一种吉非替尼的制备方法与流程
本发明属于药物化学领域,涉及药物合成,具体涉及一种吉非替尼制备方法。
背景技术:
吉非替尼(gefitinib,zd1839,商品名:iressa),如式ⅰ所示:
化学名:n-(3-氯-4-氟苯基)-7-甲氧基-6-(3-吗啉基丙氧基)喹唑啉-4-胺,由英国astra-zeneca公司开发的抗肿瘤药物,它可竞争细胞表面的表皮生长因子受体络氨酸激酶(egfr-tk)催化区域上mg-atp结合位点,该药物是一种选择性表皮生长因子受体(egfr)酪氨酸激酶抑制剂,该酶通常表达于上皮来源的实体瘤。
吉非替尼广泛抑制异种移植于裸鼠的人肿瘤细胞的生长,抑制其血管生成,在体外,可增加人肿瘤细胞株的凋亡并抑制血管生成因子的侵入和分泌。在动物试验或体外研究中已证实吉非替尼可提高化疗、放疗及激素治疗的抗肿瘤活性。
吉非替尼是一种抗受体分子,它通过竞争细胞表面的表皮生长因子受体酪氨酸激酶(egfr—tk)催化区域mg—atp结合位点,阻断egfr生成信号传递至细胞内抑制egfr与配体结合后受体发生磷酸化,抑制与其他受体分子形成各种同源或异源二聚体,从而引起下游一系列信号通路如ip13k/akt和ras/raf/mapk激酶通路等活化的下调,从而阻碍肿瘤的生长、转移和血管生成,并可诱导肿瘤细胞的调亡。
该药物目前已被fda批准用于晚期非细胞肺癌(nsclc)患者经化疗后继续恶化的单用疗法,文献报道的吉非替尼的合成路线主要有:
路线一:根据k.n吉布森的报道(文献来源wo9633980,cn96193526):该路线以6,7-二甲氧基喹唑啉酮为原料,用甲磺酸和l-蛋氨酸选择性脱掉6-位甲氧基上的甲基,用乙酰基保护羟基后,接着采用氯化亚砜氯代,3-氯-4-氟苯胺氨化,脱保护,最后和卤代-3-吗啡啉基丙烷反应得到吉非替尼。
这条路线的缺点是选择性的脱掉6-位甲氧基需使用大量的甲磺酸和l-蛋氨酸,这两种试剂无法回收,环境污染大。同时,反应原料不易获得,收率也比较低。
路线二:根据j.p.吉尔岱和d.莫笛的报道(文献来源wo2004024703):该路线以异香兰素为原料,先将醛基转化为氰基,然后接上3-吗啉丙基,再硝化,还原硝基为胺基,水解氰基为酰胺,关环,氯代,最后和3-氯-4-氟苯胺对接得到吉非替尼。
该方法虽有改进,可以进行工业化生产,但其氰基的水解比较难控制,不可避免有水解到酸的产物,同时反应步骤长,副反应较多,杂质控制难度较大,而且原料比较贵。
路线三:文献来源cn1733738:
该路线以3,4-二甲氧基苯甲酸为原料,经过硝化、脱甲基、还原、环化、氯代、引入卤代芳香胺和烷基侧链后得到目标产物。
该方法虽然以简单的3,4-二甲氧基苯甲酸为起始原料,但存在以下两点不足之处:(1)该方法在脱去甲基后未对羟基进行保护,由于羟基是活性基团,致使还原、环化、氯代等步骤副反应多,产率低。(2)该合成路线第四步反应,2-氨基-4-甲氧基-5-羟基苯甲酸直接与甲酰胺反应构建4-羰基喹啉母体环,该步骤副产物多,产率低。
其中以路线三(参见专利文献cn1733738)与本发明申请最为接近,但它是以中间体6-羟基-7-甲氧基-3,4-二氢喹唑啉-4-酮进行氯代,在该步氯代中的氯化试剂主要采用氯化亚砜,dmf为催化剂。
该专利文献介绍的制备方法存在以下不足:
1)由于其苯环上存在未经保护的羟基,易被氯代,与3-氯-4-氟苯胺进行胺化反应易产生二聚体杂质等其他杂质,如下式所示:
2)采用氯化亚砜为氯代试剂,在dmf作催化氯代反应情况下,容易生成杂质ⅵ,该杂质与3-氯-4-氟苯胺反应易生成杂质ⅶ:
由于杂质ⅶ其结构与吉非替尼的结构高度接近,在后续的精制处理中极度难以去除,经多次结晶,仍难以符合现行质量要求≤0.1%标准,故应在前面的中间步骤加以控制和改变合成路径及方法加以规避,二者结构比较如下:
3)由于氯化亚砜的强腐蚀性和挥发性,故使用二氯亚砜作为氯化试剂,对生产设备腐蚀和环境都具有较大的破坏。
综上所述,采用现有技术中报道的制备方法获得的吉非替尼化合物,存在副产物较多,后处理复杂或多次精制才能达到ich规定的原料药技术质量要求的问题,且环境污染较大,不适用于工业化大生产,为此亟待一种新的技术方案,以提供更优的工艺技术条件,解决产品质量,使之能连续稳定的生产出符合现行技术质量要求的原料药,同时将生产过程中的环境污染降到最低程度。
技术实现要素:
为克服现有技术的缺陷,本发明提供一种吉非替尼,即n-(3-氯-4-氟苯基)-7-甲氧基-6-(3-吗啉基丙氧基)喹唑啉-4-胺(式ⅰ)的制备方法,其特征在于包括以下步骤:
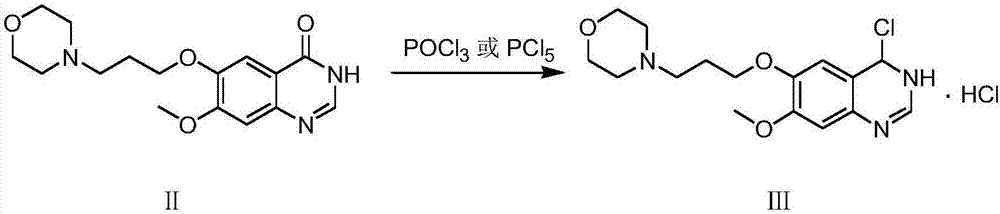
(1)以7-甲氧基-6-(3-吗啉-4-基丙氧基)喹唑啉-4(3h)-酮为起始(式ⅱ)原料,与氯代试剂发生氯代反应,得到中间体一(式ⅲ),所述的氯代试剂为pocl3或者pcl5,
(2)中间体一(式ⅲ)与3-氯-4-氟苯胺(如式ⅳ所示)进行取代反应,制备得吉非替尼粗品,
(3)吉非替尼粗品与有机溶剂经重结晶得到吉非替尼纯品。
优选的,步骤(1)中,溶剂为乙腈或甲苯;
优选的,步骤(1)中,反应温度优选60~90℃;进一步优选的,反应温度优选80~90℃;
优选的,步骤(1)中,7-甲氧基-6-(3-吗啉-4-基丙氧基)喹唑啉-4(3h)-酮(式ⅱ)与氯代试剂的用量摩尔比为1:3~10,更优选用量摩尔比为1:4;
优选的,步骤(2)中,溶剂为异丙醇;
优选的,步骤(2)中,反应温度为60~90℃;进一步优选的,反应温度优选80~90℃;
优选的,步骤(2)中,中间体一(式ⅲ)与3-氯-4-氟苯胺的摩尔用量比为1:0.9~1.5;更优选的,中间体一与3-氯-4-氟苯胺的摩尔用量比为1:1.05;
进一步优选的,步骤(2)中,中间体一(式ⅲ)与3-氯-4-氟苯胺进行取代反应还要加入缚酸剂,缚酸剂的用量与3-氯-4-氟苯胺的用量的摩尔比为1~5:1;更优选的,缚酸剂的用量与3-氯-4-氟苯胺的用量的摩尔比2:1;
更优选的,步骤(2)中,所述的缚酸剂为碳酸钾、碳酸钠、三乙胺或其他碱性试剂;
优选的,步骤(3)中,所述的重结晶有机溶剂为甲基异丁基酮、乙酸乙酯中的一种或二者混合物。
有益效果
1.本发明给出的反应步骤和操作非常简便,降低了工艺复杂性及纯化次数,易于扩大化生产;
2.采用本发明制备的产品纯度极高,纯度≥99.7%,避免产生杂质二聚物、以及化合物ⅷ,保证了产品质量;
3.本发明采用的制备方法避免使用氯化亚砜等强腐蚀性和挥发性溶剂,最大程度地减少了环境污染。
附图说明
图1吉非替尼的化学结构式;
图2中间体一的制备工艺路线图;
图3吉非替尼粗品的制备工艺路线图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚明了,下面结合具体实施方式并参照附图,对本发明进一步详细说明。应该理解,这些描述只是示例性的,而并非要限制本发明的范围。
实施例1:中间体一(式ⅲ):4-氯-7-甲氧基-6-[3-(吗啉-4-基)丙氧基]喹唑啉盐酸盐的制备
向反应罐中投入甲苯1.35l、三氯氧磷466.0ml,搅拌均匀后加入7-甲氧基-6-(3-吗啉-4-基丙氧基)喹唑啉-4(3h)-酮319.3g,加入5ml的tea,升温至80~90℃反应1h,tlc监测(meoh:dcm=1:5,原料rf=0.4,产物rf=0.6),原料基本消失,55~60℃减压蒸馏,蒸出大部分溶剂,蒸馏完毕降温,控温20~25℃下加入3l异丙醇打浆1h,室温过滤,滤饼甩干,50-60℃减压干燥8h,得到浅黄色固体268.2g,即为吉非替尼中间体1,收率:79.4%。hplc纯度为98.5%。
实施例2:吉非替尼粗品的制备
反应罐中加入3-氯-4-氟苯胺121.3g、异丙醇1.8l搅拌至全溶,加入碳酸钾219.6g,加入268.2g中间体1,搅拌升温至80~85℃回流反应1h,tlc监测(meoh:dcm=1:5,zj1的rf=0.6,产物rf=0.8),原料点消失。趁热过滤,滤液降温至室温,得固体,过滤,滤饼用异丙醇1.5l淋洗后甩干,50-60℃减压干燥6h,得到吉非替尼粗品,淡黄色固体333.5g,收率:94%,hplc纯度为99.3%。
实施例3:吉非替尼粗品的纯化
向反应罐中加入吉非替尼粗品333.5g,甲基异丁基酮2l和乙酸乙酯6l,开搅拌,升温回流3小时,溶清,趁热过滤,滤液自然降温至20~25℃,保温析晶24h,过滤,滤饼88~93℃减压(-0.08mpa)干燥6小时得到白色结晶性粉末273.5g,即为吉非替尼精品。mp:193-195℃,收率:82%。hplc显示单杂小于0.1%。
实施例4:中间体一(式ⅲ):4-氯-7-甲氧基-6-[3-(吗啉-4-基)丙氧基]喹唑啉盐酸盐的制备
向反应罐中投入乙腈1.35l、三氯氧磷466.0ml,搅拌均匀后加入7-甲氧基-6-(3-吗啉-4-基丙氧基)喹唑啉-4(3h)-酮319.3g,加入5ml的tea,升温至80~90℃反应1h,tlc监测(meoh:dcm=1:5,原料rf=0.4,产物rf=0.6),原料基本消失,55~60℃减压蒸馏,蒸出大部分溶剂,蒸馏完毕降温,控温20~25℃下加入3l异丙醇打浆1h,室温过滤,滤饼甩干,50-60℃减压干燥8h,得到浅黄色固体274.6g,即为吉非替尼中间体1,收率:81.3%。hplc纯度为98.0%。
实施例5:中间体一(式ⅲ):4-氯-7-甲氧基-6-[3-(吗啉-4-基)丙氧基]喹唑啉盐酸盐的制备
向反应罐中投入乙腈1.35l、五氯化磷832g,搅拌均匀后加入7-甲氧基-6-(3-吗啉-4-基丙氧基)喹唑啉-4(3h)-酮319.3g,加入5ml的tea,升温至80~90℃反应1h,tlc监测(meoh:dcm=1:5,原料rf=0.4,产物rf=0.6),原料基本消失,55~60℃减压蒸馏,蒸出大部分溶剂,蒸馏完毕降温,控温20~25℃下加入3l异丙醇打浆1h,室温过滤,滤饼甩干,50-60℃减压干燥8h,得到浅黄色固体284.0g,即为吉非替尼中间体1,收率为84.1%,hplc纯度为98.3%。
实施例6:中间体一(式ⅲ):4-氯-7-甲氧基-6-[3-(吗啉-4-基)丙氧基]喹唑啉盐酸盐的制备-对照组
向反应罐中投入二氯亚砜328g、dmf2.86g,搅拌均匀后加入7-甲氧基-6-(3-吗啉-4-基丙氧基)喹唑啉-4(3h)-酮30.0g,升温至75~80℃回流反应6h,tlc监测(meoh:dcm=1:5,原料rf=0.4,产物rf=0.6),原料基本消失,55~60℃减压蒸馏,蒸出大部分溶剂后加入61g甲苯继续浓缩,重复此操作两次,蒸馏完毕降温,控温20~25℃下加入119g异丙醇打浆1h,室温过滤,滤饼甩干得到黄色固体24.3g,即为吉非替尼中间体1,收率为69.3%。hplc纯度为94.5%。
实施例7:吉非替尼粗品的制备-对照组
反应罐中加入3-氯-4-氟苯胺10.4g、异丙醇163ml,搅拌至全溶,加入碳酸钾19.8g,加入24.3g中间体1(实施例6所得中间体1),搅拌升温至80~85℃回流反应1h,tlc监测(meoh:dcm=1:5,zj1的rf=0.6,产物rf=0.8),原料点消失。趁热过滤,滤液降温至室温,得固体,过滤,滤饼用异丙醇150ml淋洗后甩干,50-60℃减压干燥6h,得到吉非替尼粗品,黄色固体29.6g,收率:83.5%。hplc纯度为93.3%,杂质ⅷ含量为1.7%。
实施例8:吉非替尼粗品的纯化
向反应罐中加入吉非替尼粗品(实施例7所得粗品)29.6g,和乙酸乙酯880ml,开搅拌,升温回流3小时,溶清,趁热过滤,滤液自然降温至20~25℃,保温析晶24h,过滤,滤饼88~93℃减压(-0.08mpa)干燥6小时得到白色结晶性粉末22.7g,即为吉非替尼精品。收率为76.8%,hplc纯度为98.4%,杂质ⅷ含量为0.6%。
- 还没有人留言评论。精彩留言会获得点赞!