一种含苯并噁唑哌嗪酮类衍生物及其合成方法和应用与流程
本发明涉及合成医药化工领域,主要涉及一种含苯并噁唑哌嗪酮类衍生物及其快速、高选择性的化学合成方法和应用。
背景技术:
苯并噁唑衍生物是构建天然产物和合成一些重要药物的中心骨架,其具有较好的杀菌、防腐、除草、抗癌等生物活性,因此,其化合物的合成及其生物活性研究在合成化学和药物化学界受到研究人员的极大关注。Farfán以邻氨基苯酚为起始原料,经四步合成苯并噁唑哌嗪类衍生物(Tetrahedron:Asymmetry 1999 10 799-811);Muthusubramanian发现分子间发生环化作用也能得到苯并噁唑哌嗪类衍生物(Indian J.Chem.,SecB,2007,46,1047-1050)。之后陆续也有人发现了新合成方法,但这些合成方法和路线均有步骤长、反应条件苛刻,成本高、耗时长、产率低、操作及后处理烦琐等缺点,在工业化方面很难大规模的应用,经济价值有限。因此,上述方法都不利于含合成苯并噁唑哌嗪类衍生物在有机合成中的应用及其工业化合成。
技术实现要素:
本发明提出了一种含苯并噁唑哌嗪酮类衍生物及其合成方法。本发明的方法以α-芳基-α,β-二氨基酸酯类化合物(制备方法见专利申请:201710004520.3)为原料,在金属催化剂的催化作用下,制备得到所述含苯并噁唑哌嗪酮类衍生物。本发明使用金属催化剂来活化炔基,一步构建得到多环的苯并噁唑哌嗪酮类衍生物。本发明以廉价易得的化合物为原料,具有反应条件温和、反应路线短、反应快、效率高、成本低、废物少、操作简单、原子经济性高等特点,本发明的合成方法在药物合成领域具有广阔应用前景。
本发明提出的含苯并噁唑哌嗪酮类衍生物,如式(II)所示,
其中,
Ar为芳基,选自于苯基、卤素取代的苯基、硝基取代的苯基、甲氧基取代的苯基、三氟甲基取代的苯基、C1-C10烷基取代的苯基;
R1为C1-C10烷基、苯基、C1-C10烷基取代的苯基、苄基;
R2为带有卤素、C1-C10烷基、硝基、甲氧基、三氟甲基、酯基取代的苯环,呋喃环,噻吩环,吡咯环;
R3为氢、卤素、硝基、三氟甲基、甲氧基、C1-C10烷基;
R4为氢、C1-C10烷基、苯基、苄基。
优选地,
Ar为苯基、4-氯苯基、3-氯苯基、2-氯苯基、4-硝基苯基、3-氟苯基、2-甲氧基苯基、4-甲氧基苯基或4-溴苯基;
R1为甲基、乙基、甲苯或苄基;
R2为4-硝基苯基、4-氟苯基、3-氟苯基、2-氟苯基、4-溴苯基、3-溴苯基、2-溴苯基、4-氯苯基、3-氯苯基、3-溴-4-氟苯基、1,2-二氯苯基、4-甲氧基苯基、4-三氟甲基苯基、呋喃环或2-苯甲酸甲酯基;
R3为氢、2-氯、3-氯、4-氯、4-甲氧基、4-溴、4-氟、4-甲基。
R4为氢、甲基、乙基。
进一步优选地,
Ar为苯基;
R1为甲基;
R2为4-溴苯基、4-甲氧基苯基、4-三氟甲基苯基;
R3为氢;
R4为氢。
本发明提出的含苯并噁唑哌嗪酮类衍生物的合成方法,以α-芳基-α,β-二氨基酸酯类化合物为原料,在有机溶剂中,在分子筛存在下和氮气保护下,在金属催化剂的催化作用下,经一步反应制备得到式(II)所示含苯并噁唑哌嗪酮类衍生物。所述合成反应如式(1)所示:
其中,
Ar为芳基,选自于苯基、卤素取代的苯基、硝基取代的苯基、甲氧基取代的苯基、三氟甲基取代的苯基、C1-C10烷基取代的苯基;
R1为C1-C10烷基、苯基、C1-C10烷基取代的苯基、苄基;
R2为带有卤素、C1-C10烷基、硝基、甲氧基、三氟甲基、酯基取代的苯环、呋喃环,噻吩环,吡咯环;
R3为氢、卤素、硝基、三氟甲基、甲氧基、C1-C10烷基;
R4为氢、C1-C10烷基、苯基、苄基。
优选地,
Ar为苯基、4-氯苯基、3-氯苯基、2-氯苯基、4-硝基苯基、3-氟苯基、2-甲氧基苯基、4-甲氧基苯基或4-溴基苯基;
R1为甲基、乙基、甲苯或苄基;
R2为4-硝基苯基、4-氟苯基、3-氟苯基、2-氟苯基、4-溴苯基、3-溴苯基、2-溴苯基、4-氯苯基、3-氯苯基、3-溴-4-氟苯基、1,2-二氯苯基、4-甲氧基苯基、4-三氟甲基苯基、呋喃环或2-苯甲酸甲酯基;
R3为氢、2-氯、3-氯、4-氯、4-甲氧基、4-溴、4-氟、4-甲基。
R4为氢、甲基、乙基。
进一步优选地,
Ar为苯基;
R1为甲基;
R2为4-溴苯基、4-甲氧基苯基、4-三氟甲基苯基;
R3为氢;
R4为氢。
本发明方法中,所述α-芳基-α,β-二氨基酸酯类化合物和金属催化剂的投料量摩尔比为α-芳基-α,β-二氨基酸酯类化合物:金属催化剂=1.0:0.10~0.20,优选地,α-芳基-α,β-二氨基酸酯类化合物:金属催化剂的摩尔比为1.0:0.20。
本发明方法中,有机溶剂用量与α-芳基-α,β-二氨基酸酯类化合物的比例为5mL~10mL:1mmol。优选地,有机溶剂用量与α-芳基-α,β-二氨基酸酯类化合物用量的比例为10mL:1mmol。
本发明方法中,分子筛与α-芳基-α,β-二氨基酸酯类化合物的用量比为100mg-200mg:1mmol。优选地,分子筛与α-芳基-α,β-二氨基酸酯类化合物的用量比为150mg:1mmol。
本发明方法中,所述反应温度为-20~40℃。优选地,所述反应温度为0~25℃。
本发明方法中,所述反应时间为1~6h。优选地,所述反应时间为2~4h;进一步优选地,先在0℃条件下反应1h,然后在25℃条件下反应2h。
本发明方法中,所述式(I)所示α-芳基-α,β-二氨基酸酯类化合物为原料。所述原料为邻羟基苯基取代的α-芳基-α,β-二氨基酸酯类化合物。
本发明方法中,所述有机溶剂包括二氯甲烷、三氯甲烷、甲苯、四氢呋喃、乙酸乙酯、1,2-二氯乙烷、乙腈。优选地,所述有机溶剂为无水二氯甲烷。
本发明方法中,采用的金属催化剂包括三氟甲磺酸银(AgOTf)、三氟甲磺酸铜。优选地,所述金属催化剂为三氟甲磺酸银(AgOTf)。
本发明方法中,所述反应需在无水环境下进行,分子筛的作用是除水。
本发明方法中,优选地,所述反应在氮气保护下进行,通过油泵换气,防止空气进入反应体系;
本发明一个具体实施方式中,所述方法为:将α-芳基-α,β-二氨基酸酯类化合物、分子筛溶于有机溶剂,于0℃加入三氟甲磺酸银后。室温继续反应。经反应、纯化得到高非对映选择性的式(II)所示含苯并噁唑哌嗪酮类衍生物,
本发明另一个具体实施方式中,所述含苯并噁唑哌嗪酮类衍生物的合成方法,包括以下步骤:α-芳基-α,β-二氨基酸酯类化合物:三氟甲磺酸银=1.0:0.2摩尔比(以α-芳基-α,β-二氨基酸酯类化合物用量为基准),称取原料。将α-芳基-α,β-二氨基酸酯类化合物和分子筛混合于有机溶剂,氮气保护下冷却至0℃,将AgOTf加入上述混合溶液中,0℃继续反应1h,然后室温下继续搅拌1~5小时,直至α-芳基-α,β-二氨基酸酯类化合物消耗完全;将粗产物进行柱层析(以石油醚:乙酸乙酯=20:1~15:1~10:1为洗脱剂),得到高非对应选择性的含苯并噁唑哌嗪酮类衍生物。
本发明还提出了依本发明合成方法制备得到的式(II)含苯并噁唑哌嗪酮类衍生物。
本发明还提出了式(II)含苯并噁唑哌嗪酮类衍生物在制备抗肿瘤的药物中的应用。
本发明还提出了式(II)所示含苯并噁唑哌嗪酮类衍生物在制备抗结肠癌药物中的应用。
本发明还提出了式(II)所示含苯并噁唑哌嗪酮类衍生物在制备抗人结肠癌HCT116细胞的药物中的应用。
本发明含苯并噁唑哌嗪酮类衍生物是重要的化工和医药中间体,在医药化工领域广泛应用,具有很大应用前景。本发明的合成方法以廉价易得的化合物为原料,具有反应条件温和、反应步骤少、反应快、成本低、产生的废物少、操作简单安全、原子经济性高、选择性高等有益效果。
附图说明
图1为实施例13所得产物的1H NMR示意图。
图2为实施例13所得产物的13C NMR示意图。
图3为实施例13所得产物的19F NMR示意图。
图4为实施例14所得产物的1H NMR示意图。
图5为实施例14所得产物的13C NMR示意图。
图6为实施例15所得产物的1H NMR示意图。
图7为实施例15所得产物的13C NMR示意图。
具体实施方式
结合以下具体实施例和附图,对本发明作进一步的详细说明,本发明的保护内容不局限于以下实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。实施本发明的过程、条件、试剂、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。
本发明合成含苯并噁唑哌嗪酮类衍生物的制备方法,将α-芳基-α,β-二氨基酸酯类化合物(0.2mmol)(或和分子筛(300mg))混合于2mL有机溶剂,在一定温度下,将AgOTf或Cu(OTf)2(0.04mmol)加入上述混合溶液中,继续反应1h,而后室温下继续搅拌1~5小时,直至α-芳基-α,β-二氨基酸酯类化合物消耗完全;将粗产物进行柱层析(以石油醚:乙酸乙酯=20:1~15:1~10:1为洗脱剂),得到高非对应选择性含苯并噁唑哌嗪酮类衍生物。
实施例1~12不同实验条件下合成含苯并噁唑哌嗪酮类衍生物
本发明在不同实验条件下所得实验结果如下表1所示a:
表1
a除有说明,I:[M]=1:0.2(摩尔比);I:溶剂=1mmol:10mL。
bII通过薄层色谱法纯化得到。
c先在不同温度反应1h,之后室温反应3h。
d40℃反应1小时发现出现杂质点,即原料变质,说明温度太高不利于反应。
本发明根据如上表1结果,最终确定实验方法为12,具体制备方法为:将α-芳基-α,β-二氨基酸酯类化合物(0.2mmol)和分子筛(300mg))混合于2mLDCM溶剂中,在氮气保护下和0℃下,将AgOTf(0.04mmol)加入上述混合溶液中,0℃继续反应1h,而后室温下继续搅拌1~5小时,直至α-芳基-α,β-二氨基酸酯类化合物消耗完全;将粗产物进行柱层析(以石油醚:乙酸乙酯=20:1~15:1~10:1为洗脱剂),得到高非对应选择性含苯并噁唑哌嗪酮类衍生物。
合成反应过程如式(3):
Ar为芳基,选自于苯基;
R1为甲基;
R2为4-溴苯基、4-甲氧基苯基、4-三氟甲基苯基;
R3为氢;
R4为氢。
实施例13
将丙炔酰基二氨酯类化合物(0.2mmol)和分子筛(300mg)混合于2mLDCM溶剂中,于0℃氮气保护下加入三氟甲磺酸银(0.04mmol),继续反应1h。1h后升温至室温(25℃)下,继续搅拌2h。反应混合物通过柱层析进行纯化,得到纯产品,其结构如式(a)所示,为甲基-10a-甲基-1-氧-3-苯基-4-(4-(三氟甲基)苯基)-1,3,4,10a-四氢-2H-苯并[4,5]噁唑酮[3,2-a]吡嗪-3-羧酸酯。产率为65%,dr值等于>95:5。式(a)所示化合物的1H NMR示意图如图1所示,其13C NMR示意图如图2所示,其19F NMR示意图如图3所示。
1H NMR(400MHz,CDCl3)δ7.76(d,J=7.7Hz,2H),7.49(t,J=7.3Hz,2H),7.43(m,4H),7.32(d,J=8.1Hz,2H),6.72(dd,J=18.1,7.5Hz,2H),6.63(m,2H),5.91(s,1H),3.54(s,3H),1.05(s,3H).13C NMR(100MHz,CDCl3)δ168.36,149.95,135.46(J=250.1Hz),129.36,129.18,129.12,128.59(J=27.0Hz),126.47,126.16(J=3.78Hz),125.18,123.57,121.51,114.58,109.84,95.86,67.18,63.47,53.61,24.90.HRMS(ESI)m/z calcd for C26H21F3N2O4Na([M+Na]+)505.1353,found 505.1351.
实施例14
将丙炔酰基二氨酯类化合物(0.2mmol)和分子筛(300mg)混合于2mLDCM溶剂中,于0℃氮气保护下加入三氟甲磺酸银(0.04mmol),继续反应1h。1h后升温至室温下,继续搅拌2h。反应混合物通过柱层析进行纯化,得到纯产品,其结构如式(b)所示,为甲基-10a-甲基-1-氧-3-苯基-4-(4-溴苯基)-1,3,4,10a-四氢-2H-苯并[4,5]噁唑酮[3,2-a]吡嗪-3-羧酸酯。产率为60%,dr值等于>95:5。式(b)所示化合物的1H NMR示意图如图4所示,其13C NMR示意图如图5所示。
1H NMR(400MHz,CDCl3)δ7.67(d,J=7.5Hz,2H),7.43-7.37(m,3H),7.19(m,2H),6.99(d,J=8.1Hz,3H),6.69-6.61(m,2H),6.61-6.50(m,2H),5.73(s,1H),3.46(s,3H),0.97(s,3H).13C NMR(100MHz,CDCl3)δ167.47,166.93,148.50,137.10,134.62,132.54,130.91,130.44,129.55,128.05,127.39,125.48,122.42,120.41,113.69,108.73,94.87,66.37,62.29,52.53,23.93.HRMS(ESI)m/z calcd for C25H21BrN2O4Na([M+Na]+)515.0582,found 515.0569.
实施例15
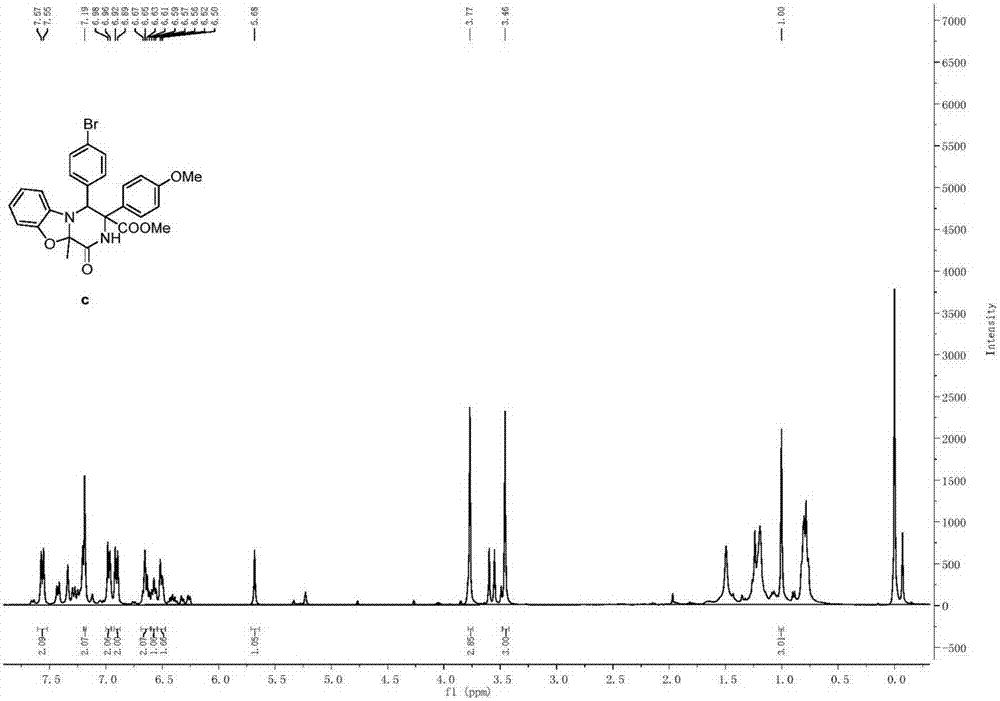
将丙炔酰基二氨酯类化合物(0.2mmol)和分子筛(300mg)混合于2mL DCM溶剂中,于0℃氮气保护下加入三氟甲磺酸银(0.04mmol),继续反应1h。1h后升温至室温下,继续搅拌2h。反应混合物通过柱层析进行纯化,得到纯产品,其结构如式(c)所示,为甲基-10a-甲基-1-氧-3-(4-甲氧基苯基)-4-(4-溴苯基)-1,3,4,10a-四氢-2H-苯并[4,5]噁唑酮[3,2-a]吡嗪-3-羧酸酯。产率为50%,dr值等于>95:5。式(c)所示化合物的1H NMR示意图如图6所示,其13C NMR示意图如图7所示。
1H NMR(400MHz,CDCl3)δ7.56(d,J=8.7Hz,2H),7.19(s,2H),6.97(d,J=8.3Hz,2H),6.91(d,J=8.7Hz,2H),6.64(q,J=7.6Hz,2H),6.57(t,J=7.2Hz,1H),6.51(d,J=7.1Hz,2H),5.68(s,1H),3.77(s,3H),3.46(s,3H),1.00(s,3H).13C NMR(101MHz,CDCl3)δ167.70,166.98,159.02,148.49,134.67,132.64,130.41,129.54,128.71,126.78,122.33,120.39,113.60,113.34,108.70,94.91,65.95,62.35,54.36,52.43,24.02.HRMS(ESI)m/z calcd for C26H24BrN2O5([M+H]+)523.0869,found 523.0851.
实施例16抗肿瘤活性测试
IC50值测试
人结肠癌HCT116细胞接种于McCoy,s5A培养液(10%血清,1%青-链霉素)和1640培养液中。置于37℃,5%CO2培养箱中,每2-3天传代一次,试验取对数生长期细胞。CCK-8法测定IC50值。
取对数生长期细胞,以配置好的新鲜培养液调整细胞悬液至2500~4000/ml,取100ul(2千细胞/孔)细胞悬液接种到96孔培养板。置于5%CO2,37℃培养箱中过夜孵育培养后,更换新鲜细胞培养液,每孔加入200ul DMSO等体积稀释的浓度梯度药物与细胞共孵育72h,更换新鲜细胞培养液,每孔加100ul+10ulCCK-8溶液继续孵育1-4小时,终止培养,用多功能酶标仪(Molecular Devices M5)检测450nm的吸光度,620nm的吸光度校正细胞数差异。
将待测化合物(实施例13~实施例15)溶解在DMSO中,并用培养液进一步稀释。DMSO最终浓度不超过0.1%(v/v)。对照组为加入等体积DMSO的肿瘤细胞;空白组无细胞,在培养液中加入等体积DMSO。在一次实验内,每个实验条件均设3个复孔。计算各浓度的化合物对细胞生长的抑制率,计算公式为:抑制率(%)={1-[(加药组)-(空白组)]/[(对照组)-(空白组)]}×100%,用GraphPad Prim6计算出IC50(为将细胞生长降至对照组的50%所需的药物浓度)。其检测结果如下表2。
表2:本发明含苯并噁唑哌嗪酮类衍生物在HCT116细胞中的生物活性数据
“---”表示无活性;“/”表示未测该项实验。a表示阳性药在肿瘤细胞中的抑制率(%)。
综上,实验结果表明,含苯并噁唑哌嗪酮类衍生物对人结肠癌细胞具有显著的抑制作用,实施例14中,活性可达5.541μM。因此,本发明为开发治疗结肠癌药物提供了非常广阔的发展空间。
本发明的保护内容不局限于以上实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 苯并双(噻二唑)衍生物、包含其的油墨、以及使用了其的有机电子装置的制造方法
- 具有含硫取代基的杀有害生物活性杂环衍生物的制造方法与工艺
- 具有含硫取代基的杀有害生物活性杂环衍生物的制造方法与工艺
- 一种苯并噻咯类衍生物及其制备方法和有机发光器件与制造工艺
- 苯并噻吩并咔唑类衍生物及其制备方法和有机发光器件与制造工艺
- 苯并二氮杂*衍生物的2-萘磺酸盐及晶型和它们的制备方法与制造工艺
- 靛红杂合喹唑啉类化合物合成的靛红衍生物及其在制备抗肿瘤药物中的应用的制造方法与工艺
- 一种杂环衍生物及使用该杂环衍生物的有机发光器件的制造方法与工艺
- 一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法与制造工艺
- 一种含氮杂环衍生物及使用该含氮杂环衍生物的有机发光器件的制造方法与工艺
- 苯并二氮杂*衍生物的2-萘磺酸盐及晶型和它们的制备方法与制造工艺
- 一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法与制造工艺
- 苯并吲唑类衍生物及其制备方法、应用与制造工艺
- 一种二苯并环庚烷衍生物的制备方法与制造工艺
- Atropurpuran的苯并咪唑基和吗啉基衍生物组合物用于防治胰腺纤维化的制造方法与工艺
- 一种苯并咪唑衍生物镉配合物及其制备方法和应用
- 一种咪唑并吡啶巯乙酸类衍生物及其制备方法与应用
- Artalbic acid的O?(苯并咪唑基)乙基与O?(二羟乙胺基)乙基衍生物的组合物在抗病毒 ...的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备抗骨质疏松药物的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备防治胰腺纤维化药物的制作方法