆菌dh5α感受态细胞中,再涂布于含葡萄糖和卡那霉素的lb平板上筛选克隆,测序获得限制性内切酶的重组表达质粒。进一步优选地,所述的葡萄糖的浓度为0.5-2g/100ml,卡那霉素的浓度为37.5-150μg/ml。进一步优选地,所述的葡萄糖的浓度为1-1.5g/100ml,卡那霉素的浓度为50-120μg/ml。本发明所要解决的技术问题还可以通过以下的技术方案来进一步实现。以上所述的方法的步骤(2)中,将限制性内切酶的重组表达质粒转化至bl21(de3)plyss感受态细胞中,涂布于含葡萄糖、卡那霉素和氯霉素的lb平板上筛选培养,获得限制性内切酶的重组表达菌株。进一步优选地,所述的葡萄糖的浓度为0.5-2g/100ml,卡那霉素的浓度为37.5-150μg/ml,氯霉素的浓度为17.5-70μg/ml。进一步优选地,所述的葡萄糖的浓度为1-1.5g/100ml,卡那霉素的浓度为50-120μg/ml,氯霉素的浓度为20-50μg/ml。本发明所要解决的技术问题还可以通过以下的技术方案来进一步实现。以上所述的方法的步骤(3)中,将限制性内切酶的重组表达菌株划线培养并挑取单菌落接种于含葡萄糖、卡那霉素和氯霉素的lb培养液中,振荡培养过夜,将得到的培养物接种于葡萄糖、卡那霉素和氯霉素的lb培养液中,摇动培养一段时间,加入iptg诱导表达限制性内切酶。本发明所要解决的技术问题还可以通过以下的技术方案来进一步实现。以上所述的方法的步骤(3)中,将限制性内切酶的重组表达菌株划线培养并挑取单菌落接种于含0.5-2g/100ml葡萄糖、37.5-150μg/ml卡那霉素和17.5-70μg/ml氯霉素的lb培养液中,振荡培养过夜,将得到的培养物按接种量2-5%接种于含0.5-2g/100ml葡萄糖、37.5-150μg/ml卡那霉素和17.5-70μg/ml氯霉素的lb培养液中,于37℃摇动培养,至菌密度达到3×108-4×108/ml时,加入iptg至终浓度为0.2-0.5mm,并在15-18℃下以200-250rpm摇动培养12-18h,诱导表达限制性内切酶。进一步优选地,将限制性内切酶的重组表达菌株划线培养并挑取单菌落接种于含1-1.5g/100ml葡萄糖、50-120μg/ml卡那霉素和20-50μg/ml氯霉素的lb培养液中,振荡培养过夜,将得到的培养物按接种量2-3%接种于含1-1.5g/100ml葡萄糖、50-90μg/ml卡那霉素和20-50μg/ml氯霉素的lb培养液中,于35-37℃摇动培养,至菌密度达到3.2×108-3.8×108/ml时,加入iptg至终浓度为0.3-0.4mm,并在16-17℃下以210-240rpm摇动培养14-16h,诱导表达限制性内切酶。本发明方法将限制酶基因克隆至利用乳糖操纵子和t7启动子调控、iptg诱导的pet系列表达载体上,通过乳糖操纵子和t7启动子调控重组蛋白的表达。由于重组蛋白基因在乳糖或乳糖类似物(如iptg)存在的情况下大量表达,而在无乳糖或乳糖类似物存在的情况下则不表达,因此使用iptg可以人为控制重组蛋白的表达,而在细菌培养基中可能含有的少量乳糖,会造成重组蛋白的基因在乳糖或乳糖类似物加入前微量表达,即本底表达,如果重组蛋白毒性较强,就会杀死宿主细胞,使细胞不能繁殖到适合诱导的浓度。本发明方法结合大肠杆菌自身的特点,在培养基中添加了葡萄糖,使宿主细胞优先利用葡萄糖不利用乳糖,从而使加入诱导剂iptg前限制性内切酶的本底表达受到抑制,保护宿主dna不被限制性内切酶切割,保证了宿主细胞能够繁殖到适合诱导的细胞浓度;当宿主细胞的浓度达到适合诱导时,培养基中的葡萄糖已被消耗,且iptg的加入量相对于培养基中的葡萄糖或乳糖都是极大量,加入iptg即诱导限制性内切酶开始大量表达。与现有技术相比,本发明方法利用葡萄糖对乳糖操纵子表达系统本底表达的抑制作用,以及bl21(de3)plyss菌株自身对乳糖操纵子表达系统本底表达的严苛控制作用,省略了对宿主dna的甲基化保护步骤,无需针对某一限制性内切酶进行繁琐的甲基化酶基因筛选,不仅大大简化了重组表达过程,还可以应用于多种限制酶的重组表达,具有较高的应用推广价值。附图说明图1为本发明实施例22中kpni重组表达质粒图谱;图2为本发明实施例22中kpni重组表达的检测电泳图;其中:1:蛋白粗酶液特异性酶切条带;2:λdna(hindiiidigest)(未酶切);m:dnamarker。图3为本发明实施例22中kpni经镍亲和柱纯化后的电泳图;其中:1:5mm咪唑;2-3:20mm咪唑;4:400mm咪唑;m:蛋白marker。图4为本发明实施例22中kpni经阴离子交换层析住纯化后的电泳图;其中:1:100mmkcl;2:300mmkcl;3:500mmkcl;4:700mmkcl;5:1mkcl;m:蛋白marker。图5为本发明实施例22中kpni酶活力检测电泳图;其中:1:kpni原液稀释16倍;2:kpni原液稀释32倍;3:kpni原液稀释64倍;4:kpni原液稀释128倍;5:kpni原液稀释256倍;6:kpni原液稀释512倍;7:λdna(hindiiidigest);m:dnamarker。图6为本发明实施例22中kpni星号活性及非特异性dna酶污染检测电泳图;其中:1-6为kpni酶切底物1h的电泳图,7-12为kpni酶切底物16h的电泳图;1、7:kpni原液稀释16倍;2、8:kpni原液稀释32倍;3、9:kpni原液稀释64倍;4、10:kpni原液稀释128倍;5、11:kpni原液稀释256倍;6、12:kpni原液稀释512倍;13:λdna(hindiiidigest);m:dnamarker。图7为本发明实施例22中酶切-连接-再酶切检测电泳图;其中:1:kpni酶切commonfragment;2:t4dna连接酶连接后片段;4:kpni再次酶切连接片段;m:dnamarker。具体实施方式以下参照附图,进一步描述本发明的具体技术方案,以便于本领域的技术人员进一步地理解本发明,而不构成对其权利的限制。实施例1,一种高效重组表达限制性内切酶的方法,包括如下步骤:(1)先将限制性内切酶基因插入,再将载体转化导入大肠杆菌,筛选培养、测序,获得限制性内切酶的重组表达质粒;所述质粒载体为利用乳糖操纵子和t7启动子调控,iptg诱导的pet系列表达载体;(2)将限制性内切酶的重组表达质粒转化至bl21(de3)plyss感受态细胞中,筛选获得限制性内切酶的重组表达菌株;(3)扩大培养限制性内切酶的重组表达菌株并诱导表达限制性内切酶。实施例2,实施例1所述的高效重组表达限制性内切酶的方法,所述的限制性内切酶选自:bcli,dpni,dpnii,ecori,hindiii,kpni,mlui,msei,ncoi,ndei,nhei,noti,nsii,nspv,psti,sali,sbfi,sgei,sspi,stui,taqi,xbai,xhoi。实施例3,实施例1所述的高效重组表达限制性内切酶的方法,所述的质粒载体为pet-28b。实施例4,实施例1所述的高效重组表达限制性内切酶的方法的步骤(1)中,将限制性内切酶基因片段连接至质粒载体上后,将连接产物转化至大肠杆菌dh5α感受态细胞中,再涂布于含葡萄糖和卡那霉素的lb平板上筛选克隆,测序获得限制性内切酶的重组表达质粒。实施例5,实施例4所述的高效重组表达限制性内切酶的方法中,所述的葡萄糖的浓度为0.5g/100ml,卡那霉素的浓度为37.5μg/ml。实施例6,实施例4所述的高效重组表达限制性内切酶的方法中,所述的葡萄糖的浓度为2g/100ml,卡那霉素的浓度为150μg/ml。实施例7,实施例4所述的高效重组表达限制性内切酶的方法中,所述的葡萄糖的浓度为1g/100ml,卡那霉素的浓度为75μg/ml。实施例8,实施例4所述的高效重组表达限制性内切酶的方法中,所述的葡萄糖的浓度为1.5g/100ml,卡那霉素的浓度为120μg/ml。实施例9,实施例4所述的高效重组表达限制性内切酶的方法中,所述的葡萄糖的浓度为1g/100ml,卡那霉素的浓度为50μg/ml。实施例10,实施例1所述的高效重组表达限制性内切酶的方法的步骤(2)中,将限制性内切酶的重组表达质粒转化至bl21(de3)plyss感受态细胞中,涂布于含葡萄糖、卡那霉素和氯霉素的lb平板上筛选培养,获得限制性内切酶的重组表达菌株。实施例11,实施例10所述的高效重组表达限制性内切酶的方法中,所述的葡萄糖的浓度为0.5g/100ml,卡那霉素的浓度为37.5μg/ml,氯霉素的浓度为17.5μg/ml。实施例12,实施例10所述的高效重组表达限制性内切酶的方法中,所述的葡萄糖的浓度为2g/100ml,卡那霉素的浓度为150μg/ml,氯霉素的浓度为70μg/ml。实施例13,实施例10所述的高效重组表达限制性内切酶的方法中,所述的葡萄糖的浓度为1g/100ml,卡那霉素的浓度为75μg/ml,氯霉素的浓度为35μg/ml。实施例14,实施例10所述的高效重组表达限制性内切酶的方法中,所述的葡萄糖的浓度为1g/100ml,卡那霉素的浓度为50μg/ml,氯霉素的浓度为20μg/ml。实施例15,实施例10所述的高效重组表达限制性内切酶的方法中,所述的葡萄糖的浓度为1.5g/100ml,卡那霉素的浓度为120μg/ml,氯霉素的浓度为50μg/ml。实施例16,实施例1所述的高效重组表达限制性内切酶的方法的步骤(3)中,将限制性内切酶的重组表达菌株划线培养并挑取单菌落接种于含葡萄糖、卡那霉素和氯霉素的lb培养液中,振荡培养过夜,将得到的培养物接种于葡萄糖、卡那霉素和氯霉素的lb培养液中,摇动培养一段时间,加入iptg诱导表达限制性内切酶。实施例17,实施例14所述的高效重组表达限制性内切酶的方法中,将限制性内切酶的重组表达菌株划线培养并挑取单菌落接种于含0.5g/100ml葡萄糖、37.5μg/ml卡那霉素和17.5μg/ml氯霉素的lb培养液中,振荡培养过夜,将得到的培养物按接种量2%接种于含0.5g/100ml葡萄糖、37.5μg/ml卡那霉素和17.5μg/ml氯霉素的lb培养液中,于35℃摇动培养,至菌密度达到3×108/ml时,加入iptg至终浓度为0.2mm,并在15℃下以200rpm摇动培养12h,诱导表达限制性内切酶。实施例18,实施例14所述的高效重组表达限制性内切酶的方法中,将限制性内切酶的重组表达菌株划线培养并挑取单菌落接种于含2g/100ml葡萄糖、150μg/ml卡那霉素和70μg/ml氯霉素的lb培养液中,振荡培养过夜,将得到的培养物按接种量5%接种于含2g/100ml葡萄糖、150μg/ml卡那霉素和70μg/ml氯霉素的lb培养液中,于37℃摇动培养,至菌密度达到4×108/ml时,加入iptg至终浓度为0.5mm,并在18℃下以250rpm摇动培养18h,诱导表达限制性内切酶。实施例19,实施例14所述的高效重组表达限制性内切酶的方法中,将限制性内切酶的重组表达菌株划线培养并挑取单菌落接种于含1g/100ml葡萄糖、75μg/ml卡那霉素和35μg/ml氯霉素的lb培养液中,振荡培养过夜,将得到的培养物按接种量2%接种于含1g/100ml葡萄糖、75μg/ml卡那霉素和35μg/ml氯霉素的lb培养液中,于37℃摇动培养,至菌密度达到3.5×108/ml时,加入iptg至终浓度为0.5mm,并在16℃下以230rpm摇动培养16h,诱导表达限制性内切酶。实施例20,实施例14所述的高效重组表达限制性内切酶的方法中,将限制性内切酶的重组表达菌株划线培养并挑取单菌落接种于含1g/100ml葡萄糖、50μg/ml卡那霉素和20μg/ml氯霉素的lb培养液中,振荡培养过夜,将得到的培养物按接种量2%接种于含1g/100ml葡萄糖、50μg/ml卡那霉素和20μg/ml氯霉素的lb培养液中,于36℃摇动培养,至菌密度达到3.2×108/ml时,加入iptg至终浓度为0.3mm,并在16℃下以210rpm摇动培养16h,诱导表达限制性内切酶。实施例21,实施例14所述的高效重组表达限制性内切酶的方法中,将限制性内切酶的重组表达菌株划线培养并挑取单菌落接种于含1.5g/100ml葡萄糖、120μg/ml卡那霉素和50μg/ml氯霉素的lb培养液中,振荡培养过夜,将得到的培养物按接种量3%接种于含1.5g/100ml葡萄糖、120μg/ml卡那霉素和50μg/ml氯霉素的lb培养液中,于37℃摇动培养,至菌密度达到3.8×108/ml时,加入iptg至终浓度为0.4mm,并在17℃下以240rpm摇动培养16h,诱导表达限制性内切酶。实施例22,实验例,本实施例以限制性内切酶kpni和pet-28b载体为例来描述本发明所述的高效重组表达限制性内切酶的方法。1、材料1.1菌株与质粒肺炎克雷伯氏菌(klebsiellapneumoniae0k8)菌株:购自美国菌种保藏中心(atcc);bl21(de3)plyss菌株、λdna(hindiiidigest):购自赛默飞世尔科技有限公司;pet-28b、puc19和共同片段(commonfragment)(大小为894bp的dna片段,核苷酸序列见seqidno.3):购自江苏愚公生命科技有限公司。1.2仪器与试剂细胞高压破碎仪:购自广州聚能纳米生物科技股份有限公司;蛋白分子量标准和核酸分子量标准:购自金斯瑞生物科技有限公司;限制性内切酶、kod高保真dna聚合酶、t4dna连接酶和1×cutonebuffer(51mm乙酸钾(potassiumacetate)、22mm三(羟甲基)氨基甲烷-乙酸(tris-acetate)、10mm镁离子(magnesium)、0.1mg/mlbsa、ph8.0@25℃):购自江苏愚公生命科技有限公司;ni亲和层析纯化柱(toyopearlaf-chelate-650m)和阴离子交换层析柱(toyopearlgigacapq-650m):购自日本tosohcorporation;b-per细菌蛋白提试剂:购自赛默飞世尔科技有限公司;其余试剂均为国产分析纯。2、实验方法2.1kpni基因的pcr扩增根据ncbi数据库上肺炎克雷伯氏菌菌株的kpni基因序列(genbank:wp_004176755.1)设计一对引物,在上游引物的5’端引入bamhi酶切识别序列和保护碱基,在下游引物的5’端引入xhoi酶切识别序列和保护碱基,上游引物设计为kpni基因的表达框与质粒载体上的n-端6×his纯化标签的表达框一致:引物f:5’-ccgcggatccatggatgtgtttgataaagt-3’(下划线部分为bamhi识别序列);引物r:5’-tatgctcgagttaacgtttcaggccatagtagt-3’(下划线部分为xhoi识别序列)。将肺炎克雷伯氏菌菌株平板培养菌落以去离子水重悬后,95℃温育10min,作为pcr模板,用上述引物pcr扩增kpni基因,获得了与理论值大小一致的kpni基因dna片段,pcr扩增反应使用了kod高保真dna聚合酶。2.2限制性内切酶kpni的重组表达质粒的构建kpni的pcr产物纯化后用bamhi和xhoi双酶切,酶切产物用t4连接酶连接至由bamhi和xhoi双酶切后的pet-28b载体上,得到环状dna连接产物,其中,kpni基因dna的插入部位处于t7启动子的调控之下;通过化学转化法将得到的连接产物转化至大肠杆菌dh5α感受态细胞中,并涂布于含1g/100ml葡萄糖和75μg/ml卡那霉素的lb平板上筛选克隆,经测序获得kpni的重组表达质粒,并将其命名为pet-kpni(见附图1)。2.3限制性内切酶kpni的重组表达菌株的构建用pet-kpni转化bl21(de3)plyss感受态细胞,涂布于含1g/100ml葡萄糖、75μg/ml卡那霉素、35µg/ml氯霉素的lb平板上筛选培养,获得kpni的重组表达菌株,将该菌株命名为plyss-kpni。2.4目的蛋白的诱导表达及琼脂糖凝胶电泳检测将plyss-kpni划线培养并挑取单菌落接种于含1g/100ml葡萄糖、75μg/ml卡那霉素和35μg/ml氯霉素的lb培养液中,振荡培养过夜,将得到的培养物按接种量2%接种于含1g/100ml葡萄糖、75μg/ml卡那霉素和35μg/ml氯霉素的lb培养液中,于37℃摇动培养,至菌密度达到3×108-4×108/ml时,加入iptg至终浓度为0.5mm,并在16℃下以200-250rpm摇动培养16h,诱导表达kpni。为了验证目的蛋白是否存在,进行了琼脂糖凝胶电泳检测,检测过程如下:取1ml蛋白表达后的菌液,以4000g离心10min,弃去上清,收集菌体,然后用b-per细菌蛋白提取试剂对菌体总蛋白进行提取,得细菌蛋白粗体液;取1-3μl细菌蛋白粗提液,在1×cutonebuffer缓冲条件下、20μl反应体系中,与底物λdna(hindiiidigest)在37℃温育15min,用1%琼脂糖凝胶电泳检测底物λdna(hindiiidigest)被细菌蛋白粗提液消化的情况(见附图2)。结果显示,底物λdna(hindiiidigest)被细菌蛋白粗提液消化后形成了限制性内切酶kpni的特异性酶切条带,即大小为1700bp的片段(以λdna(hindiiidigest)为底物时,该条带只能由kpni酶切获得),与理论值一致,表明细菌蛋白粗提液具有限制酶kpni的活性,限制酶kpni的重组表达成功。2.5目的蛋白的纯化取适量蛋白表达后的plyss-kpni菌液,以4000g离心10min收集菌体,菌体以缓冲液(10mmtris-hclph7.4@25℃,50mmkcl,5%甘油,5mm咪唑)充分重悬后,以细胞高压破碎仪破碎,以43000g4℃离心30min,收集上清液。上清液分别通过ni亲和层析柱、阴离子交换层析柱进行纯化,整个纯化过程在4˚c或冰浴上进行。2.5.1ni亲和层析纯化取上清液过ni亲和层析纯化柱,分步收集洗脱液,将单管收集的洗脱液进行sds-page电泳检测(见附图3)。其中,ni亲和层析过程中使用的缓冲液为:平衡缓冲液(10mmtris-hclph7.4@25℃,50mmkcl,5%甘油,5mm咪唑)、漂洗缓冲液(10mmtris-hclph7.4@25℃,50mmkcl,5%甘油,20mm咪唑)(漂洗2次)和洗脱缓冲液(10mmtris-hclph7.4@25℃,50mmkcl,5%甘油,400mm咪唑)。由图3可知,泳道4的蛋白量要普遍高于其他泳道,因此将该泳道对应的缓冲液洗脱下的几管洗脱液混合、透析。2.5.2阴离子交换层析纯化将上一步透析后的洗脱液过阴离子交换层析柱进一步纯化,梯度洗脱,分步收集,对不同浓度洗脱液洗脱到的样液进行sds-page电泳检测(见附图4)。其中,阴离子交换层析过程中使用的缓冲液为:平衡缓冲液(10mmtris-hclph7.4@25℃,100mmkcl,5%甘油,1mmdtt,0.1mmedta)和梯度洗脱液(10mmtris-hclph7.4@25℃,5%甘油,1mmdtt,0.1mmedta及kcl,其中,kcl的浓度自100mm→300mm→500mm→700mm→1m,呈梯度变化)。由图4可知,泳道1和2,即通过含100mmkcl和300mmkcl的洗脱液洗脱得到的样液中蛋白含量和纯度要高于其他浓度洗脱得到的样液,将这两种浓度的洗脱液洗脱下的样液合并后,透析至贮存缓冲液(10mmtris-hclph7.4@25℃,50mmkcl,50%甘油,1mmdtt,0.1mmedta),并添加bsa至终浓度0.2mg/ml,得到kpni原液,保存于-20℃。2.6酶活性检测限制酶kpni的酶活定义为:37℃下,在20μl反应体系中,1μl酶能够在15min内完全消化1μgλdna(hindiiidigest)。对得到的kpni原液进行梯度稀释后,在1×cutonebuffer缓冲条件下、20μl反应体系中,与底物λdna(hindiiidigest)在37℃温育15min,然后以1%琼脂糖凝胶电泳检测底物λdna(hindiiidigest)的酶切情况(见附图5)。由检测结果可知,根据本发明方法纯化获得的限制性内切酶kpni具有良好的酶切活力,比活力为128000u/mg,产率为128000u/l诱导菌液。2.7限制性内切酶kpni的质量检测2.7.1星号活性/非特异性dna酶污染检测对kpni原液进行梯度稀释后,在1×cutonebuffer缓冲条件下、20μl反应体系中,底物λdna(hindiiidigest)在37˚c温育60min及16h,然后以1%琼脂糖凝胶电泳检测底物λdna(hindiiidigest)的酶切情况(见附图6)。结果显示,高酶量(8u)1h及超长时间(16h)酶切均未观察到底物λdna产生非特异性降解条带,说明测试的酶中不存在非特异性的dna酶,表明根据本实施例生产的酶合格。2.7.2酶切-连接-再酶切检测20倍酶量的kpni在1×cutonebuffer缓冲条件下、20μl反应体系中,与底物commonfragment(见seqidno.3)在37℃温育15min获得酶切片段,将这些片段用t4dna连接酶连接,所得连接产物超过95%可以被限制性内切酶kpni再次切开,再酶切结果中无杂带、无拖尾,表明本实施例所纯化的kpni完全达到了限制性内切酶的分子生物学实验要求(见附图7),2.7.3蓝白斑筛选鉴定使用8倍或16倍酶量的限制酶kpni线性化puc19载体后,连接并转化该载体进入表达laczbetafragment的大肠杆菌感受态细胞中,并涂x-gal/iptg/amp平板,结果见表1。在蓝白斑鉴定中,本发明实施例生产的限制性内切酶产生的白斑(白色菌落)比例仅为0.1-0.34%,结果合格。表1.蓝白斑筛选鉴定数据统计表编号酶量白斑数总斑数白斑比例背景蓝斑18u3396330.340%19136216u27251790.107%3904sequencelisting<110>淮海工学院<120>一种高效重组表达限制性内切酶的方法<130>2017<160>3<170>patentinversion3.5<210>1<211>30<212>dna<213>人工序列<220><223>引物f<400>1ccgcggatccatggatgtgtttgataaagt30<210>2<211>33<212>dna<213>人工序列<220><223>引物r<400>2tatgctcgagttaacgtttcaggccatagtagt33<210>3<211>894<212>dna<213>人工序列<400>3ggcctttttacggttcctggccttttgctggccttttgctcacatgttctttcctgcgtt60atcccctgattctgtggataaccgtattaccgcctttgagtgagctgataccgctcgccg120cagccgaacgaccgagcgcagcgagtcagtgagcgaggaagcggaagagcgcccaatacg180caaaccgcctctccccgcgcgttggccgattcattaatgcagctggcacgacaggtttcc240cgactggaaagcgggcagtgagcgcaacgcaattaatgtgagttagctcactcattaggc300accccaggctttacactttatgcttccggctcgtatgttgtgtggaattgtgagcggata360acaatttcacacaggaaacagctatgaccatgattacgccaagcttgcatgcgcggccgc420tgcaggctagcgtcgactctagagatatcccatggggatccccgggtacccatatggagc480tcctcgaggaattctcactggccgtcgttttacaacgtcgtgactgggaaaaccctggcg540ttacccaacttaatcgccttgcagcacatccccctttcgccagctggcgtaatagcgaag600aggcccgcaccgatcgcccttcccaacagttgcgcagcctgaatggcgaatggcgcctga660tgcggtattttctccttacgcatctgtgcggtatttcacaccgcatttggtgcactctca720gtacaatctgctctgatgccgcatagttaagccagccccgacacccgccaacacccgctg780acgcgccctgaccaggtcgggcttgtctgctcccggcatccgcttacagacaagctgtga840ccgtctccgggagctgcatgtgtcagaggttttcaccgtcatcaccgaaacgcg894当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1