一种三元苯并噁嗪中间体、制备方法及其聚合物与流程
本发明涉及高性能热固性树脂制备技术领域,具体涉及一种三元苯并噁嗪中间体、制备方法及其聚合物。
背景技术:
苯并噁嗪是一类由酚类化合物、胺类化合物和醛类化合物经缩合反应合成的六元杂环化合物,最初在mannich反应中发现,在加热或催化剂作用下开环聚合,生成含氮且类似酚醛树脂的网状结构。苯并噁嗪树脂作为一种新型的酚醛树脂,其不仅具有传统酚醛树脂的热性能、力学性能、电学性能,且固化成型时收缩率小,无小分子释放,吸水率低,稳定性好,玻璃化转变温度高,加之单体原料来源广泛,分子设计灵活,因而受到广泛关注。
中国发明专利cn104356084a向反应物胺类化合物、甲醛水溶液及单酚或二元酚类化合物中加入少量溶剂,升温反应后冷却结成块状物即为苯并噁嗪中间体,并称该方法用少量溶剂代替悬浮剂,结合了悬浮反应法、熔融反应法及溶剂反应法的优点,该方法制备的苯并噁嗪中间体未经纯化即直接使用,产物中不可避免地含有杂质,势必影响固化物的性能,尤其是热性能和加工特性,其公开的按此方法制备的双酚a型苯并噁嗪中间体固化后失重15%对应的温度仅为356℃,耐热性并不理想。
中国发明专利cn101220153a以一元胺、含磷二元酚和甲醛为反应原料,在苯并噁嗪中间体中引入含磷化合物,合成了含磷阻燃性苯并噁嗪中间体,经固化得到含磷阻燃性苯并噁嗪树脂。该方法利用含磷二元酚代替传统的双酚作为反应原料,成本较高;固化得到的树脂在氮气和空气中失重5%对应的温度分别为287℃,293℃,耐热性能并不优异,在氮气中800℃的残炭率为47%,极限氧指数为37.9。如何提高苯并噁嗪树脂的树脂玻璃化转变温度(tg)、热性能、力学性能和阻燃性能,成为本领域亟待解决的技术问题。
技术实现要素:
本发明的目的在于针对现有技术的不足,提供一种三元苯并噁嗪中间体、制备方法及其聚合物,该三元苯并噁嗪中间体加工性良好,固化得到的聚合物综合性能优异。
本发明解决上述技术问题所提供的技术方案为,一种三元苯并噁嗪中间体,结构式如下:
其中,r1~r6各自独立地选自氢或c1~c4烷基;r选自芳香族基团、脂肪族基团或脂环族基团。
进一步,所述芳香族基团优选为苯基、邻甲基苯基、间甲基苯基、对甲基苯基或间乙炔基苯基;所述脂肪族基团优选为烯丙基;所述脂环族基团优选为环己基。
本发明还提供一种三元苯并噁嗪中间体的制备方法,包括如下步骤:
1)向反应器中加入多聚甲醛、一元伯胺和反应溶剂,0~10℃搅拌反应;所述一元伯胺为芳香胺、脂肪胺、脂环胺中的至少一种;
2)继续加入式ⅱ化合物,升温至60~90℃反应,除去反应溶剂,得到剩余物;
其中,r1~r6各自独立地选自氢或c1~c4烷基,且酚羟基邻位不能同时为烷基;
3)将剩余物溶解在溶解溶剂中,并倾倒至碱液中,析出固体,过滤,洗涤,得到三元苯并噁嗪中间体。
所涉及的反应方程式如下:
其中,r1~r6如上所述;rnh2为一元伯胺;(ch2o)n为多聚甲醛。
上述技术方案中,选用三元酚类化合物(式ⅱ化合物)作为起始原料,向终产物三元苯并噁嗪中间体(式ⅰ化合物)中引入烷基柔性链段,同时增加单分子中苯并噁嗪结构的数量,从而降低中间体的熔点并且增加了苯并噁嗪树脂的交联度,能够显著拓宽加工窗口和提高树脂玻璃化转变温度(tg)、热性能、力学性能和阻燃性能。
所述步骤1)和2)中反应为避光反应。由于一元伯胺见光易变质,故应采取避光反应,减少副产物的生成,同时提高收率。
优选的,所述步骤1)中避光反应时间为10~60min。
优选的,所述步骤2)中避光反应时间为2~10h。所述除去反应溶剂采用旋蒸。
所述步骤1)中多聚甲醛与一元伯胺按醛基和氨基的摩尔配比为2:0.6~1.2投料。
所述步骤2)中式ⅱ化合物与一元伯胺按摩尔配比为1:2.5~3.3投料。
所述步骤2)中r1~r6均为氢。所述式ⅱ化合物优选为1,1,1-三(4-羟基苯基)乙烷(thpe)。
所述步骤1)中芳香胺优选为:苯胺、邻甲基苯胺、间甲基苯胺、对甲基苯胺、间乙炔基苯胺;脂肪胺优选为:烯丙基胺;脂环胺优选为:环己胺。
进一步优选,所述步骤1)中一元伯胺为苯胺。
所述步骤1)中反应溶剂选自二氧六环、甲苯、丁酮、丙酮、乙醇、甲醇、乙酸乙酯中的至少一种;所述步骤3)中溶解溶剂选自n,n-二甲基乙酰胺、n,n-二甲基甲酰胺、n-甲基吡咯烷酮中的至少一种。
所述步骤3)中碱液选自氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸氢钠和碳酸氢钾中的一种。
优选的,所述步骤3)中碱液的浓度为0.1~1mol/l。
本发明还提供一种三元苯并噁嗪聚合物,由上述的三元苯并噁嗪中间体经加热固化制得。
同现有技术相比,本发明的有益效果体现在:
(1)本发明制备的三元苯并噁嗪中间体增加了单分子中苯并噁嗪结构的数量,从而增加开环固化反应点和树脂交联度,能够显著提高树脂tg、热性能、力学性能、阻燃性能和残炭率。
(2)本发明制备的三元苯并噁嗪中间体引入烷基柔性链段,降低其熔点,显著拓宽加工窗口,大大拓宽了加工的适宜范围,更有利于加工成型,且固化放热效应发生范围较大,使放热不集中,有利于降低材料的内应力,提高材料强度。
(3)本发明反应条件温和,步骤少,溶剂可回收利用,成本低,副产物少,产率可达87%,适于工业化生产。
附图说明
图1为三元苯并噁嗪中间体的结构式;
图2为实施例1制备的三元苯并噁嗪中间体的ftir谱图;
图3为实施例1制备的三元苯并噁嗪中间体的dsc谱图;
图4为实施例1制备的三元苯并噁嗪中间体经固化得到的聚合物tga谱图。
具体实施方式
下面结合具体的实施例和附图对本发明作进一步说明。
三元苯并噁嗪中间体的结构式如图1所示。
实施例1
向反应器中依次加入18.00g(0.60mol,按甲醛单体计算,下同)多聚甲醛、27.90g(0.30mol)苯胺和200ml丁酮,5℃避光搅拌反应30min后加入30.60g(0.10mol)式ⅱ1化合物(r1~r6均为氢,具体为thpe),升温至75℃避光反应6h,除去反应溶剂丁酮,加150mln,n-二甲基乙酰胺溶解剩余物后倾倒至2000ml0.5mol/l氢氧化钠溶液中,析出固体,过滤,水洗,60℃真空干燥8h得三元苯并噁嗪中间体粉末产品,产率89%。
式ⅱ1化合物(thpe)结构式如下:
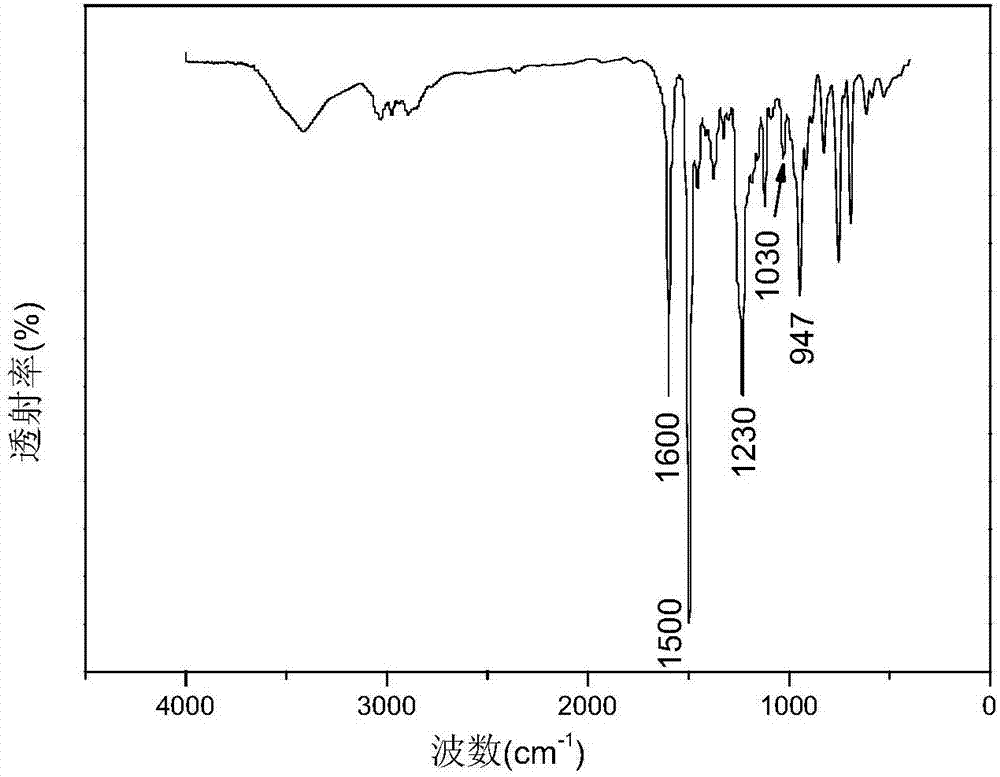
针对实施例1所制备的三元苯并噁嗪中间体进行傅里叶红外光谱测试,结果如图2所示,947cm-1处信号为噁嗪环特征峰;1030cm-1和1230cm-1处信号分别为噁嗪环c-o-c对称与非对称伸缩振动峰;1500cm-1、1600cm-1处信号为苯环骨架振动峰。
将三元苯并噁嗪中间体粉末置于铝制长方形模具内,于马弗炉中梯度升温,分段固化得到三元苯并噁嗪聚合物,固化程序为:140℃/1h,160℃/1h,180℃/2h,200℃/1h,220℃/3h,240℃/2h,得三元苯并噁嗪聚合物。
实施例2~14
重复实施例1的过程,改变反应条件,得到表1所列结果:
表1实施例2~23的反应条件及产率
注:第3列为thpe与rnh2的摩尔比;第4列为醛基与氨基的摩尔比;第6列为第i阶段温度:0~10℃,时间:10~60min;第7列为第ii阶段温度:60~90℃,时间:2~10h。
分别将实施例2~14所制备得到的三元苯并噁嗪中间体粉末置于铝制长方形模具内,于马弗炉中梯度升温,分段固化得到三元苯并噁嗪聚合物,固化程序为:140℃/1h,160℃/1h,180℃/2h,200℃/1h,220℃/3h,240℃/2h,得三元苯并噁嗪聚合物。
实施例15
向反应器中依次加入18.00g(0.60mol)多聚甲醛、27.90g(0.30mol)苯胺和200ml丁酮,0℃避光搅拌反应60min后加入34.80g(0.10mol)式ⅱ2化合物,升温至75℃避光反应6h,除去反应溶剂丁酮,加150mln,n-二甲基乙酰胺溶解剩余物后倾倒至1500ml1mol/l碳酸钠溶液中,析出固体,过滤,水洗,60℃真空干燥8h得三元苯并噁嗪中间体粉末产品,产率88%。
式ⅱ2化合物结构式如下:
将苯并噁嗪中间体粉末置于铝制长方形模具内,于马弗炉中梯度升温,分段固化得到三元苯并噁嗪聚合物,固化程序为:140℃/1h,160℃/1h,180℃/2h,200℃/1h,220℃/3h,240℃/2h,得三元苯并噁嗪聚合物。
实施例16
向反应器中依次加入18.00g(0.60mol)多聚甲醛、27.90g(0.30mol)苯胺和250ml乙酸乙酯,0℃避光搅拌反应60min后加入43.20g(0.10mol)式ⅱ3化合物,升温至80℃避光反应5h,除去反应溶剂乙酸乙酯,加200mln,n-二甲基甲酰胺溶解剩余物后倾倒至2000ml0.5mol/l氢氧化钠溶液中,析出固体,过滤,水洗,60℃真空干燥8h得三元苯并噁嗪中间体粉末产品,产率87%。
式ⅱ3化合物结构式如下:
将苯并噁嗪中间体粉末置于铝制长方形模具内,于马弗炉中梯度升温,分段固化得到三元苯并噁嗪聚合物,固化程序为:140℃/1h,160℃/1h,180℃/2h,200℃/1h,220℃/3h,240℃/2h,得三元苯并噁嗪聚合物。
对比例1
向反应器中依次加入18.00g(0.60mol)多聚甲醛、27.90g(0.30mol)苯胺和200ml丁酮,5℃避光搅拌反应30min后加入34.20g(0.15mol)双酚a,升温至75℃避光反应6h,除去反应溶剂丁酮,加150mln,n-二甲基乙酰胺溶解剩余物后倾倒至2000ml0.5mol/l氢氧化钠溶液中,析出固体,过滤,水洗,60℃真空干燥8h得双酚a型苯并噁嗪中间体粉末产品,产率86%。
将苯并噁嗪中间体粉末置于铝制长方形模具内,于马弗炉中梯度升温,分段固化得到聚合物,固化程序为:140℃/1h,160℃/1h,180℃/2h,200℃/1h,220℃/3h,240℃/2h,得双酚a型苯并噁嗪聚合物。
性能测试
1、针对实施例1、10和15所制备的三元苯并噁嗪中间体进行示差扫描量热测试,其中实施例1结果如图3所示,具体数据如表2所示。
表2、三元苯并噁嗪中间体dsc数据
依表2可知,实施例1中三元苯并噁嗪中间体的熔点低至65℃,开环聚合起始温度为152℃,加工窗口为87℃,大大拓宽了加工的适宜范围,更有利于加工成型,且固化放热效应发生在138℃范围内,使放热不集中,有利于降低材料的内应力,提高材料强度。
实施例10中三元苯并噁嗪中间体的熔点低至63℃,开环聚合起始温度为156℃,加工窗口为93℃,固化放热效应发生在131℃范围内。
实施例15中三元苯并噁嗪中间体的熔点低至61℃,开环聚合起始温度为151℃,加工窗口为90℃,固化放热效应发生在128℃范围内。
2、针对实施例1、10、15和对比例1中的聚合物进行热失重测试,其中实施例1结果如图4所示。具体数据(tg、热性能和极限氧指数)如表3所示。
表3、实施例与对比例制备的苯并噁嗪聚合物的tg、热性能与极限氧指数比较
依表3可知,实施例1、10和15中的聚合物的tg、热(氧)稳定性、残炭率和阻燃性能均优于对比例1。相较于对比例1中双酚a型二元苯并噁嗪中间体及其聚合物,实施例中三元苯并噁嗪中间体增加了单分子中苯并噁嗪结构的数量,从而增加开环固化反应点和树脂交联度,能够显著提高聚合物tg、热(氧)稳定性、残炭率和阻燃性能。
- 还没有人留言评论。精彩留言会获得点赞!