一种硫酸氢氯吡格雷晶型II制备方法与流程
本发明涉及一种硫酸氢氯吡格雷晶型ⅱ制备方法,属于化学制药领域。
背景技术:
心脑血管血栓性疾病是一种严重影响人类健康的重大疾病,然而抑制血小板聚集是治疗该疾病的有效途径。硫酸氢氯吡格雷(clopidogrelhydrogensulfate,如下图)是新一代的血小板聚集抑制剂,化学名为(s)-α-(2-氯苯基)-6,7-二氢噻吩并[3,2-c]吡啶-5(4h)-乙酸甲酯硫酸氢盐,商品名为波立维(plavix)。该产品由法国赛诺菲研发,1998年3月率先在美国上市,2001年8月进入中国市场。该药具有疗效强、安全性高、副作用小、费用低等优点,目前已成为治疗血栓性疾病的一线药物。
目前,国内的硫酸氢氯吡格雷制剂产品主要有赛诺菲公司的波立维(plavix)(晶型ⅱ)和深圳信立泰药业股份有限公司的泰嘉(晶型ⅰ)。
ep0281459a最先描述了氯吡格雷硫酸氢盐及其制备方法,但没有提及晶型。
cn1305483a公开了氯吡格雷硫酸氢盐的两种药用晶型—晶型ⅰ和晶型ⅱ。晶型ⅰ为亚稳态晶型,具有较高的溶解度和生物利用度,但是其对湿热不稳定,容易降解产生杂质a(如下图)。晶型ⅱ为热力学稳定性晶型,但溶解度和生物利用度比晶型ⅰ差。该专利报道晶型ⅰ熔点为184±3℃,晶型ⅱ熔点为176±3℃。
专利报道了多种晶型ⅱ的制备方法,
专利wo099/65915,报道以丙酮为溶剂制备晶型ⅱ的方法,此方法需要低温,并且收率低,不适合工业化生产。
专利wo2003/051362,报道以乙酸乙酯为溶剂制备晶型ⅱ的方法,此方法需要回流,并且晶型ⅱ产品结成硬块,不利于工业化生产。
专利cn101643476a,报道将硫酸氢氯吡格雷晶型ⅰ溶解于低级醇(甲醇、乙醇、异丙醇),然后加入反溶剂,制备硫酸氢氯吡格雷晶型ⅱ。此方法需要先制备晶型ⅰ,然后在转晶,操作繁琐,并且收率只有80%左右,收率较低。
由以上专利方法可以看出,同样是以乙酸乙酯为溶剂改变不同的条件即可以制备出晶型ⅰ也可以制备出晶型ⅱ,以上都说明没有很稳定的制备硫酸氢氯吡格雷晶型ⅱ的工业化方法。因此,急需开发一种能得到高纯度的硫酸氢氯比格雷晶型ⅱ产品,收率又高,对环境友好又适合工业化大生产的制备方法。
我们经过大量的实验工作,最后惊奇的发现,在乙酸乙酯为溶剂制备晶型ⅱ时,乙酸乙酯中残留的低级醇(甲醇、乙醇、异丙醇)的含量,对晶型的制备起着至关重要的作用,这一意外发现是难以预见的。具体实验结果如表(1)所示,可见当乙酸乙酯中加入0.5%的甲醇或乙醇或异丙醇,就会导致产生硫酸氢氯吡格雷晶型ⅱ;我们将得到的结果,应用乙酸甲酯,乙酸异丙酯,甲酸乙酯时,同样会得到上面结果,就是酯类溶剂中含有微量的低级醇,就会导致制备硫酸氢氯吡格雷时得到晶型ⅱ,并且此方法收率高,操作简单;
表1不同质量的乙酸乙酯对制备硫酸氢氯吡格雷的影响。
我们将自制的硫酸氢氯吡格雷晶型ⅱ,进行加速考核,考核结果见表(2),结果证明自制的硫酸氢氯吡格雷晶型ⅱ稳定性好,加速6月后,杂质a小于0.15%,并且晶型ⅱ没有发生改变,仍为晶型ⅱ符合药典标准;
表2自制硫酸氢氯吡格雷加速稳定性实验杂质a变化结果(40℃,湿度75%)
技术实现要素:
本发明所要解决的技术问题为提供一种简单的、高收率的、环境友好的制备稳定的硫酸氢氯吡格雷晶型ⅱ方法。
本发明提供的技术方案,一种ⅱ型氯吡格雷硫酸氢盐的制备方法,包括如下步骤:
步骤1:常温条件下,将氯吡格雷盐与有机溶剂a混合,加入水,搅拌条件下,向溶剂中加入缚酸剂或缚酸剂的水溶液,控制水层ph值在7~8,保留有机相,生成氯吡格雷游离碱,静置分层,水相用有机溶剂a萃取,合并有机相,有机相水洗一次,得到有机相,减压回收溶剂a,得油状物氯吡格雷游离碱(式1);检测油状物氯吡格雷游离碱水份,如果水份≧0.5%,则向油状物氯吡格雷游离碱中加入有机溶剂a搅拌溶解,继续减压浓缩掉溶剂a,得油状物氯吡格雷游离碱,检测油状物氯吡格雷游离碱水份,如果水份≦0.5%,停止浓缩,得到油状物氯吡格雷游离碱(式1);
步骤2:干燥气体或惰性气体保护下,在浓缩后得到的氯吡格雷游离碱中加入有机溶剂b和低级醇混合物,搅拌,使氯吡格雷游离碱完全溶解,加入或不加入硫酸氢氯吡格雷晶型ⅱ晶种条件下,一定温度下,向反应液中滴加浓硫酸,滴完后,于此温度继续搅拌3~10小时,过滤,得硫酸氢氯吡格雷(式2)晶型ⅱ湿品;
步骤3:干燥气体或惰性气体保护下,将硫酸氢氯吡格雷晶型ⅱ湿品,加于有机溶剂c中,搅拌2~5小时,过滤,得固体于60℃~90℃进行干燥,得到硫酸氢氯吡格雷(式2)晶型ⅱ;
其中步骤(1)中所述有机溶剂a选自二氯甲烷、二氯乙烷、三氯甲烷、异丙醚、甲基叔丁基醚中的一种或几种;所述缚酸剂选自氨水、碳酸钠、碳酸钾、碳酸氢钠或碳酸氢钾中的一种或几种;
其中步骤(1)中氯吡格雷盐为氯吡格雷樟脑磺酸盐、氯吡格雷硫酸盐、氯吡格雷盐酸盐、氯吡格雷氢溴酸盐的一种或几种的混合物;
其中步骤(1)中缚酸剂包括碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾,其与氯吡格雷盐摩尔比为1.2~1.5∶1;
其中步骤(1)中油状物氯吡格雷碱(式1)水份≦0.5%,优选油状物氯吡格雷碱(式1)水份≦0.3%,最优选油状物氯吡格雷碱(式1)水份≦0.1%;
其中步骤(1)中有机溶剂a的加入量为氯吡格雷盐质量的5~10倍;
其中步骤(2)中所述有机溶剂b为乙酸乙酯、乙酸甲酯、甲酸乙酯、乙酸异丙酯中的一种或几种;
其中步骤(2)中所述有机溶剂b中水份含量≦0.1%;
其中步骤(2)中所述低级醇选自甲醇、乙醇或异丙醇;
其中步骤(2)中有机溶剂b的加入量为氯吡格雷盐质量的5~10倍;
其中步骤(2)中有机溶剂c的加入量为氯吡格雷盐质量的5~10倍;
其中步骤(2)中有机溶剂b和低级醇,的质量比为10:1~100:0.5,优选20:1~100:1;
其中步骤(2)中使用的浓硫酸和氯吡格雷游离碱(式1)的摩尔比为0.95~1.05:1,优选0.98~1.02:1,最优选1:1;
其中步骤(2)中滴加浓硫酸温度为-5℃~40℃,优选温度为10℃~25℃;
其中步骤(3)中所述有机溶剂c选自异丙醚、甲基叔丁基醚、乙酸乙酯、乙酸甲酯、甲酸乙酯、乙酸异丙酯中的一种或几种。
附图说明
图1:自制硫酸氢氯吡格雷晶型ⅰxrpd典型图谱
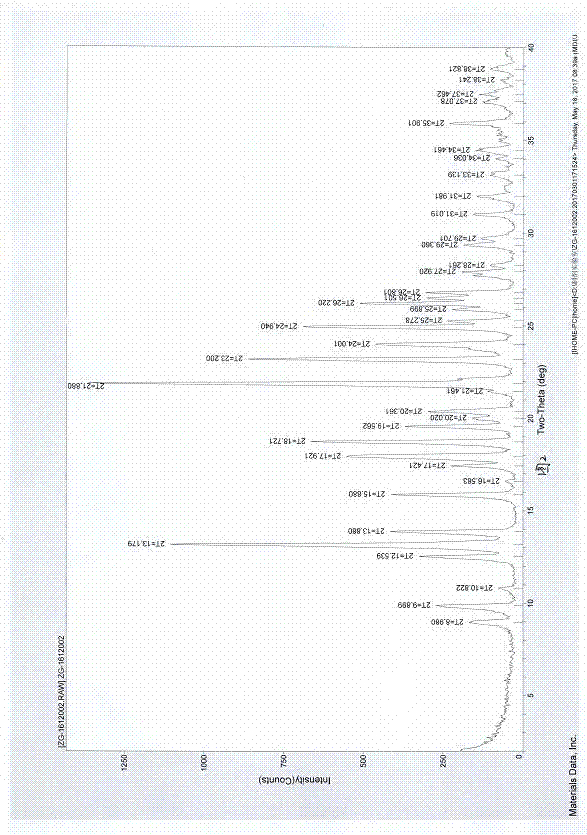
图2:自制硫酸氢氯吡格雷晶型ⅱxrpd典型图谱。
具体实施方式
实施例1
常温条件下,将氯吡格雷樟脑磺酸盐500g,氯仿4.0kg,4%碳酸钠水溶液(100g碳酸钠2.5kg水)加于反应瓶中,搅拌30min,分液,水层ph为7-8,水层用2.0kg氯仿萃取一次,合并有机层,有机层用2kg水洗一次,有机相减压浓缩至干,得油状物氯吡格雷游离碱335g。(经检测油状物含氯吡格雷游离碱80.5%,水分2.0%)收率:93%(折溶剂后)。
实施例2
常温条件下,将氯吡格雷樟脑磺酸盐500g,异丙醚2.5kg,4%碳酸钠水溶液(100g碳酸钠2.5kg水)加于反应瓶中,搅拌30min,分液,水层ph为7-8,水层用0.5kg异丙醚萃取一次,合并有机层,有机层用2kg水洗一次,有机相减压浓缩至干,得油状物氯吡格雷游离碱312g。(经检测油状物含氯吡格雷游离碱90%,水分1.0%)收率:97%(折溶剂后)。
实施例3
常温条件下,将氯吡格雷樟脑磺酸盐500g,甲基叔丁基醚2.5kg,4%碳酸钠水溶液(100g碳酸钠2.5kg水)加于反应瓶中,搅拌30min,分液,水层ph为7-8,水层用0.5kg甲基叔丁基醚萃取一次,合并有机层,有机层用2kg水洗一次,有机相减压浓缩至干,得油状物氯吡格雷游离碱322g。(经检测油状物含氯吡格雷游离碱90%,水分0.5%)收率:100%(折溶剂后)。
实施例4
常温条件下,将氯吡格雷硫酸盐5kg,二氯甲烷50kg,4%碳酸钠水溶液(1kg碳酸钠25kg水)加于反应瓶中,搅拌30min,分液,水层ph为7-8,水层用5kg二氯甲烷萃取一次,合并有机层,有机层用20kg水洗一次,有机相减压浓缩至干,得油状物氯吡格雷游离碱4kg,向油状物中加入32kg二氯甲烷,搅拌溶解,减压浓缩至干,得油状物4.1kg。(经检测油状物含氯吡格雷游离碱88%,水分0.3%)收率:92%(折溶剂后)。
实施例5
氮气保护下,向50g(125mmol)氯吡格雷游离碱(实施例1制备),加入乙酸乙酯500g,搅拌溶解,温度将至10℃,滴加98%浓硫酸12.5g(125mmol),滴完后,于10℃~25℃,搅拌10小时,反应分层下层油状物。
实施例6
氮气保护下,向50g(140mmol)氯吡格雷游离碱(实施例2制备),加入乙酸乙酯500g,搅拌溶解,温度将至-5℃,滴加98%浓硫酸14.0g(140mmol),滴完后,于25℃~30℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
氮气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸乙酯中,于40℃搅拌5小时,过滤,得固体于60℃进行真空干燥24h,得到硫酸氢氯吡格雷晶型i,58.2g,收率99%。(水份:0.10%)。
实施例7
氮气保护下,向50g(140mmol)氯吡格雷游离碱(实施例3制备),加入乙酸乙酯500g,搅拌溶解,温度将至10℃,滴加98%浓硫酸13.3g(133mmol),滴完后,于35℃~40℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
氮气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸乙酯中,于40℃搅拌5小时,过滤,得固体于60℃进行真空干燥24h,得到硫酸氢氯吡格雷晶型i,55.8g,收率95%。(水份:0.03%)。
实施例8
氮气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸乙酯500g,搅拌溶解,温度将至-5℃,加入硫酸氢氯吡格雷晶型i晶种0.5g,滴加98%浓硫酸14.4g(144mmol),滴完后,于30℃~40℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
氮气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸乙酯中,于40℃搅拌5小时,过滤,得固体于85℃进行真空干燥15h,得到硫酸氢氯吡格雷晶型i,52.9g,收率92%。(水份:0.01%)。
实施例9
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸乙酯500g,甲醇2.5g,搅拌溶解,温度将至15℃,滴加98%浓硫酸13.7g(137mmol),滴完后,于15℃~20℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸乙酯中,于40℃搅拌5小时,过滤,得固体于85℃进行真空干燥15h,得到硫酸氢氯吡格雷晶型ⅱ,51.7g,收率90%。(水份:0.01%)。
实施例10
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸乙酯500g,甲醇5.0g,搅拌溶解,温度降至15℃,滴加98%浓硫酸13.7g(137mmol),滴完后,于15℃~20℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸乙酯500ml中,于40℃搅拌5小时,过滤,得固体于85℃进行真空干燥15h,得到硫酸氢氯吡格雷晶型ⅱ,51.7g,收率90%。(水份:0.01%)。
实施例11
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸乙酯250g,甲醇12.5g,搅拌溶解,温度降至15℃,滴加98%浓硫酸13.7g(137mmol),滴完后,于15℃~20℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸乙酯中,于40℃搅拌5小时,过滤,得固体于85℃进行真空干燥15h,得到硫酸氢氯吡格雷晶型ⅱ,54.4g,收率95%。(水份:0.01%)。
实施例12
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸乙酯250g,甲醇25g,搅拌溶解,温度将至15℃,滴加98%浓硫酸13.7g(137mmol),滴完后,于15℃~20℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于甲基叔丁基醚250ml中,于40℃搅拌5小时,过滤,得固体于85℃进行真空干燥15h,得到硫酸氢氯吡格雷晶型ⅱ,收率90%。(水份:0.01%)。
实施例13
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸乙酯500g,乙醇2.5g,搅拌溶解,温度将至15℃,滴加98%浓硫酸13.7g(137mmol),滴完后,于15℃~20℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于500ml异丙醚中,于40℃搅拌5小时,过滤,得固体于85℃进行真空干燥15h,得到硫酸氢氯吡格雷晶型ⅱ,收率90%。(水份:0.01%)。
实施例14
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸乙酯500g,乙醇50g,搅拌溶解,温度将至15℃,滴加98%浓硫酸14.4g(144mmol),滴完后,于15℃~20℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸乙酯500ml中,于40℃搅拌5小时,过滤,得固体于85℃进行真空干燥15h,得到硫酸氢氯吡格雷晶型ⅱ,收率88%。(水份:0.01%)。
实施例15
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸乙酯500g,乙醇50g,搅拌溶解,温度将至-5℃,滴加98%浓硫酸13.0g(130mmol),滴完后,于15℃~20℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸乙酯中,于40℃搅拌5小时,过滤,得固体于85℃进行真空干燥15h,得到硫酸氢氯吡格雷晶型ⅱ,收率92%。(水份:0.01%)。
实施例16
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸乙酯500g,异丙醇50g,搅拌溶解,温度将至25℃,滴加98%浓硫酸13.0g(130mmol),滴完后,于15℃~40℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸乙酯中,于40℃搅拌5小时,过滤,得固体于85℃进行真空干燥15h,得到硫酸氢氯吡格雷晶型ⅱ,收率94%。(水份:0.01%)。
实施例17
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸乙酯500g,异丙醇2.5g,搅拌溶解,温度将至40℃,滴加98%浓硫酸13.0g(130mmol),滴完后,于30℃~40℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于甲叔醚中,于40℃搅拌5小时,过滤,得固体于65℃进行真空干燥24h,得到硫酸氢氯吡格雷晶型ⅱ,收率96%。(水份:0.01%)。
实施例18
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸乙酯500g,异丙醇5g,搅拌溶解,温度将至25℃,滴加98%浓硫酸13.7g(137mmol),滴完后,于15℃~30℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸甲酯500ml中,于20℃搅拌5小时,过滤,得固体于85℃进行真空干燥15h,得到硫酸氢氯吡格雷晶型ⅱ,收率94%。(水份:0.01%)。
实施例19
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸乙酯500g,异丙醇25g,搅拌溶解,温度将至40℃,滴加98%浓硫酸13.7g(137mmol),滴完后,于30℃~40℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于甲叔醚中,于40℃搅拌5小时,过滤,得固体于65℃进行真空干燥24h,得到硫酸氢氯吡格雷晶型ⅱ,收率96%。(水份:0.01%)。
实施例20
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸乙酯500g,异丙醇5g,搅拌溶解,温度将至10℃,滴加98%浓硫酸13.7g(137mmol),滴完后,于20℃~25℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸异丙酯中,于20℃搅拌5小时,过滤,得固体于65℃进行真空干燥24h,得到硫酸氢氯吡格雷晶型ⅱ,收率97%。(水份:0.01%)。
实施例21
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入甲酸乙酯500g,异丙醇5g,搅拌溶解,温度将至10℃,滴加98%浓硫酸13.7g(137mmol),滴完后,于20℃~25℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸异丙酯中,于20℃搅拌5小时,过滤,得固体于65℃进行真空干燥24h,得到硫酸氢氯吡格雷晶型ⅱ,收率97%。(水份:0.01%)。
实施例22
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸甲酯500g,异丙醇5g,搅拌溶解,温度将至10℃,滴加98%浓硫酸13.7g(137mmol),滴完后,于20℃~25℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸异丙酯中,于20℃搅拌5小时,过滤,得固体于65℃进行真空干燥24h,得到硫酸氢氯吡格雷晶型ⅱ,收率96%。(水份:0.01%)。
实施例23
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入乙酸异丙酯500g,甲醇5g,搅拌溶解,温度将至10℃,滴加98%浓硫酸13.7g(137mmol),滴完后,于20℃~25℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸异丙酯中,于20℃搅拌5小时,过滤,得固体于65℃进行真空干燥24h,得到硫酸氢氯吡格雷晶型ⅱ,收率97%。(水份:0.01%)。
实施例24
干燥空气保护下,向50g(137mmol)氯吡格雷游离碱(实施例4制备),加入甲酸乙酯500g,乙醇5g,搅拌溶解,温度将至10℃,滴加98%浓硫酸13.7g(137mmol),滴完后,于20℃~25℃,搅拌3小时,过滤,得硫酸氢氯吡格雷湿品;
干燥空气保护下,将上述硫酸氢氯吡格雷湿品,加于乙酸异丙酯中,于20℃搅拌5小时,过滤,得固体于65℃进行真空干燥24h,得到硫酸氢氯吡格雷晶型ⅱ,收率97%。(水份:0.01%)。
本发明提出的一种硫酸氢氯吡格雷晶型i的制备方法已通过实施例进行了描述,相关技术人员明显能在不脱离本发明内容、精神和范围内对本文所述的硫酸氢氯吡格雷晶型i的制备方法进行改动或适当变更与组合,来实现本发明技术。特别需要指出的是,所有相类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明的精神、范围和内容中。
- 还没有人留言评论。精彩留言会获得点赞!