一种氧化偶氮四唑的制备方法与流程
本发明涉及一种氧化偶氮四唑的制备方法,具体地说,涉及一种2,2'-二取代-5,5'-氧化偶氮四唑的制备方法,属于有机合成和含能材料领域。
背景技术:
氧化偶氮结构应用于含能化合物不仅能保持偶氮化合物良好的热稳定性与机械感度,还通过引入氧原子提高密度并改善氧平衡(zhangj,shreevejm.nitroaminofurazanswithazoandazoxylinkages:acomparativestudyofstructural,electronic,physicochemical,andenergeticproperties[j],journalofphysicalchemistryc,2015,119,12887–12895.)。其中,氧化偶氮四唑具有较高的能量和良好的热稳定性,是除了氧化偶氮呋咱外少有的氧化偶氮含能化合物(fischern,etal.5,5'-azoxytetrazolates–anewnitrogen-richdianionanditscomparisonto5,5'-azotetrazolate[j].daltontransactions,2012,41,11201–11211.)。而限制氧化偶氮结构在含能化合物中应用的最大障碍是其合成方法的匮乏。
现有的几种合成氧化偶氮结构的方法,包括氨基氧化(coburnmd.oxidationofheterocyclicnitrogenylidestonitroheterocycles.comparisonofasulfilimineandaphosphineimine[j].journalofheterocyclicchemistry,1986,23,421–423.)、硝基还原(cravottog,etal.aneasyaccesstoaromaticazocompoundsunderultrasound/microwaveirradiation[j].synlett,2006,2605–2608.)或叠氮基脱氮气氧化(shaforostakv,etal.4-azidoquinolinen-oxide:synthesisandphotolysis[j].russianjournaloforganicchemistry,2008,44,728–730.)等,最终都要经过亚硝基结构的中间体,进一步偶联得到氧化偶氮化合物。由于氨基化合物是相对最容易获得的原料,所以氨基氧化法也是目前合成氧化偶氮化合物最主要的方法,但由于氨基氧化反应总是伴随硝基化合物和偶氮化合物的形成,且多数反应氧化偶氮化合物仅为副产物,因此大多数反应产率都很低。目前文献报道的以取代-5-氨基四唑为原料通过直接氧化法制备2,2'-二取代-5,5'-氧化偶氮四唑的最高产率为35%(churakovam,etal.theoxidationofheterocyclicaminestonitrocompoundsusingdinitrogenpentoxide[j].mendeleevcommunications,1995,5,102–103.)。
技术实现要素:
针对现有技术存在的缺陷,本发明的目的在于提供一种氧化偶氮四唑的制备方法,所述方法简单、高效,能够显著提高制备2,2'-二取代-5,5'-氧化偶氮四唑的收率。
本发明的目的是通过以下技术方案实现的。
一种氧化偶氮四唑的制备方法,所述方法步骤为:
方法一
在-15℃~10℃下将1,3-二(取代四唑基)三氮烯与质量分数为80%~100%的发烟硝酸按照1:10~30的摩尔比进行混合得到反应溶液,将反应溶液在0℃~25℃搅拌反应6h~48h;随后在-15℃~10℃加入酸酐,发烟硝酸与酸酐的摩尔比为1:1~3;在0℃~25℃继续反应1h~12h后,将反应液倒入冰水,萃取,干燥,减压蒸馏,再采用硅胶柱层析分离法进行分离,再次减压蒸馏,得到所述氧化偶氮四唑,为2,2'-二取代-5,5'-氧化偶氮四唑;或
方法二
在-15℃~10℃下将1,3-二(取代四唑基)三氮烯、质量分数为80%~100%的发烟硝酸以及酸酐按照1:10~30:10~90的摩尔比,且发烟硝酸与酸酐摩尔比不大于1:1进行混合得到反应溶液;将反应溶液在0℃~25℃搅拌反应4h~24h后,将反应液倒入冰水,萃取,干燥,减压蒸馏,再采用硅胶柱层析分离法进行分离,再次减压蒸馏,得到所述氧化偶氮四唑,为2,2'-二取代-5,5'-氧化偶氮四唑。
所述2,2'-二取代-5,5'-氧化偶氮四唑为2,2'-二甲基-5,5'-氧化偶氮四唑、2,2'-二乙基-5,5'-氧化偶氮四唑、2,2'-二异丙基-5,5'-氧化偶氮四唑、2,2'-二叔丁基-5,5'-氧化偶氮四唑、2,2'-二氰甲基-5,5'-氧化偶氮四唑、2,2'-二溴乙基-5,5'-氧化偶氮四唑、2,2'-二甲氧酰甲基-5,5'-氧化偶氮四唑、2,2'-二乙酰甲基-5,5'-氧化偶氮四唑、2,2'-二羧甲基-5,5'-氧化偶氮四唑或2,2'-二硝酸酯基乙基-5,5'-氧化偶氮四唑。
所述1,3-二(取代四唑基)三氮烯为1,3-二(2-甲基四唑-5-基)三氮烯、1,3-二(2-乙基四唑-5-基)三氮烯、1,3-二(2-异丙基四唑-5-基)三氮烯、1,3-二(2-叔丁基四唑-5-基)三氮烯、1,3-二(2-氰甲基四唑-5-基)三氮烯、1,3-二(2-溴乙基四唑-5-基)三氮烯、1,3-二(2-甲氧酰甲基四唑-5-基)-三氮烯、1,3-二(2-乙酰甲基四唑-5-基)三氮烯、1,3-二(2-羧甲基四唑-5-基)三氮烯或1,3-二(2-羟乙基四唑-5-基)三氮烯。
优选所述酸酐为乙酸酐、丙酸酐、正丁酸酐、异丁酸酐、戊酸酐、异戊酸酐、己酸酐、丁二酸酐、戊二酸酐、苯甲酸酐和三氟乙酸酐中的一种以上。
有益效果
本发明提供了一种氧化偶氮四唑的制备方法,所述方法以1,3-二(取代四唑基)三氮烯为原料,利用发烟硝酸和酸酐制备得到2,2'-二取代-5,5'-氧化偶氮四唑,简单、高效,能够显著提高制备2,2'-二取代-5,5'-氧化偶氮四唑的收率,产率可达到53~82%,或从取代-5-氨基四唑为原料计算产率为44%~71%。
附图说明
图1为实施例1中制备的2,2'-二甲基-5,5'-氧化偶氮四唑的单晶结构图。
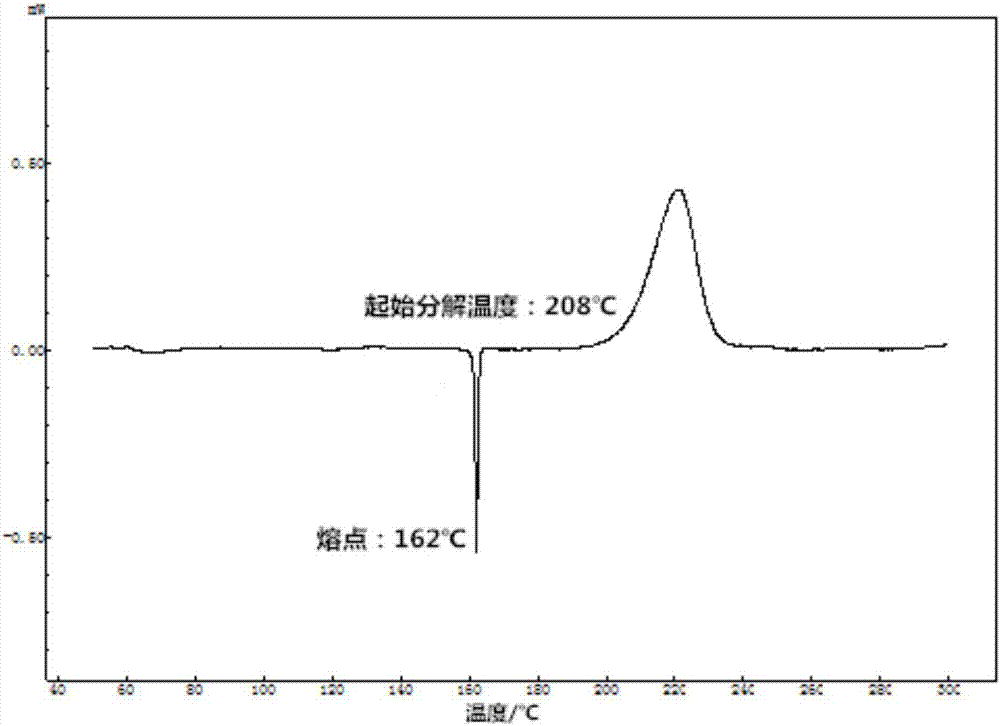
图2为实施例1中制备的2,2'-二甲基-5,5'-氧化偶氮四唑的差示扫描量热(dsc)图。
图3为实施例1中制备的2,2'-二甲基-5,5'-氧化偶氮四唑的红外谱图。
图4为实施例1中制备的2,2'-二甲基-5,5'-氧化偶氮四唑的核磁氢谱图。
图5为实施例1中制备的2,2'-二甲基-5,5'-氧化偶氮四唑的核磁碳谱图。
图6为实施例2中制备的2,2'-二氰甲基-5,5'-氧化偶氮四唑的dsc图。
图7为实施例2中制备的2,2'-二氰甲基-5,5'-氧化偶氮四唑的红外谱图。
图8为实施例2中制备的2,2'-二氰甲基-5,5'-氧化偶氮四唑的核磁氢谱图。
图9为实施例2中制备的2,2'-二氰甲基-5,5'-氧化偶氮四唑的核磁碳谱图。
图10为实施例2中制备的2,2'-二氰甲基-5,5'-氧化偶氮四唑的高分辨质谱图。
图11为实施例3中制备的2,2'-二硝酸酯基乙基-5,5'-氧化偶氮四唑的核磁氢谱图。
图12为实施例3中制备的2,2'-二硝酸酯基乙基-5,5'-氧化偶氮四唑的核磁碳谱图。
具体实施方式
下面结合附图和具体实施方式对本发明做进一步说明。
以下实施例中:
硅胶柱层析分离法中,所用硅胶为市售200目~300目的柱层析硅胶,流动相为石油醚与乙酸乙酯的混合溶液;
旋转蒸发仪:型号n1001,厂家eyela;
显微熔点仪:xt4a(温度计未校正),厂家北京福凯仪器有限公司;
差示扫描量热仪:型号dsc-1,厂家mettlertoledo,测试时的升温速率为5℃/min;
红外光谱仪:型号alphaft-ir-spektrometer,厂家bruker;
核磁共振波谱仪:型号avanceⅲ400m,厂家buruker;
元素分析仪:型号varioel,厂家elementar;
傅立叶离子回旋变换质谱:型号apexiv,厂家bruker;
x-射线单晶衍射仪:型号saturn724ccd,厂家rigaku。
理论产量=1,3-二(取代四唑基)三氮烯的摩尔数×2,2'-二取代-5,5'-氧化偶氮四唑的摩尔质量;产率=实际产量/理论产量。
从取代-5-氨基四唑为原料计算产率=产率×取代-5-氨基四唑合成1,3-二(取代四唑基)三氮烯的文献产率(文献产率参照wangq,etal.pentazadiene:ahigh-nitrogenlinkageinenergeticmaterials[j].chemicalcommunications,2017,53,2327–2330.)。
实施例1
2,2'-二甲基-5,5'-氧化偶氮四唑的合成
在0℃下,将0.5mmol1,3-二(2-甲基四唑-5-基)三氮烯加入0.5ml质量分数为92%的发烟硝酸(10.9mmol)中混合得到反应溶液,将反应溶液在0℃搅拌反应12h;随后在0℃下向反应溶液滴加1.5ml乙酸酐(15.9mmol),在0℃继续反应12h,之后将反应液倒入15ml冰水中,用乙酸乙酯萃取两次,分离后的有机相合并,之后依次用水萃取一次,饱和食盐水萃取一次,然后用无水硫酸钠对分离的有机相进行干燥,再将干燥后的有机相使用旋转蒸发仪在0.01mpa、30℃下进行减压蒸馏除去溶剂,最后用硅胶柱层析分离,流动相为石油醚与乙酸乙酯体积比为1:1的混合溶液,再使用旋转蒸发仪在0.01mpa、30℃下将分离后的有机相进行减压蒸馏除去溶剂,即可得到终产物。
对本实施例所制备的终产物进行如下表征和测试:
所制备的终产物质量为56mg,经计算可得产率为53%,或从2-甲基-5-氨基四唑为原料计算产率为44%。
采用x-射线单晶衍射仪对所制备的终产物进行表征,得到其单晶结构图,如图1所示。
采用差示扫描量热仪对所制备的终产物进行测试,从图2中可以得知其熔点为162℃,起始分解温度为208℃。
用红外光谱仪对所制备的终产物进行表征,图3的红外图谱中,1513.22cm-1和1491.04cm-1为氧化偶氮结构的特征峰,1448.60cm-1和1287.54cm-1为四唑环的特征峰。
图4和图5是通过核磁共振波谱仪表征得到的,图4的核磁共振氢谱(400mhz,dmso-d6)中4.55ppm和4.50ppm处有一组1:1的单峰,分别对应终产物中两个四唑环上的甲基取代基;图5的核磁共振碳谱(100mhz,dmso-d6)中165.5ppm和162.9ppm对应四唑环中的两个碳,41.1ppm和40.3ppm对应四唑环上的甲基取代基的两个碳。
采用元素分析仪对所制备的终产物进行测试,测试结果为c的质量分数为23.17%,h的质量分数为2.75%,n的质量分数为66.47%与2,2'-二甲基-5,5'-氧化偶氮四唑中各元素的理论值(c22.86%,h2.88%,n66.65%)非常接近。
采用傅立叶离子回旋变换质谱对所制备的终产物进行表征,测得的[m+na]+分子量为233.0613与2,2'-二甲基-5,5'-氧化偶氮四唑的[m+na]+理论分子量233.0618几乎相等。
由上述表征结果可知,本实施例所制备的终产物为2,2'-二甲基-5,5'-氧化偶氮四唑。
通过gaussian09(revisionb.01)软件计算,可以预测所制备的2,2'-二甲基-5,5'-氧化偶氮四唑的生成焓为519.8kj·mol-1,密度为1.57g·cm-3,运用软件explo5可以预估2,2'-二甲基-5,5'-氧化偶氮四唑的预测爆速为7465m·s-1,爆压为19.0gpa,爆热为3945j·g-1。
实施例2
2,2'-二氰甲基-5,5'-氧化偶氮四唑的合成
在-15℃下,将0.5mmol1,3-二(2-氰甲基四唑-5-基)三氮烯加入0.25ml质量分数为100%的发烟硝酸(5.9mmol)中混合得到反应溶液,将反应溶液在25℃搅拌反应6h;随后在10℃下向反应溶液滴加0.8ml丙酸酐(6.2mmol),在25℃继续反应1h,之后将反应液倒入15ml冰水中,用乙酸乙酯萃取两次,分离后的有机相合并,之后依次用水萃取一次,饱和食盐水萃取一次,然后用无水硫酸钠对分离的有机相进行干燥,再将干燥后的有机相使用旋转蒸发仪在0.01mpa、30℃下进行减压蒸馏除去溶剂,最后用硅胶柱层析分离,流动相为石油醚与乙酸乙酯体积比为1:1的混合溶液,再使用旋转蒸发仪在0.01mpa、30℃下将分离后的有机相进行减压蒸馏除去溶剂,即可得到终产物。
对本实施例所制备的终产物进行如下表征和测试:
所制备的终产物质量为98mg,经计算可得产率为76%,或从2-氰甲基-5-氨基四唑为原料计算产率为65%。
采用差示扫描量热仪对所制备的终产物进行测试,从图6中可以得知其起始分解温度为182℃。
用红外光谱仪对所制备的终产物进行表征,图7的红外图谱中,1518.04cm-1和1491.04cm-1为氧化偶氮结构的特征峰,1413.88cm-1和1347.34cm-1为四唑环的特征峰。
图8和图9是通过核磁共振波谱仪表征得到的,图8的核磁共振氢谱(400mhz,dmso-d6)中6.46ppm和6.40ppm处有一组1:1的单峰,分别对应终产物中两个四唑环上的亚甲基取代基;图9的核磁共振碳谱(100mhz,dmso-d6)中165.7ppm和163.2ppm对应四唑环中的两个碳,113.2ppm和112.8ppm对应四唑环上的氰基取代基的两个碳,42.3ppm和41.5ppm对应四唑环上的亚甲基取代基的两个碳。
采用傅立叶离子回旋变换质谱对所制备的终产物进行表征,结果如图10所示,测得的[m-h]ˉ分子量为259.0557与2,2'-二氰甲基-5,5'-氧化偶氮四唑的[m-h]ˉ理论分子量259.0558几乎相等。
由上述表征结果可知,本实施例所制备的终产物为2,2'-二氰甲基-5,5'-氧化偶氮四唑。
通过gaussian09(revisionb.01)软件计算,可以预测所制备的2,2'-二氰甲基-5,5'-氧化偶氮四唑的生成焓为869.0kj·mol-1,密度为1.58g·cm-3,运用软件explo5可以预估2,2'-二氰甲基-5,5'-氧化偶氮四唑的预测爆速为7262m·s-1,爆压为18.7gpa,爆热为4379j·g-1。
实施例3
2,2'-二硝酸酯基乙基-5,5'-氧化偶氮四唑的合成
在10℃下,将0.5mmol1,3-二(2-羟乙基四唑-5-基)三氮烯加入0.75ml质量分数为80%的发烟硝酸(13.9mmol)中混合得到反应溶液,将反应溶液在25℃搅拌反应48h;随后在-15℃下向反应溶液滴加5.5ml三氟乙酸酐(39.0mmol),在0℃继续反应6h,之后将反应液倒入15ml冰水中,用乙酸乙酯萃取两次,分离后的有机相合并,之后依次用水萃取一次,饱和食盐水萃取一次,然后用无水硫酸钠对分离的有机相进行干燥,再将干燥后的有机相使用旋转蒸发仪在0.01mpa、30℃下进行减压蒸馏除去溶剂,最后用硅胶柱层析分离,流动相为石油醚与乙酸乙酯体积比为2:1的混合溶液,再使用旋转蒸发仪在0.01mpa、30℃下将分离后的有机相进行减压蒸馏除去溶剂,即可得到终产物。
对本实施例所制备的终产物进行如下表征和测试:
所制备的终产物质量为148mg,经计算可得产率为82%,或从2-羟乙基-5-氨基四唑为原料计算产率为57%。
采用差示扫描量热仪对所制备的终产物进行测试,由结果可知其熔点为114℃,起始分解温度为175℃。
用红外光谱仪对所制备的终产物进行表征,1635.71cm-1为硝酸酯基的特征峰,1521.90cm-1和1507.43cm-1为氧化偶氮结构的特征峰,1457.28cm-1和1283.68cm-1为四唑环的特征峰。
图11和图12是通过核磁共振波谱仪表征得到的,图11的核磁共振氢谱(400mhz,dmso-d6)中5.34ppm和5.29ppm处有一组1:1的单峰,分别对应终产物中乙基取代基中靠近四唑环的亚甲基,本应为三重峰,但由于裂分不明显故以类似单峰出现,而5.12ppm处的单峰为两个乙基取代基中靠近硝酸酯基的亚甲基重合产生;图12的核磁共振碳谱(100mhz,dmso-d6)中165.7ppm和163.2ppm对应四唑环中的两个碳,而70.2ppm、69.9ppm、52.0ppm和51.2ppm对应两侧乙基取代基的四个碳。
采用傅立叶离子回旋变换质谱对所制备的终产物进行表征,测得的[m+h]+分子量为361.0712与2,2'-二硝酸酯基乙基-5,5'-氧化偶氮四唑的[m+h]+理论分子量361.0712几乎相等。
由上述表征结果可知,本实施例所制备的终产物为2,2'-二硝酸酯基乙基-5,5'-氧化偶氮四唑。
通过gaussian09(revisionb.01)软件计算,可以预测所制备的2,2'-二硝酸酯基乙基-5,5'-氧化偶氮四唑的生成焓为345.4kj·mol-1,密度为1.68g·cm-3,运用软件explo5可以预估2,2'-二硝酸酯基乙基-5,5'-氧化偶氮四唑的预测爆速为8066m·s-1,爆压为25.8gpa,爆热为4944j·g-1。
实施例4
2,2'-二溴乙基-5,5'-氧化偶氮四唑的合成
在-15℃下,将0.5mmol1,3-二(2-溴乙基四唑-5-基)三氮烯加入1.201g丁二酸酐(12.0mmol)和0.5ml质量分数为95%的发烟硝酸(11.3mmol)的溶液中进行混合得到反应溶液;将反应溶液在0℃搅拌反应24h,之后将反应液倒入15ml冰水中,用乙酸乙酯萃取两次,分离后的有机相合并,之后依次用水萃取一次,饱和食盐水萃取一次,然后用无水硫酸钠对分离的有机相进行干燥,再将干燥后的有机相使用旋转蒸发仪在0.01mpa、30℃下进行减压蒸馏除去溶剂,最后用硅胶柱层析分离,流动相为石油醚与乙酸乙酯体积比为2:1的混合溶液,再使用旋转蒸发仪在0.01mpa、30℃下将分离后的有机相进行减压蒸馏除去溶剂,即可得到终产物。
对本实施例所制备的终产物进行如下表征和测试:
所制备的终产物质量为158mg,经计算可得产率为80%,或从2-溴乙基-5-氨基四唑为原料计算产率为71%。
采用显微熔点仪对所制备的终产物进行测试,得到其熔点为124℃~125℃。
用红外光谱仪对所制备的终产物进行表征,1513.22cm-1和1491.04cm-1为氧化偶氮结构的特征峰,1437.99cm-1和1302.97cm-1为四唑环的特征峰。
核磁共振氢谱(400mhz,dmso-d6)中5.35ppm和5.29ppm处有一组1:1的三重峰,分别对应终产物中乙基取代基中靠近四唑环的亚甲基,而4.11ppm和4.09ppm处的一组1:1的三重峰为乙基取代基中靠近溴原子的亚甲基;核磁共振碳谱(100mhz,dmso-d6)中165.6ppm和163.1ppm对应四唑环中的两个碳,而55.9ppm、55.0ppm、29.8ppm和29.3ppm对应两侧乙基取代基的四个碳。
采用傅立叶离子回旋变换质谱对所制备的终产物进行表征,测得的[m+h]+分子量为394.9320与2,2'-二溴乙基-5,5'-氧化偶氮四唑的[m+h]+理论分子量394.9322几乎相等。
由上述表征结果可知,本实施例所制备的物质为2,2'-二溴乙基-5,5'-氧化偶氮四唑。
实施例5
2,2'-二甲氧酰甲基-5,5'-氧化偶氮四唑的合成
在10℃下,将0.5mmol1,3-二(2-甲氧酰甲基四唑-5-基)三氮烯加入8ml异戊酸酐(41.0mmol)和0.75ml质量分数为86%的发烟硝酸(15.0mmol)的溶液中进行混合得到反应溶液;将反应溶液在25℃搅拌反应4h,之后将反应液倒入15ml冰水中,用乙酸乙酯萃取两次,分离后的有机相合并,之后依次用水萃取一次,饱和食盐水萃取一次,然后用无水硫酸钠对分离的有机相进行干燥,再将干燥后的有机相使用旋转蒸发仪在0.01mpa、30℃下进行减压蒸馏除去溶剂,最后用硅胶柱层析分离,流动相为石油醚与乙酸乙酯体积比为2:1的混合溶液,再使用旋转蒸发仪在0.01mpa、30℃下将分离后的有机相进行减压蒸馏除去溶剂,即可得到终产物。
对本实施例所制备的终产物进行如下表征和测试:
所制备的终产物质量为124mg,经计算可得产率为76%,或从2-甲氧酰甲基-5-氨基四唑为原料计算产率为63%。
采用显微熔点仪对所制备的终产物进行测试,得到其熔点为133℃~134℃。
用红外光谱仪对所制备的终产物进行表征,1746.62cm-1为酯基的特征峰,1514.19cm-1和1495.86cm-1为氧化偶氮结构的特征峰,1441.85cm-1和1244.14cm-1为四唑环的特征峰。
核磁共振氢谱(400mhz,dmso-d6)中6.12ppm和6.04ppm以及3.79ppm和3.77ppm处有两组1:1的三重峰,分别对应终产物中亚甲基和甲基取代基;核磁共振碳谱(100mhz,dmso-d6)中166.3ppm和163.7ppm对应四唑环中的两个碳,166.8ppm和166.3ppm对应甲氧酰甲基的羰基碳,而55.3ppm、54.5ppm、53.7ppm和53.6ppm对应两侧甲氧酰甲基的亚甲基和甲基的四个碳。
采用傅立叶离子回旋变换质谱对所制备的终产物进行表征,测得的[m+h]+分子量为327.0904与2,2'-二甲氧酰甲基-5,5'-氧化偶氮四唑的[m+h]+理论分子量327.0908几乎相等。
由上述表征结果可知,本实施例所制备的终产物为2,2'-二甲氧酰甲基-5,5'-氧化偶氮四唑。
综上所述,以上仅为本发明的较佳实施例而已,并非用于限定本发明的保护范围。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一类用于全固态量子点敏化太阳能电池的双电荷有机空穴传输材料的制作方法
- 一种双偶氮苯聚合物作为双光子光存储介质用途及结构的制作方法
- 电解还原生产对氨基苯酚的工艺及其装置的制作方法
- 液晶化合物—4'-正己氧基-4-偶氮苯羧酸-3-氟-4-氰基苯酯及其制备方法
- 液晶化合物—4'-正戊氧基-4-偶氮苯羧酸-3-氟-4-氰基苯酯及其制备方法
- 液晶化合物—4'-正庚氧基-4-偶氮苯羧酸-3-氟-4-氰基苯酯及其制备方法
- 液晶化合物—4'-正辛氧基-4-偶氮苯羧酸-3-氟-4-氰基苯酯及其制备方法
- 液晶化合物—4'-烯丙氧基-4-偶氮苯羧酸-3-氟-4-氰基苯酯及其制备方法
- 液晶化合物—4'-正癸氧基-4-偶氮苯羧酸-3-氟-4-氰基苯酯及其制备方法
- 一种五偶氮苯及其合成方法