一种3,4‑二氰基氧化呋咱的制备方法与流程
本发明属于有机化合物的合成技术领域,特别是一种3,4-二氰基氧化呋咱的制备方法。
背景技术:
自1896年氧化呋咱被发现以来,人们对氧化呋咱及其衍生物的研究从来没有间断过,氧化呋咱是具有配位氧原子的呋咱环结构,是构筑新型高能化合物的重要结构单元。以3,4-二氰基氧化呋咱(dcfo)为原料合成的多硝基氧化呋咱衍生物是一类绿色环保型炸药,具有较高的有效氧含量、标准生成焓及能量密度,具有良好的爆炸性能。为了合成该类含能化合物,其中间体的合成不容忽视,改进3,4-二氰基氧化呋咱的制备方法,提高其工艺安全性及反应产率具有重要的意义。
大多数文献报道的3,4-二氰基氧化呋咱合成方法是以氰基乙酸为原料,在三氟乙酸反应介质中以浓硝酸为硝化剂的进行硝化反应。该方法首先将氰基乙酸溶于三氟乙酸中,然后滴加浓硝酸在40~50℃反应一段时间,将反应液倒入冰水中进行降温稀释,再经萃取、水洗、干燥后得到3,4-二氰基氧化呋咱目标产物(angew.chem.2016,128,782-785)。其合成反应式如下:
此方法的不足之处在于氰基乙酸虽然能在三氟乙酸中溶解,可能由于副反应的发生,导致反应最终产物产率低。此外,以三氟乙酸或三氟乙酸酐作为反应介质,不仅原料成本高,而且反应物料自身的危险性较大,产生的大量含三氟乙酸的废酸回收处理工艺复杂,制备成本相对较高。
技术实现要素:
本发明的目的是提供一种提高3,4-二氰基氧化呋咱产率的简便安全的制备方法。
实现本发明目的技术解决方案为:一种3,4-二氰基氧化呋咱的制备方法,包括以下步骤:
步骤1、将硝硫混酸硝化剂滴加到氰基乙酸和二氯甲烷的悬浮液中;所述的二氯甲烷用量为氰基乙酸质量的5~10倍;所述的硝硫混酸硝化剂为浓度98%的浓硫酸或浓度20%的发烟硫酸与浓度68~98%浓硝酸的混合液,硝酸与硫酸的体积之比为(1:2)~(4:1),硝酸用量为氰基乙酸质量的1~3倍;在硝硫混酸硝化剂滴加过程中反应物料的温度控制到0~20℃范围。
步骤2、将步骤1得到的反应物持续搅拌,完成硝化反应;上述反应的反应温度为15~35℃,搅拌时间为90~240min。
步骤3、将步骤2得到的反应物降温,之后滴加去离子水到上述反应物中;将反应物进行降温时,是将温度降到15℃以下;上述滴加的去离子水的温度为15℃以下;所述去离子水的体积为硝硫混酸硝化剂体积的5~10倍。
步骤4、将步骤3得到的反应物采用二氯甲烷进行萃取,合并溶剂相,用饱和食盐水洗涤,之后用无水硫酸镁干燥后过滤,旋转蒸馏回收溶剂,得到3,4-二氰基氧化呋咱产物;所述采用二氯甲烷进行萃取的次数为2~4次,用饱和食盐水洗涤时,将其洗涤至ph值为5~6时为止。
本方法与现有技术相比,其显著优点为:1)本发明采用惰性的二氯甲烷溶剂替代文献中报道的三氟乙酸等强腐蚀性反应介质,起到分散作用的同时,大幅度提高了产率,而且制备成本降低,安全性也得到了提升;2)二氯甲烷容易回收利用,比三氟乙酸等强腐蚀性反应介质更容易回收;3)采用本发明方法制备目标化合物产生的废酸少,且废的硝硫混酸回收处理工艺成熟,回收处理更方便。
附图说明
图1为实施例1制备的dcfo样品的光学显微镜测试的形貌图。
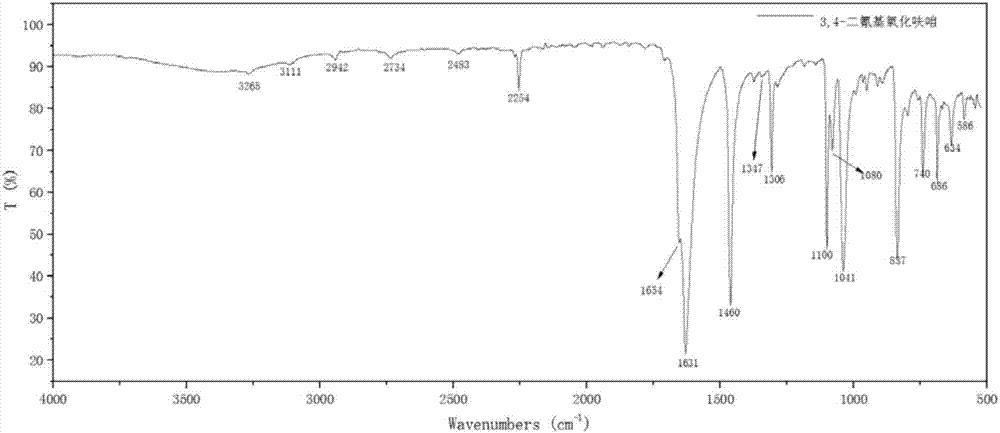
图2为实施例1制备的dcfo样品的红外光谱图。
图3为实施例1制备的dcfo样品的核磁共振谱图。
图4为实施例2制备的dcfo样品的光学显微镜测试的形貌图。
图5为实施例2制备的dcfo样品的x射线衍射图谱。
具体实施方法
结合附图,本发明的一种3,4-二氰基氧化呋咱的制备方法,包括以下步骤:
步骤1、将硝硫混酸硝化剂滴加到氰基乙酸和二氯甲烷的悬浮液中;所述的二氯甲烷用量为氰基乙酸质量的5~10倍;所述的硝硫混酸硝化剂为浓度98%的浓硫酸或浓度20%的发烟硫酸与浓度68~98%浓硝酸的混合液,硝酸与硫酸的体积之比为(1:2)~(4:1),硝酸用量为氰基乙酸质量的1~3倍。在硝硫混酸硝化剂滴加过程中反应物料的温度控制在0~20℃范围内。
步骤2、将步骤1得到的反应物持续搅拌,完成硝化反应;反应温度为15~35℃,搅拌时间为90~240min。
步骤3、将步骤2得到的反应物降温,之后滴加去离子水到上述反应物中;将反应物进行降温处理时,是将温度降到15℃以下;滴加的去离子水的温度为15℃以下;去离子水的体积为硝硫混酸硝化剂体积的5~10倍。
步骤4、将步骤3得到的反应物采用二氯甲烷进行萃取,合并溶剂相,用饱和食盐水洗涤,之后用无水硫酸镁干燥后过滤,旋转蒸馏回收溶剂,得到3,4-二氰基氧化呋咱产物。具体是采用二氯甲烷进行萃取的次数为2~4次,用饱和食盐水洗涤时,将其洗涤至ph值为5~6时为止。
本发明采用惰性的二氯甲烷溶剂替代文献中报道的三氟乙酸等强腐蚀性反应介质,起到分散作用的同时,大幅度提高了产率,而且制备成本降低,安全性也得到了提升。
下面通过实施例的方式进一步说明。
实施例1
预先按照20%的发烟硫酸与98%浓硝酸的体积比为1:1配制硝硫混酸硝化剂,并冷却到室温。称取纯度大于99%的氰基乙酸10.0g加到500ml的反应器中,加入50ml二氯甲烷,开动搅拌器,将反应体系物料的温度降到0℃,以0.5ml/min的速率滴加入14ml预先准备好的硝硫混酸硝化剂,在滴加过程中监测物料的温度,通过降低滴加速度来防止物料温度的上升,确保物料的温度不超过5℃。在搅拌条件下,将反应体系的温度提升至28℃,继续反应180min。将体系温度降至15℃以下,再滴加70ml温度为10℃的去离子水对反应物进行稀释,加水速度由1ml/min逐步提高到5ml/min,体系温度维持在15℃以下,去离子水滴加完后,继续搅拌5~10min。接着,将反应液转入分液漏斗,分离出溶剂相,水相采用二氯甲烷萃取四次,每次20ml。合并溶剂相,用饱和食盐水洗涤三次,每次加食盐水15ml,洗涤过程中测试上层饱和食盐水的酸度,第一次水洗液显强酸性(ph<2),第二次水洗液显弱酸性,第三次水洗液接近中性,ph达到5~6,水洗完毕后加入10.0g无水硫酸镁对溶剂相干燥。过滤得到溶解有目标产物的溶剂,旋转蒸馏回收溶剂得到微黄色的3,4-二氰基氧化呋咱6.35g,产率75.4%,经液相色谱测试,纯度为90.4%。分离出来的酸性水溶液集中处理,防止造成环境污染。图1为本实施例制备的dcfo样品的光学显微镜测试的形貌图,图2为dcfo样品的红外光谱图,图3为dcfo样品的核磁共振谱图。
作为对比,采用上述工艺过程和工艺参数,仅将二氯甲烷反应介质用相同体积的三氟乙酸替代,最后得到的目标产物3,4-二氰基氧化呋咱2.86g,产率35.7%,经液相色谱测试,纯度为91.6%。
由上述实验数据可知,在相同工艺过程和工艺参数的条件下,以二氯甲烷作为反应介质得到目标产物的产量是三氟乙酸为反应介质产量的2.2倍,产率显著提高,且操作过程更加安全可靠。
实施例2
预先按照98%的浓硫酸与90%浓硝酸的体积比为1:1配制硝硫混酸硝化剂,并冷却到室温。称取氰基乙酸10.0g加到500ml的反应器中,加入70ml二氯甲烷,开动搅拌器,将反应体系物料的温度降到2℃,以0.5ml/min的速率滴加入14ml预先准备好的硝硫混酸硝化剂,在滴加过程中监测物料的温度,通过降低滴加速度来防止物料温度的上升,确保物料的温度不超过5℃。在搅拌条件下,将反应体系的温度提升至15℃,继续反应180min。将体系温度降至10℃以下,再滴加加入100ml温度为12℃的去离子水进行稀释,加水速度由1ml/min逐步提高到5ml/min,体系温度维持在15℃以下,滴加完水后,继续搅拌5~10min。接着,将反应液转入分液漏斗,分离出溶剂相,水相采用二氯甲烷萃取四次,每次20ml。合并溶剂相,用饱和食盐水洗涤三次,每次加食盐水15ml,洗涤过程中测试上层饱和食盐水的酸度,第一次水洗液显强酸性(ph<2),第二次水洗液显弱酸性,第三次水洗液接近中性,ph5~6,水洗完毕后加入10.0g无水硫酸镁干燥。过滤得到溶解有目标产物的溶剂相,旋转蒸馏回收溶剂得到白色略显黄色目标产物3,4-二氰基氧化呋咱6.43g,产率80.4%,经液相色谱测试,纯度为91.8%。分离出来的酸性水溶液集中处理,防止造成环境污染。图4为本实施例所制备的dcfo样品光学显微镜测试的形貌图,图5为本实施例所制备的dcfo样品x射线衍射图谱结果。
作为对比,采用上述工艺过程和工艺参数,仅将二氯甲烷反应介质用相同体积的三氟乙酸替代,最后得到的目标产物3,4-二氰基氧化呋咱2.05g,产率25.6%,经液相色谱测试,纯度为92.1%。
由上述实验验证可知,在相同工艺过程和工艺参数的条件下,以二氯甲烷作为反应介质得到目标产物的产量是三氟乙酸为反应介质产量的3.1倍,产率显著提高,且操作过程更加安全可靠。
实施例3
预先按照98%的浓硫酸与68%浓硝酸的体积比为1:2配制硝硫混酸硝化剂,并冷却到室温。称取氰基乙酸10.0g加到500ml的反应器中,加入100ml二氯甲烷,开动搅拌器,将反应体系物料的温度降到20℃,以0.5ml/min的速率加入21ml硝硫混酸硝化剂,在滴加过程中监测物料的温度,确保物料的温度不超过20℃。在搅拌条件下反应体系升温至25℃硝化反应150min。将体系温度降至15℃以下,再滴加入210ml温度为5℃的去离子水进行稀释,加水速度由1ml/min逐步提高到5ml/min,体系温度维持在15℃以下。加完水后继续搅拌5~10min使酸充分溶解于水相。接着,将反应液转入分液漏斗,分离出溶剂相,水相采用二氯甲烷萃取四次,每次20ml。合并溶剂相,用饱和食盐水洗涤三次,每次加食盐水15ml,洗涤过程中测试上层饱和食盐水的酸度,第一次水洗液显强酸性(ph<2),第二次水洗液显弱酸性,第三次水洗液接近中性,水洗完毕后加入10.0g无水硫酸镁干燥。过滤得到溶解有目标产物的溶剂相,旋转蒸馏回收溶剂得到淡黄色目标产物3,4-二氰基氧化呋咱6.32g,产率79.0%,经液相色谱测试,纯度为92.0%。分离出来的酸性水溶液倒入回收瓶中集中处理,防止造成环境污染。
作为对比,采用上述工艺过程和工艺参数,仅将二氯甲烷反应介质用相同体积的三氟乙酸酐替代,最后得到的目标产物3,4-二氰基氧化呋咱3.46g,产率43.2%,经液相色谱测试,纯度为92.1%。
由上述实验验证可知,在相同工艺过程和工艺参数的条件下,以二氯甲烷作为反应介质得到目标产物的产量是三氟乙酸酐为反应介质产量的1.8倍,产率显著提高,且操作过程更加安全可靠。
实施例4~12
采用与实施例1相同的工艺过程,原料及惰性反应介质加入量不变,仅改变部分工艺参数和物料比例进行对比试验,分别改变硝硫混酸硝化剂中硫酸与硝酸的体积比、硝酸浓度、硝化剂加入量、硝化反应温度及硝化反应时间,通过连续输送装置控制加料的速度来保证体系安全稳定,工艺参数和试验结果如下表。
从以上实施例可以看出,以二氯甲烷作为惰性反应介质,无论是采用浓硫酸还是发烟硫酸与不同浓度硝酸混合作为硝化试剂,目标产物的得率都比采用三氟乙酸或三氟乙酸酐做反应介质的合成方法有显著提高,而且反应条件温和,操作过程简便,制备成本较低,适用于大批量的合成制备。
综上所述,以上仅为本发明的较佳实施例,并非用于限定本发明的保护范围。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!