一种用于无创产前检测的文库构建方法及试剂盒与流程
本发明涉及分子生物学
技术领域:
,尤其涉及文库构建
技术领域:
,特别涉及一种用于无创产前检测的文库构建方法及试剂盒。
背景技术:
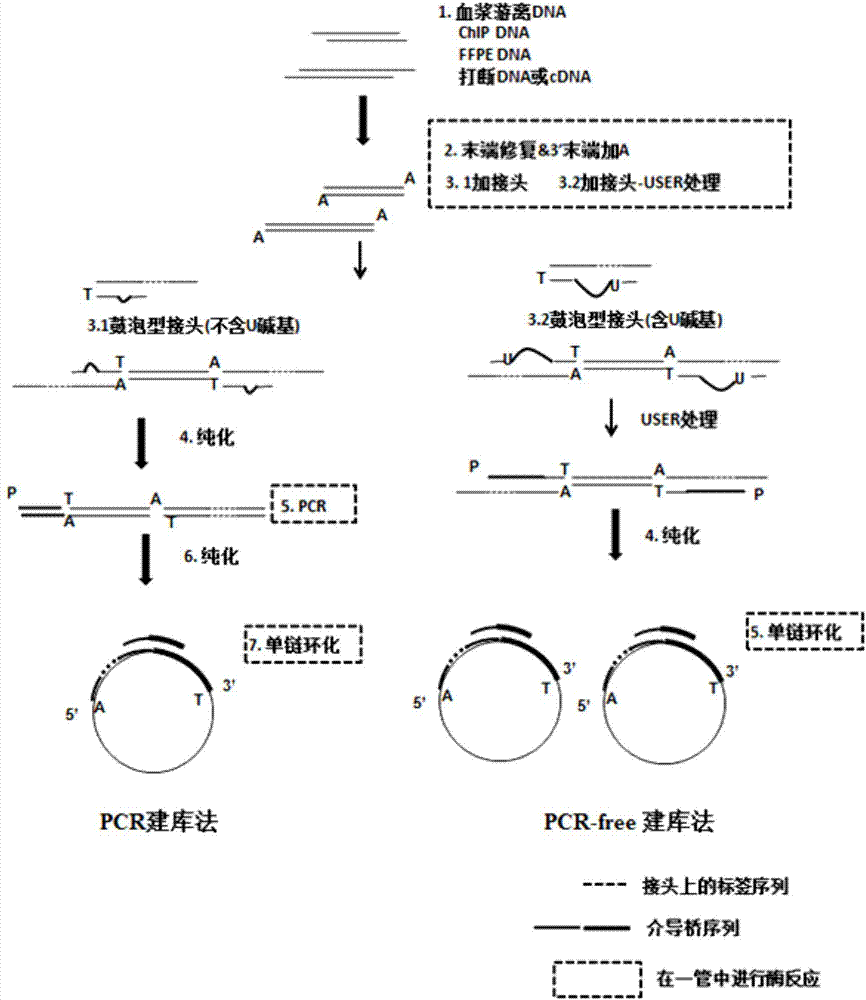
:目前,高通量测序技术已广泛应用到医学研究和检测领域,如无创产前检测(nipt),有限的样本量和检测报告输出的时效性对该技术提出了更高的要求。简化文库构建流程、提高低起始量样本的建库成功率势在必行。通过优化和改进文库构建方法,缩短酶反应时间和建库步骤、提高样本转化成文库的效率成为高通量测序技术拓展应用领域的一大关键点。经典的高通量文库构建步骤包括:1.基因组打断(包括物理打断法和化学打断法,如covaris超声打断、fragmentase酶切打断);2.双链dna(deoxyribonucleicacid,脱氧核糖核酸)末端修复;3.3’末端加datp(deoxyadenosinetriphosphate,三磷酸脱氧腺苷);4.加与测序平台匹配的接头;5.pcr(polymerasechainreaction,聚合酶链式反应)扩增。每个步骤都需要进行纯化反应,有时还需要在酶反应之后进行一次片段选择(步骤1、4、5其中之一),整个流程需要大概8-9个小时才能完成。为简化流程,一些生物公司推出了新的建库技术,如illumina公司提出了基于转座酶的打断与加接头一步完成的快速建库方法,但该方法存在明显的测序数据碱基偏向性问题,且无法应用到血浆游离dna这类不需要打断的样本。此外,illumina和biooscientific等公司提出了无需pcr(pcr-free)的方法,节省pcr扩增步骤,但对样品的起始量要求较高(500ng-2μg)。技术实现要素:本发明提供一种用于无创产前检测的文库构建方法,该方法能够在同一个反应体系中实现dna末端修复与加a反应,并且接着进行接头连接和纯化处理,简化酶反应和纯化步骤,提高起始dna转化成可连接接头dna的效率,降低对起始dna总量的要求,也可降低pcr扩增的循环数和提高低起始量dna实现无需pcr的建库,可以提高上机文库的可用数据比率并且降低数据碱基偏向性,本发明的方法适用于不同类型微量起始dna的快速建库。本发明还提供一种用于无创产前检测的文库构建试剂盒,以及构建得到的可以用于无创产前检测的文库。根据本发明的第一方面,本发明提供一种用于无创产前检测的文库构建方法,该方法包括:在同一反应体系中,在dntp存在下,利用聚合酶将片段化dna的末端补平或切平,例如利用聚合酶的5’-3’聚合活性将5’突出末端补平和/或聚合酶的3’-5’外切活性将3’突出末端切平,利用多聚核苷酸激酶将5’羟基转变成5’磷酸基团且将3’磷酸基团转变成3’羟基;在过量datp存在下,利用不具有3’-5’外切活性的聚合酶在双链dna3’末端加上datp;在上述反应体系中,直接加入鼓泡型接头和连接反应混合液,使上一步产生的3’a突出的双链dna与上述接头连接;纯化上一步接头连接的产物后,加入与上述接头序列两端互补的核酸单链作为引物进行pcr扩增,其中一条引物5’末端有磷酸化修饰,得到大量双链dna,其中一条链的5’端有磷酸基团;纯化上一步pcr扩增的产物后,利用热变性将上述双链dna变成单链脱氧核酸,并加入一段与接头首尾序列互补的单链核酸作为介导桥序列,与变性后的单链脱氧核酸杂交及连接反应,使目的单链核酸环化,得到上述文库。根据本发明的第一方面,本发明还提供一种用于无创产前检测的文库构建方法,该方法包括:在同一反应体系中,在dntp存在下,利用聚合酶将片段化dna的末端补平或切平,例如利用聚合酶的5’-3’聚合活性将5’突出末端补平和/或聚合酶的3’-5’外切活性将3’突出末端切平,利用多聚核苷酸激酶将5’羟基转变成5’磷酸基团且将3’磷酸基团转变成3’羟基;在过量datp存在下,利用不具有3’-5’外切活性的聚合酶在双链dna3’末端加上datp;在上述反应体系中,直接加入含u的鼓泡型接头和连接反应混合液,使上一步产生的3’a突出的双链dna与上述接头连接,同时加入user酶,进行消化反应,使连接上上述接头的dna的5’端经user酶切后产生5’磷酸基团;纯化上一步接头连接的产物后,利用热变性将上述双链dna变成单链脱氧核酸,并加入一段与接头首尾序列互补的单链核酸作为介导桥序列,与变性后的单链脱氧核酸杂交及连接反应,使目的单链核酸环化,得到上述文库。根据本发明的第一方面,本发明还提供一种用于无创产前检测的文库构建方法,该方法包括:在同一反应体系中,在dntp存在下,利用聚合酶将片段化dna的末端补平或切平,例如利用聚合酶的5’-3’聚合活性将5’突出末端补平和/或聚合酶的3’-5’外切活性将3’突出末端切平,利用多聚核苷酸激酶将5’羟基转变成5’磷酸基团且将3’磷酸基团转变成3’羟基;在过量datp存在下,利用不具有3’-5’外切活性的聚合酶在双链dna3’末端加上datp;在上述反应体系中,直接加入y型接头和连接反应混合液,使上一步产生的3’a突出的双链dna与上述接头连接;纯化上一步接头连接的产物后,加入与上述接头序列两端互补的核酸单链作为引物进行pcr扩增,其中一条引物5’末端有磷酸化修饰,得到大量双链dna,其中一条链的5’端有磷酸基团;纯化上一步pcr扩增的产物后,得到可以用于上机测序的文库。根据本发明的第一方面,本发明还提供一种用于无创产前检测的文库构建方法,该方法包括:在同一反应体系中,在dntp存在下,利用聚合酶将片段化dna的末端补平或切平,例如利用聚合酶的5’-3’聚合活性将5’突出末端补平和/或聚合酶的3’-5’外切活性将3’突出末端切平,利用多聚核苷酸激酶将5’羟基转变成5’磷酸基团且将3’磷酸基团转变成3’羟基;在过量datp存在下,利用不具有3’-5’外切活性的聚合酶在双链dna3’末端加上datp;在上述反应体系中,直接加入含标签序列的y型接头和连接反应混合液,使上一步产生的3’a突出的双链dna与上述接头连接;纯化上一步接头连接的产物后,得到可以用于上机测序的文库。作为本发明的进一步改进的方案,上述聚合酶为t4dna聚合酶和/或klenow大片段;上述多聚核苷酸激酶为t4多聚核苷酸激酶;上述不具有3’-5’外切活性的聚合酶为taq聚合酶和/或klenow片段(3′→5′exo-)。作为本发明的进一步改进的方案,上述片段化dna为血浆游离dna、染色质免疫共沉淀(chromatinimmunoprecipitation,chip)dna、福尔马林固定石蜡包埋(formalin-fixedparaffin-embedded,ffpe)dna、物理打断dna、酶切打断dna或cdna(complementarydna,互补dna,rna反转录得到的dna);优选为血浆游离dna、物理打断dna或酶切打断dna。作为本发明的进一步改进的方案,片段化dna为物理打断dna,每50μl的体系中含有,物理打断dna1-100ng,t4dna聚合酶1.2-10u,klenow大片段0-2u,t4多聚核苷酸激酶4-16u,taq聚合酶1-4u,每种dntp各0.02-0.2mm,额外的datp0.4-1mm,以及mg离子8-15mm;更进一步优选地,反应条件为15-37℃反应10-30min,然后65-75℃反应10-30min。作为本发明的进一步改进的方案,片段化dna为血浆游离dna或酶切打断dna,每50μl的体系中含有,血浆游离dna或酶切打断dna1-100ng,t4dna聚合酶0-3u,klenow大片段0-2u,t4多聚核苷酸激酶4-10u,taq聚合酶1-2u,每种dntp各0.02-0.2mm,额外的datp0.4-1mm,以及mg离子8-10mm,条件是t4dna聚合酶和klenow大片段的含量不同时为0;更进一步优选地,反应条件为15-37℃反应10-30min,然后65-75℃反应10-30min。根据本发明的第二方面,本发明提供一种用于无创产前检测的文库构建试剂盒,该试剂盒按照t4dna聚合酶1.2-10u,klenow大片段0-2u,t4多聚核苷酸激酶4-16u,taq聚合酶1-4u,每种dntp各1-10nmol,额外的datp20-50nmol,以及mg离子400-750nmol,形成用于稀释成50μl反应体系的试剂盒单位;或者,该试剂盒按照t4dna聚合酶0-3u,klenow大片段0-2u,t4多聚核苷酸激酶4-10u,taq聚合酶1-2u,每种dntp各1-10nmol,额外的datp20-50nmol,以及mg离子400-500nmol,条件是t4dna聚合酶和klenow大片段的含量不同时为0,形成用于稀释成50μl反应体系的试剂盒单位。需要说明的是,本发明的试剂盒按照上述定义,并不意味着本发明的试剂盒局限于用于稀释成50μl反应体系的试剂盒,而是根据上述定义来确定本发明的试剂盒中各种组分的比例关系和浓度。本领域的技术人员可以根据上述用于稀释成50μl反应体系的试剂盒单位中各种组分的比例关系和浓度来平行地放大或者缩小试剂盒的容量大小,只要各种组分的比例关系和浓度与上述定义一致即可认为是本发明的试剂盒。上述试剂盒单位是人为定义的,本领域的技术人员可以理解,平行地放大或者缩小的试剂盒的容量均可以称为一个试剂盒单位。根据本发明的第三方面,本发明提供一种用于无创产前检测的文库,该文库使用第一方面的方法或者第二方面的试剂盒构建得到。本发明方法的优势体现在:首先,本发明的dna末端修复与加a在同一反应体系中进行,可以将经典建库方法的步骤2至4从原来的3步酶反应、3步纯化反应,简化为2步酶反应、1步纯化反应,大大缩短建库时间,节省纯化步骤的建库成本,提升高通量建库技术应用到医学临床检测项目(如无创产前诊断、游离肿瘤dna基因检测等)的实效。其次,本发明在同一个反应体系中进行从dna末端修复至加a到加接头的反应,能够降低起始dna在建库过程的损耗,提高dna回收效率。另外,本发明通过优化反应条件,提高起始dna转化成可连接接头dna的效率,降低pcr扩增的循环数,从而降低pcr循环数过多导致重复数据的产出,提高可用数据比率,降低数据碱基偏向性。例如,无创产前诊断项目,未优化的反应法pcr扩增的循环数为13,优化之后循环数只需要11。此外,起始dna转化效率的提高使低起始量dna(如20-100ng)进行无需pcr(pcr-free)的建库成为可能。此外,本发明的方法可以广泛实现不同类型微量起始dna的快速建库,如血浆游离dna、染色质免疫共沉淀dna、福尔马林固定石蜡包埋dna、1-100ng低起始量的物理打断dna或酶切打断dna,以及cdna等;尤其是血浆游离dna、物理打断dna或酶切打断dna。本发明的方法可以应用于多种测序平台(如bgiseq、illumina等)。附图说明图1为本发明一个实施方案的基于bgiseq-50/500/1000测序平台的末端修复-加a-加接头反应法(即图中“一管法”)的建库流程示意图;其中,1:起始dna;2:末端修复和加a在同一反应体系中进行;3.1:采用pcr策略时,dna两端加上鼓泡型接头(不含u碱基);3.2:采用pcr-free策略时,dna两端加上含u碱基的鼓泡型接头,同时进行user处理,user处理的加上接头的dna5’末端为磷酸基团,便于后续单链环化;4:纯化反应体系,去除接头污染;5:对于pcr建库策略,连接产物进行pcr扩增;对于pcr-free建库策略,连接产物直接用于单链环化;6:纯化pcr反应体系,去除引物二聚体污染;7:将pcr产物用于单链环化。图2为本发明另一个实施方案的基于illumina测序平台的末端修复-加a-加接头反应法的建库流程示意图;其中,1:起始dna;2:末端修复和加a在同一反应体系中进行;3.1:采用pcr策略时,dna两端加上不含标签序列的y型接头;3.2:采用pcr-free策略时,dna两端加上含标签序列的y型接头;4:纯化反应体系,去除接头污染;对于pcr-free建库策略,纯化的连接产物经q-pcr定量后可直接用于上机;5:对于pcr建库策略,连接产物进行pcr扩增;6:纯化pcr反应体系,去除引物二聚体污染,纯化的pcr产物经q-pcr定量后可直接用于上机。图3示出了本发明一个实施例的无创产前诊断项目bgiseq-50/500/1000建库方法对比,以无创产前诊断项目的文库构建为例,展示了经典建库方法与末端修复-加a-加接头反应法的建库流程和建库时间差异。图4示出了本发明一个实施例的pcrfree文库dna纳米球电泳结果,样品大部分都在胶孔里,无法电泳出来,符合dna纳米球在聚丙烯酰胺凝胶电泳时无法跑出胶孔的特点。图5示出了本发明一个实施例的1例基因组样品的测序深度分布图,测序深度符合泊松分布,测序深度集中在30×左右,符合人基因组重测序的数据需求。图6示出了本发明一个实施例的1例基因组样品的gc含量累积比例,虽然高gc含量的片段的覆盖度有所下降,但覆盖率接近1,说明大部分高gc含量的片段可以被检测到,所有不同gc含量的片段均一化后的覆盖度基本保持水平。图7示出了本发明一个实施例的8例酶打断基因组样品的pcr产物电泳图,pcr产物的片段大小在250bp左右,符合bgiseq-50/500/1000测序平台的上机要求。具体实施方式下面通过具体实施方式结合附图对本发明作进一步详细说明。本发明提出了一种dna末端修复与加a的反应方法,使用本发明的方法能够实现dna末端修复与加a在同一个反应体系(一管)中完成,并且可以直接在反应管中加入接头和连接反应混合液,进而实现一管式加接头。本发明的dna末端修复与加a的反应方法得到的末端修复补平和加a的dna产物可以应用于多种应用场景之中,最常见的应用是用于文库构建。采用本发明的dna末端修复与加a反应法,能够实现低起始量打断样本或血浆游离dna等特殊临床样本的快速建库,缩短文库制备的时间,降低文库制备的成本,提高文库的可用数据产出,并且不仅可应用到bgiseq平台,也可推广到illumina平台。下面将以不同的平台为例,并结合附图,详细说明本发明的技术方案。需要说明的是,以下涉及到的平台仅是示例性的,本发明并不局限于这些平台,而是可以应用于更广泛的平台上。并且本发明的技术方案也不局限于以下参考附图所说明的技术方案的细节上。请参考图1,以bgiseq-50/500/1000测序平台的文库构建为示例,本方案的文库构建过程具体步骤如下:1、模板dna的准备:用制备好的模板dna进行建库反应,模板dna包括但不限于血浆游离dna、chipdna、ffpedna、超声打断或酶切打断纯化好的基因组dna或rna反转录得到的cdna(如图1中编号1所示)。2、dna末端修复与加a反应:在同一反应体系中,在dntp存在下,利用聚合酶将片段化dna的末端补平或切平,例如利用聚合酶的5’-3’聚合活性将5’突出末端补平和/或聚合酶的3’-5’外切活性将3’突出末端切平,利用多聚核苷酸激酶将5’羟基转变成5’磷酸基团且将3’磷酸基团转变成3’羟基;在过量datp存在下,利用不具有3’-5’外切活性的聚合酶在双链dna3’末端加上datp(如图1中编号2所示)。由机体自身酶切产生的游离dna片段或被物理或化学方法人为打断的基因组dna片段末端的完整性会受到不同程度的破环,形成5’突出末端、3’突出末端、5’羟基、3’磷酸基团等构型,不利于后续与接头发生连接反应。末端修复反应主要利用聚合酶(如t4dna聚合酶、klenow大片段等)在dntp(deoxy-ribonucleosidetriphosphate,脱氧核糖核苷三磷酸)存在时的5’-3’聚合活性将5’突出末端补平,3’-5’外切酶活性将3’突出末端切平;利用多聚核苷酸激酶(如t4多聚核苷酸激酶等)的5’羟基激酶活性将5’羟基转变成5’磷酸基团,多聚核苷酸激酶的3’磷酸酯酶活性将3’磷酸基团转变成3’羟基。3’末端加a反应则是在反应体系中存在datp时,利用不具有3’-5’外切活性的聚合酶(如去除了3’-5’外切活性的klenow大片段(又称“klenow片段(3′→5′exo-)”)、不具有3’-5’校正活性的taqdna聚合酶等),在双链dna3’末端加上datp,以利于双链dna后续与5’t突出构型的接头序列进行a-t连接。基于上述原理,末端修复和加a反应在同一反应体系中进行需要具备的成分有聚合酶、多聚核苷酸激酶、无3’-5’外切活性的聚合酶、dntp(其中datp需多于dttp、dctp、dgtp)、兼容三种酶的反应缓冲液。在本发明的一个优选的实施例中,从酶量、反应缓冲液成分、反应温度、反应时间多个方面进行了系统地优化,得到了一系列适合不同起始量和不同来源模板dna的末端修复与加a的反应试剂配方和建库条件。如此,加入优化的酶反应混合液,通过控制反应温度,使双链dna末端修复反应和3’末端加a反应在一个反应体系中有序地进行。具体来讲,对于物理打断dna,每50μl的体系中含有,物理打断dna1-100ng,t4dna聚合酶1.2-10u,klenow大片段0-2u,t4多聚核苷酸激酶4-16u,taq聚合酶1-4u,每种dntp各0.02-0.2mm,额外的datp0.4-1mm,以及mg离子8-15mm;优选地,反应条件为15-37℃反应10-30min,然后65-75℃反应10-30min。对于血浆游离dna或酶切打断dna,每50μl的体系中含有,血浆游离dna或酶切打断dna1-100ng,t4dna聚合酶0-3u,klenow大片段0-2u,t4多聚核苷酸激酶4-10u,taq聚合酶1-2u,每种dntp各0.02-0.2mm,额外的datp0.4-1mm,以及mg离子8-10mm,条件是t4dna聚合酶和klenow大片段的含量不同时为0;优选地,反应条件为15-37℃反应10-30min,然后65-75℃反应10-30min。需要说明的是,额外的datp0.4-1mm,是指除了每种dntp(包括datp、dttp、dctp、dgtp)各0.02-0.2mm以外,还含有多余的datp0.4-1mm。也就是说,datp的总量为0.42-1.2mm。还需要说明的是,本发明以50μl的体系为例进行说明,并不表明本发明的方法仅适合于50μl的体系,本领域的技术人员能够理解,以本发明的50μl的体系为基准,维持各种组分的含量不变,进行体系的平行放大或者缩小均能够实现本发明,本发明权利要求的技术方案也包括这里所说明的平行放大或者缩小的体系。在本发明的另一个更加优选的实施例中,从酶量、反应缓冲液成分、反应温度、反应时间多个方面进行了系统地优化,得到了一系列适合不同起始量和不同来源模板dna的末端修复与加a的反应试剂配方和建库条件(如表1所示)。表150μl反应体系下不同模板dna的末端修复-加a的反应优化参数需要说明:在表1中,对于酶切打断和血浆游离dna情况,t4dna聚合酶与klenow大片段的含量不同时为0。3、加接头:在步骤2的反应液中直接加入接头和连接反应混合液,使上一步3’a突出的双链dna与接头连接。针对不同测序平台,可以加入不同平台的接头。接头是一段特殊设计的脱氧核糖核酸序列,通过连接等方法固定在dna片段两端后,在测序时能被识别并作为测序的起始位点,供仪器读取其后的序列信息。由于不同平台的文库结构不同,其使用的接头结构也有差异。在此步骤使用不同接头,即可满足不同测序平台对于文库的需求。针对bgiseq平台,当采用pcr建库策略时,加入不含u的鼓泡型接头(如图1中编号3.1所示);当采用pcr-free建库策略时,加入含u的鼓泡型接头,同时加少量user酶进行消化反应,使连接上接头的dna5’端经user酶切后产生5’磷酸基团(如图1中编号3.2所示)。4、纯化接头连接产物:纯化末端修复-加a-加接头的反应体系,去除接头污染(如图1中编号4所示)。5、pcr扩增或单链环化:对于pcr建库策略,加入与目的接头序列两端互补的核酸单链作为引物进行pcr扩增,得到大量的dna产物,由于其中一条引物5’末端有磷酸化修饰,扩增得到的双链dna其中一条的5’端有磷酸基团;然而,对于pcr-free建库策略,纯化的连接产物直接用于单链环化反应,dna的两条链都可以形成环化产物(如图1中编号5所示)。6、纯化pcr反应体系:对于pcr建库策略,纯化pcr反应体系,去除引物二聚体污染(如图1中编号6所示)。7、单链环化:对于pcr建库策略,纯化的pcr扩增产物dna的其中一条链可以发生单链环化反应(如图1中编号7所示)。单链环化反应利用热变性将双链dna变成单链脱氧核酸,并加入一段与接头首尾序列互补的单链核酸(可称为介导桥序列)与变性后的单链脱氧核酸杂交和连接反应,使目的单链核酸环化。反应完的单链环化体系可直接用于后续的滚环复制,形成测序上机模板产物dnb(dnananoball,核酸纳米球),用于测序反应。请参考图2,以illumina测序平台的文库构建为示例,说明其技术方案。如图2所示,其与图1所示的bgiseq-50/500/1000测序平台的文库构建方法的差别在于:在步骤3的加接头过程中,当采用pcr建库策略时,加入不含标签序列的y型接头(如图2中编号3.1所示);当采用pcr-free建库策略时,加入含标签序列的y型接头(如图2中编号3.2所示)。此外,图2所示的方法中,不需要最后的单链环化,可以将纯化后的产物直接用于上机测序。如图3所示,本发明的建库方法将经典建库方法的步骤2至4从原来的3步酶反应、3步纯化反应,简化为2步酶反应、1步纯化反应,大大缩短建库时间,节省纯化步骤的建库成本,提升高通量建库技术应用到医学临床检测项目(如无创产前检测、游离肿瘤dna基因检测等)的实效。以下通过实施例详细说明本发明的技术方案,应当理解,实施例仅是示例性的,不能理解为对本发明保护范围的限制。实施例1:对16例孕妇血浆进行胎儿染色体三倍性变异检测1.样品收集及处理:取孕妇外周血2ml,1600g,4℃离心10分钟,将血细胞和血浆分开,血浆再以16000g,4℃离心10分钟,进一步去除残留的白细胞。孕妇血浆用qiaampcirculatingnucleicacidkit(qiagen)提取血浆dna,最后将dna溶于40μlte溶液。2.末端修复-加datp:按下表2配置体系。表2组分用量10×t4多聚核苷酸激酶缓冲液(enzymatics公司)5μlt4多聚核苷酸激酶(enzymatics公司)0.4μlklenow大片段(5u/μl)(enzymatics公司)0.2μl脱氧核糖核酸混合液(每种25mm)(enzymatics公司)0.4μltaqdna聚合酶(5u/μl)(takara公司)0.2μl腺苷酸脱氧核糖核酸(100mm)(enzymatics公司)1μlt4dna聚合酶(3u/μl)(enzymatics公司)0.4μl无酶纯水2.4μl总体积10μl将10μl反应液加入均一化至40μl的dna溶液中,混匀,置于37℃孵育10min;75℃孵育10min,以0.1℃/秒的速率降温至4℃。3.接头连接:本方案中使用的接头序列如下(本实施例中的序列从左到右为5’端至3’端,“//”示修饰基团,“phos”示磷酸化,b10示10个碱基的标签序列)。长链:/5phos/agtcggaggccaagcggtcttaggaagacaa(b10)caactccttggctcaca(seqidno:1);短链:ttgtcttcctaaggaacgacatggctacgatccgactt(seqidno:2)。接头混合液(10μm)按以下表3配方配制。表3将1μl配制好的接头混合液(10μm)加入上一步骤产物中,充分混匀。按下表4配置体系。表4组分用量三磷酸腺苷(100mm)0.8μl连接酶(600u/μl)(enzymatics公司)1.6μl10xt4多聚核苷酸激酶缓冲液(enzymatics公司)3μl聚乙二醇8000(50%)12μl无酶纯水11.6μl总体积29μl将连接反应体系与接头和产物的混合液混匀,置于23℃孵育30min。反应完之后,加入20μlte缓冲液,再加入50μlampurexp磁珠(beckman公司)进行纯化,42μlte缓冲液溶解回收产物。需要说明:反应产物的纯化有多种方式,有磁珠法、柱纯化法、凝胶回收法等,均可用于替换。4.pcr(聚合酶链式反应)引物序列如下:(本实施例中的序列从左到右为5’端至3’端,“//”示修饰基团,“phos”示磷酸化)。引物1序列:/5phos/gaacgacatggctacga(seqidno:3);引物2序列:tgtgagccaaggagttg(seqidno:4)。按下表5配制体系。表5将上步骤40μl回收产物,加入到以上体系中,混匀后按下表条件进行反应,条件如表6。表6反应完成后使用100μlampurexp磁珠进行纯化,30μlte缓冲液溶解回收产物。取1μl回收产物,用qubitdsdnahs分析试剂盒(invitrogen公司)定量产物浓度,进行下一步反应。5.单链环化:将16个带有不同标签序列的扩增产物各取10ng进行混合,用te配置成48μl体系,加入5μl20μm介导片段,混匀。将反应体系置于95℃孵育3min,立马置于冰上。其中介导片段具有相应互补序列用于连接单链两端,其序列如下:(本实施例中的序列从左到右为5’端至3’端)。gccatgtcgttctgtgagccaagg(seqidno:5)。配制如下表7的反应体系:表7组分用量10×ta缓冲液(epicentre公司)6μl三磷酸腺苷(100mm)0.6μlt4dna连接酶(快速)(600u/μl)(enzymatics公司)0.2μl总体积6.8μl将表7反应体系加入纯化的产物中,混匀,置于37℃孵育30min。6.根据bgiseq-50/500/1000的血浆文库构建要求进行文库构建,构建好的文库经浓度检测合格后利用bgiseq-50/500/1000测序仪测序。1)测序取构建的单链环状dna文库进行dna纳米球制备、bgiseq-50/500/1000上机测序。测序过程严格按照bgiseq-50/500/1000的标准操作流程进行上机操作。2)数据分析通过对测序得到的序列,对样本的染色体数目进行分析,详细步骤如下:a)对测试样本计算相对测序序列数:参考唯一比对序列统计参考唯一比对序列的数目,将人类基因组参考序列按染色体进行划分,统计落在每个染色体上的实际测序序列数rij,其中下标i和j分别代表染色体编号和样本编号;b)数据标准化:计算测试样本中多条常染色体的测序序列总数nj,则待测样本每条染色体的相对百分数为rij其中:rij=rij/nj;c)z-score统计:d)染色体数目异常判定:令z-score服从标准正态分布,取0.001与0.999处的分位点(即-3与3)作为判定染色体数目异常的阈值。当某条染色体z-score大于3时为该染色体数目增加,当z-score小于-3时,为染色体数目减少。表8示出了16例胎儿染色体三倍性变异检测样本信息及检测结果。表8实施例2:对48例孕妇血浆进行pcr-free文库构建1.样品收集及处理:取孕妇外周血2ml,1600g,4℃离心10分钟,将血细胞和血浆分开,血浆再以16000g,4℃离心10分钟,进一步去除残留的白细胞。孕妇血浆用qiaampcirculatingnucleicacidkit(qiagen)提取血浆dna,最后将dna溶于40μlte溶液。2.末端修复-加datp:按下表9配置体系。表9组分用量10×t4多聚核苷酸激酶缓冲液(enzymatics公司)5μlt4多聚核苷酸激酶(enzymatics公司)0.4μlklenow大片段(5u/μl)(enzymatics公司)0.2μl脱氧核糖核酸混合液(每种25mm)(enzymatics公司)0.4μltaqdna聚合酶(5u/μl)(takara公司)0.2μl腺苷酸脱氧核糖核酸(100mm)(enzymatics公司)1μlt4dna聚合酶(3u/μl)(enzymatics公司)0.4μl无酶纯水2.4μl总体积10μl将10μl反应液加入均一化至40μl的dna溶液中,混匀,置于37℃孵育30min;65℃孵育15min,降温至4℃。3.接头连接:本方案中使用的接头序列如下(本实施例中的序列从左到右为5’端至3’端,“//”示修饰基团,“phos”示磷酸化,b10示10个碱基的标签序列。ideoxyu示尿嘧啶核苷酸)。长链:/5phos/agtcggaggccaagcggtcttaggaagacaa(b10)caactccttggctcaca(seqidno:1)。短链:agccaaggagt/ideoxyu/gaacgacatggctacgatccgactt(seqidno:6)。接头混合液(10μm)按以下表10配方配制:表10组分用量长链(100μm)40μl短链(100μm)40μl氯化钠(5m)4μl三羟甲基氨基甲烷-盐酸(1m,ph7.8)4μl乙二胺四乙酸二钠(2mm)20μl无酶纯水292μl总体积400.0μl将1μl配制好的接头混合液(10μm)加入上一步骤产物中,充分混匀。按下表11配置体系。表11组分用量三磷酸腺苷(100mm)0.8μl连接酶(600u/μl)(enzymatics公司)1.6μl10×t4多聚核苷酸激酶缓冲液(enzymatics公司)3μl聚乙二醇8000(50%)12μl无酶纯水11.6μl总体积29μl将连接反应体系与接头和产物的混合液混匀,置于23℃孵育60min。反应完之后,将24个带不同标签序列的样品混合在一起,再加入一倍体积的ampurexp磁珠(beckman公司)进行纯化,55μlte缓冲液溶解回收产物。4.user酶切按下表12配置体系。表12组分用量10×ta缓冲液(epicentre公司)6μluser酶切(1u/μl)(neb公司)1μl总体积7μl将7μl反应液加入上一步骤产物中,混匀,置于37℃孵育15min;降温至4℃。5.单链环化:配制反应体系1:将5μl20μm介导片段加入上一步骤产物中,混匀。将反应体系置于95℃孵育3min,立马置于冰上。其中介导片段具有相应互补序列用于连接单链两端,其序列如下:(本实施例中的序列从左到右为5’端至3’端)。gccatgtcgttctgtgagccaagg(seqidno:5)。配制表13的反应体系2。表13组分用量三磷酸腺苷(100mm)0.6μlt4dna连接酶(600u/μl)(enzymatics公司)0.2μl总体积0.8μl将反应体系2加入反应体系1中,混匀,置于37℃孵育30min。6.测序取构建的单链环状dna文库进行dna纳米球制备、bgiseq-50/500/1000上机测序。测序过程严格按照的bgiseq-50/500/1000标准操作流程进行上机操作及数据分析。表14示出了本实施例的pcrfree文库dna纳米球的浓度,符合bgiseq平台上机测序的要求。表14文库浓度(ng/μl)pcrfree161.3pcrfree260.1图4示出了本实施例的pcrfree文库dna纳米球电泳结果,样品大部分都在胶孔里,无法电泳出来,符合dna纳米球在聚丙烯酰胺凝胶电泳时无法跑出胶孔的特点。实施例3:对1例超声打断的基因组进行文库构建1.dna样本片段化:取炎黄细胞系dna500ng,进行超声波打断,用1.5倍ampurexp磁珠(beckman公司)进行纯化,22μlte缓冲液溶解回收产物。2.末端修复-加datp:按下表15配置体系。表15将30μl反应液加入均一化至20μl的dna溶液中,混匀,置于37℃孵育30min;65℃孵育15min,降温至4℃。3.接头连接:本方案中使用的接头序列如下(本实施例中的序列从左到右为5’端至3’端,“//”示修饰基团,“phos”示磷酸化,b10示10个碱基的标签序列)。长链:/5phos/agtcggaggccaagcggtcttaggaagacaa(b10)caactccttggctcaca(seqidno:1)。短链:ttgtcttcctaaggaacgacatggctacgatccgactt(seqidno:2)。接头混合液(10μm)按以下表16配方配制:表16组分用量长链(100μm)40μl短链(100μm)40μl氯化钠(5m)4μl三羟甲基氨基甲烷-盐酸(1m,ph7.8)4μl乙二胺四乙酸二钠(2mm)20μl无酶纯水292μl总体积400.0μl将5μl配制好的接头混合液(10μm)加入上一步骤产物中,充分混匀。按下表17配置体系。表17将连接反应体系与接头和产物的混合液混匀,置于23℃孵育30min。反应完之后,加入20μlte缓冲液,再加入50μlampurexp磁珠(beckman公司)进行纯化,42μlte缓冲液溶解回收产物。4.pcr(聚合酶链式反应)引物序列如下:(本实施例中的序列从左到右为5’端至3’端,“//”示修饰基团,“phos”示磷酸化)。引物1序列:/5phos/gaacgacatggctacga(seqidno:3);引物2序列:tgtgagccaaggagttg(seqidno:4)。按下表18配制体系。表18组分用量10*pfx缓冲液(thermoscience公司)10μl脱氧核糖核酸缓和液(25mm)(enzymatics公司)1.6μl硫酸镁(50mm)(thermoscience公司)4μl引物1(25μm)2.5μl引物2(25μm)2.5μlpfxdna聚合酶(2.5u/μl)(thermoscience公司)2μl无酶纯水57.4μl总体积80μl将上步骤40μl回收产物,加入到以上体系中,混匀后按下表19的条件进行反应。表19反应完成后使用100μlampurexp磁珠进行纯化,30μlte缓冲液溶解回收产物。取1μl回收产物,用qubitdsdnahs分析试剂盒(invitrogen公司)定量产物浓度。进行下一步反应。5.单链环化:配制反应体系1:取扩增产物320ng进行混合,用te配置成65μl体系,加入5μl20μm介导片段,混匀。将反应体系置于95℃孵育3min,立马置于冰上。其中介导片段具有相应互补序列用于连接单链两端,其序列如下:(本实施例中的序列从左到右为5’端至3’端)。gccatgtcgttctgtgagccaagg(seqidno:5)。配制表20的反应体系2。表20组分用量10×ta缓冲液(enzymatics公司)12μl三磷酸腺苷(100mm)1.2μlt4dna连接酶(快速)(600u/μl)(enzymatics公司)0.4μl无酶纯水36.4μl总体积50μl将反应体系2加入反应体系1中,混匀,置于37℃孵育30min。6.测序取构建的单链环状dna文库进行dna纳米球制备、bgiseq-50/500/1000上机测序。测序过程严格按照bgiseq-50/500/1000的标准操作流程进行上机操作及数据分析。表21示出了1例基因组样品的pcr和测序结果,pcr产量满足打断文库上机的需求。测序数据产出量以及深度、覆盖度都达到了数据分析的需求。表21图5示出了1例基因组样品的测序深度分布图,测序深度符合泊松分布,测序深度集中在30×左右,符合人基因组重测序的数据需求。图6示出了1例基因组样品的gc含量累积比例,虽然高gc含量的片段的覆盖度有所下降,但覆盖率接近1,说明大部分高gc含量的片段可以被检测到,所有不同gc含量的片段均一化后的覆盖度基本保持水平。实施例4:对8例neb打断酶打断的基因组进行文库构建1.dna样本片段化:取炎黄细胞系dna100ng,用1.2倍ampurexp磁珠(beckman公司)进行纯化(此步骤当dna溶于无edta的溶液或者无酶纯水时不需要实施),18μl无酶纯水溶解回收产物。按下表22配置体系。表22组分用量10xneb打断酶缓冲液v2(neb公司)2μlneb打断酶(neb公司)2μl总体积4μl将4μl反应液加入均一化至16μl的dna中,混匀,置于4℃孵育5min;37℃孵育20min,65℃孵育15min,降温至4℃。反应完之后,再加入20μlampurexp磁珠(beckman公司),混匀,静置5分钟后,取上清,往上清中加入22μlampurexp磁珠(beckman公司)进行纯化,42μlte缓冲液溶解回收产物。2.末端修复-加datp:按下表23配置体系。表23组分用量10xt4多聚核苷酸激酶缓冲液(enzymatics公司)5μlt4多聚核苷酸激酶(enzymatics公司)0.6μl脱氧核糖核酸混合液(每种25mm)(enzymatics公司)0.1μltaqdna聚合酶(5u/μl)(takara公司)0.2μldatp(100mm)(enzymatics公司)0.25μlt4dna聚合酶(3u/μl)(enzymatics公司)1μl无酶纯水5.85μl总体积10μl将10μl反应液加入均一化至40μl的dna溶液中,混匀,置于15℃孵育30min;65℃孵育15min,降温至4℃。3.接头连接:本方案中使用的接头序列如下(本实施例中的序列从左到右为5’端至3’端,“//”示修饰基团,“phos”示磷酸化,b10示10个碱基的标签序列)。长链:/5phos/agtcggaggccaagcggtcttaggaagacaa(b10)caactccttggctcaca(seqidno:1);短链:ttgtcttcctaaggaacgacatggctacgatccgactt(seqidno:2)。接头混合液(10μm)按以下表24配方配制。表24组分用量长链(100μm)40μl短链(100μm)40μl氯化钠(5m)4μl三羟甲基氨基甲烷-盐酸(1m,ph7.8)4μl乙二胺四乙酸二钠(2mm)20μl无酶纯水292μl总体积400.0μl将1μl配制好的接头混合液(10μm)加入上一步骤产物中,充分混匀。按下表25配置体系。表25组分用量三磷酸腺苷(100mm)0.8μl连接酶(600u/μl)(enzymatics公司)1μl10xt4多聚核苷酸激酶缓冲液(enzymatics公司)3μl聚乙二醇8000(50%)12μl无酶纯水12.2μl总体积29μl将连接反应体系与接头和产物的混合液混匀,置于23℃孵育30min。反应完之后,加入20μlte缓冲液,再加入50μlampurexp磁珠(beckman公司)进行纯化,22μlte缓冲液溶解回收产物。4.pcr(聚合酶链式反应)引物序列如下:(本实施例中的序列从左到右为5’端至3’端,“//”示修饰基团,“phos”示磷酸化)。引物1序列:/5phos/gaacgacatggctacga(seqidno:3);引物2序列:tgtgagccaaggagttg(seqidno:4)。按下表26配制体系。表26组分用量10*pfx缓冲液(thermoscience公司)10μl脱氧核糖核酸缓和液(25mm)(enzymatics公司)1.6μl硫酸镁(50mm)(thermoscience公司)4μl引物1(25μm)2.5μl引物2(25μm)2.5μlpfxdna聚合酶(2.5u/μl)(thermoscience公司)2μl无酶纯水67.4μl总体积90μl取上步骤10μl回收产物,加入到以上体系中,混匀后按下表27条件进行反应。表27反应完成后使用100μlampurexp磁珠进行纯化,22μlte缓冲液溶解回收产物。取1μl回收产物,用qubitdsdnahs分析试剂盒(invitrogen公司)定量产物浓度。进行下一步反应。5.单链环化:配制反应体系1:将8个带有不同标签序列的扩增产物各取20ng进行混合,用te配置成48μl体系,加入5μl20μm介导片段,混匀。将反应体系置于95℃孵育3min,立马置于冰上。其中介导片段具有相应互补序列用于连接单链两端,其序列如下:(本实施例中的序列从左到右为5’端至3’端)。gccatgtcgttctgtgagccaagg(seqidno:5)。配制表28的反应体系2。表28组分用量10×ta缓冲液(enzymatics公司)6μl三磷酸腺苷(100mm)0.6μlt4dna连接酶(快速)(600u/μl)(enzymatics公司)0.2μl总体积6.8μl将反应体系2加入反应体系1中,混匀,置于37℃孵育30min。6.测序取构建的单链环状dna文库进行dna纳米球制备、bgiseq-50/500/1000上机测序。测序过程严格按照bgiseq-50/500/1000的标准操作流程进行上机操作及数据分析。表29示出了8例酶打断基因组样品的pcr产量,8个样品的产量都达到了bgiseq-50/500/1000测序平台的上机要求。表29样品浓度(ng/μl)总量(ng)15.96119.226.2512535.68113.646.41128.256.78135.665.87117.476.69133.886.4128图7示出了8例酶打断基因组样品的pcr产物电泳图,pcr产物的片段大小在250bp左右,符合bgiseq-50/500/1000测序平台的上机要求。以上内容是结合具体的实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属
技术领域:
的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。sequencelisting<110>深圳华大基因科技有限公司<120>一种用于无创产前检测的文库构建方法及试剂盒<130>15i22173<160>6<170>patentinversion3.3<210>1<211>58<212>dna<213>人工序列<220><221>标签序列<222>(32)..(41)<220><221>misc_feature<222>(32)..(41)<223>nisa,c,g,ort<400>1agtcggaggccaagcggtcttaggaagacaannnnnnnnnncaactccttggctcaca58<210>2<211>43<212>dna<213>人工序列<400>2ttgtcttcctaaggaacgacatggctacgatccgactt38<210>3<211>17<212>dna<213>人工序列<400>3gaacgacatggctacga17<210>4<211>17<212>dna<213>人工序列<400>4tgtgagccaaggagttg17<210>5<211>24<212>dna<213>人工序列<400>5gccatgtcgttctgtgagccaagg24<210>6<211>37<212>dna<213>人工序列<220><221>尿嘧啶核苷酸<222>(12)..(12)<400>6agccaaggagttgaacgacatggctacgatccgactt37当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1