用于对复杂异质群落中的完整微生物株系进行分析、确定其功能关系及相互作用以及基于此来识别和合成生物活性改性剂的方法、设备和系统与流程
本申请可能包含受版权、掩码作品和/或其他知识产权保护的材料。此类知识产权的相应所有者不反对任何人对公开内容的拓制,因为它出现在已公布的专利局文件/记录中,但在其他方面保留所有权利。
背景技术:
微生物在自然界中以群落形式共存并参与多种相互作用,从而导致各个群落成员之间的协作和竞争。微生物生态学的进展揭示了大多数群落中高度的物种多样性和复杂性。微生物在环境中无处不在,它们栖息在生物圈内的各种各样的生态系统中。各个微生物及其相应群落在诸如海洋(深海和海洋表面)、土壤和动物组织(包括人体组织)的环境中发挥着独特的作用。
技术实现要素:
公开了用于对复杂异质群落中的完整微生物株系进行分析、确定其功能关系及相互作用以及基于此来识别和/或合成生物活性改性剂(诸如微生物群集)的方法、设备和系统。还公开了基于此来识别和利用感兴趣的途径和功能的方法。
在本公开的一个方面,公开了用于从多个样品中识别活性微生物以及对识别的微生物与至少一个元数据进行分析的方法。所公开的方法的实施方案包括获得在一些情况下共有至少一个共同环境参数的至少两个样品或样品集合,以及针对每个样品和/或样品集合,检测每个样品中一种或多种微生物类型的存在及其相应的量。然后对每个样品中的dna进行测序以识别多种微生物株系的全基因组,并通过将测序读段映射到所识别的全基因组来确定样品中每种株系的相对频率。每个样品中每种微生物株系的绝对细胞计数由每种微生物类型的量和每种株系的相对频率确定。通过对每个样品中的cdna进行测序并将所测序的cdna映射到所识别的全基因组来测量每种微生物株系的rna。基于映射到每个全基因组的cdna读段的量确定每个样品中每种微生物株系的活性。根据确定的活性来过滤、筛选和/或评估每种微生物株系的绝对细胞计数,以提供所述至少两个样品和/或样品集合中每一个的活性微生物株系及其相应绝对细胞计数的集合。对所述至少两个样品和/或样品集合中每一个的活性微生物株系的经过滤的绝对细胞计数与所述至少两个样品和/或样品集合中每一个的至少一个测得元数据进行比较。根据预测的功能和/或化学性质,可以将活性微生物株系分组和/或分类成例如至少两组。在一些情况下,生物活性改性剂的识别和/或生成/合成可包括一种或多种化合物和/或至少一种微生物株系、和/或来自所述至少两组中的每一组或其他分类/识别的至少一种微生物株系、和/或经确定会影响或改变至少一种微生物株系的活性或行为和/或两种或更多种株系之间的关系和/或分类/组的化合物、以及识别的一种或多种化合物,例如选择的微生物株系,其与载体培养基组合以形成生物活性改性剂(诸如活性微生物的生物群集),该生物活性改性剂(例如,生物群集)被配置为当被引入到目标生物环境中时改变和/或调节与该目标生物环境的至少一个元数据相对应的性质。应当理解,尽管给出了使用微生物生物群集的实例,但是这些概念也可以应用于其他生物活性改性剂,因此这些实例不将本公开限制于微生物生物群集。
所公开的方法的其他实施方案包括获得共有至少一个共同环境参数的至少两个样品/样品集合,以及针对每个样品,检测每个样品中一种或多种微生物类型的存在及其相应量。然后对每个样品中的dna进行测序以识别多种微生物株系的全基因组,并通过将测序读段映射到所识别的全基因组来确定样品中每种株系的相对频率。每个样品中每种微生物株系的绝对细胞计数由每种微生物类型的量和每种株系的相对频率确定。通过处理每个样品中的蛋白质(例如,通过质谱法)并将得自该处理的氨基酸序列映射到所识别的全基因组来测量每种微生物株系的蛋白质。基于映射到每个全基因组的氨基酸序列的量确定每个样品中每种微生物株系的活性。通过过滤所确定的活性可以确定每种微生物株系的绝对细胞计数,以提供所述至少两个样品中的每一个的活性微生物株系及其相应绝对细胞计数的列表。可以对所述至少两个样品中的每一个的活性微生物株系的经过滤的绝对细胞计数与所述至少两个样品中的每一个的至少一个测得元数据进行分析或比较,并且根据预测的功能和/或化学性质将活性微生物株系分组成至少两组。可以基于此来选择和/或生成一种或多种生物活性改性剂,例如,可以选择来自所述至少两组中的每一组的至少一种微生物株系并将其与载体培养基组合以形成生物活性改性剂(这里称为活性微生物的生物群集),该生物活性改性剂被配置为当被引入到目标生物环境中时改变/调节与该目标生物环境的至少一个元数据相对应的性质。
所公开的方法的一些实施方案包括获得共有至少一个共同环境参数的至少两个样品/样品集合,以及对于每一者,检测每个样品中一种或多种微生物类型的存在及其相应量。然后对每个样品中的dna进行测序以识别多种微生物株系的全基因组,并通过将测序读段映射到所识别的全基因组来确定样品中每种株系的相对频率。每个样品中每种微生物株系的绝对细胞计数由每种微生物类型的量和每种株系的相对频率确定。通过对每个样品中的cdna进行测序并将所测序的cdna映射到所识别的全基因组来测量每种微生物株系的rna。基于映射到每个全基因组的cdna读段的量确定每个样品中每种微生物株系的活性。根据所确定的活性来过滤/筛选/评估每种微生物株系的绝对细胞计数,以提供所述至少两个样品/组中每一个的活性微生物株系及其相应绝对细胞计数的集合。对所述至少两个样品/组中每一个的活性微生物株系的经过滤的绝对细胞计数与所述至少两个样品中每一个的至少一个测得元数据进行比较。根据预测的功能和/或化学性质,将活性微生物株系分组成至少两组。然后,对于每组,确定该组中至少两种活性微生物株系之间的遗传相似性,以识别该组的至少一种感兴趣的功能。然后可以基于每组的所述至少一种识别的功能来识别一条或多条途径。然后,可以亚克隆和/或以其他方式利用一条或多条识别的途径,例如,以用于工业和/或天然产品生产目的、新化合物合成和/或新化合物选择,然后可以被合成和/或用作生物活性改性剂。另选地或另外地,可以基于所识别的至少一种感兴趣的功能而选择来自所述至少两组中的每一组的至少一种微生物株系和/或至少一种株系,并将其与载体培养基组合以形成活性微生物株系的生物群集,该生物群集被配置为当被引入到目标生物环境中时改变与该目标生物环境的至少一个元数据相对应的性质。
所公开的方法还包括确定样品中一种或多种活性微生物株系的绝对细胞计数,其中所述一种或多种活性微生物株系存在于样品中的微生物群落中。所述一种或多种微生物株系是微生物类型的亚分类单元(subtaxon)。本文提供的方法中使用的样品可以是任何环境来源的。例如,在一个实施方案中,样品来自动物、土壤(例如,根外土壤或根际土壤)、空气、盐水、淡水、污水污泥、沉积物、油、植物、农产品、植物或极端环境。在另一个实施方案中,动物样品是血液、组织、牙齿、汗液、指甲、皮肤、毛发、粪便、尿液、精液、粘液、唾液、胃肠道、瘤胃、肌肉、脑、组织或器官样品。其他应用包括用于食品,尤其是发酵食品和微生物食品,例如面包、奶酪、葡萄酒、啤酒、泡菜等。在一个实施方案中,提供了用于确定一种或多种活性微生物株系的绝对细胞计数的方法。
根据一些实施方案,提供了一种形成生物活性改性剂(诸如活性微生物株系的生物群集)的方法,该生物活性改性剂被配置为改变目标生物环境中的性质。此类方法可以包括获得共有至少一个共同环境参数(诸如样品类型、样品时间、样品位置、样品来源类型等)的至少两个样品(或样品集合),并检测每个样品中多种微生物类型的存在。然后确定每个样品中所述多种微生物类型中的每种检出微生物类型的绝对细胞数量(例如,以非限制性实例的方式,染色程序、细胞分选/facs等,如本文所讨论),并且测量每个样品中独特的第一标记物的数量及其量,每个独特的第一标记物是检出微生物类型的微生物株系的标记物。根据该实施方案,某些检出的微生物/株系可以从进一步处理/分析中省略,例如,以提高效率。基于该样品中每种检出微生物类型的数量和该样品中独特的第一标记物的数量及其量,确定存在于每个样品中的一些或每种微生物株系的绝对细胞计数。测量每种微生物株系的指示活性(例如,代谢活性)的至少一个独特的第二标记物,以确定每个样品中的活性微生物株系,并生成所述至少两个样品中的每一个的活性微生物株系及其相应绝对细胞计数的集合或列表。对所述至少两个样品中的每一个的活性微生物株系及相应绝对细胞计数与所述至少两个样品中的每一个的至少一个测得元数据进行分析,以识别每种活性微生物株系与至少一个测得元数据、每个样品的测得元数据和/或样品集合的测得元数据之间的关系。基于该分析,选择多种活性微生物株系并将其与载体培养基组合以形成活性微生物的生物群集,该活性微生物的生物群集被配置为当被引入到目标生物环境中时改变该目标生物环境的至少一个性质(对应于所述至少一个元数据)。取决于该实施方案,元数据可以是一个或多个环境参数,并且可以在样品或样品集合之间相同或相对相似,在不同样品或样品集合之间具有不同的值。例如,奶牛的元数据可以包括饲料和产奶量,并且饲料元数据值可以相同(即,向奶牛饲喂相同的饲料),而产奶量则可以变化(即,来自一头奶牛的样品或来自特定牛群的样品集合的平均产奶量不同于对应于来自第二头奶牛的样品或来自单独牛群的样品集合的平均产奶量)。
根据本公开的一些实施方案,提供了用于分析微生物群落的方法。此类方法可包括获得至少两个样品(或至少两个样品的数据),每个样品包括异质微生物群落,并检测每个样品中多种微生物类型的存在。然后,确定每个样品中所述多种微生物类型中的每种检出微生物类型的绝对细胞数量(例如,通过facs或本文所讨论的其他方法)。测量每个样品中独特的第一标记物的数量及其量,每个独特的第一标记物是检出微生物类型的微生物株系的标记物;如上所讨论,本公开的实施方案利用微生物株系的全基因组;在本公开的一些实施方案中,独特的标记物/独特的第一标记物包含全基因组;在本公开的一些实施方案中,独特的标记物/独特的第一标记物是全基因组;在一些实施方案中,独特的标记物/独特的第一标记物由全基因组组成;在一些实施方案中,独特的标记物/独特的第一标记物基本上由全基因组组成。测量一种或多种独特的第二标记物的值(活性、浓度、表达等),独特的第二标记物指示检出的微生物类型的特定微生物株系的活性(例如,代谢活性),以及基于所述一个或多个独特的第二标记物的测得值(例如,基于超过指定设定阈值的值)确定每种检出的微生物株系的活性;在本公开的一些实施方案中,独特的标记物/独特的第二标记物包含全转录组、蛋白质组或代谢组;在本公开的一些实施方案中,独特的标记物/独特的第二标记物是全转录组、蛋白质组或代谢组;在一些实施方案中,独特的标记物/独特的第二标记物由全转录组、蛋白质组或代谢组组成;在一些实施方案中,独特的标记物/独特的第二标记物基本上由全转录组、蛋白质组或代谢组组成。确定每种检出的活性微生物株系的比例存在和/或相应比率(例如,基于每种微生物类型的株系的相对量、每种微生物类型的数量/每种类型的相应绝对细胞计数、每种检出的活性微生物株系的绝对细胞计数、第一独特标记物值、第二独特标记物值等)。然后分析所述至少两个样品中的每种检出活性微生物株系(或其子集)以识别每种检出活性微生物株系与其他检出活性微生物株系之间的关系及其强度,以及每种检出活性微生物株系与至少一个测得元数据之间的关系及其强度。然后显示或以其他方式输出所识别的关系,并且可以将其用于识别和/或生成一种或多种生物活性改性剂(例如生物群集和/或组合物、化合物、代谢物、抗生素等,其被配置为更改、改变、修改、影响和/或调节(通常“调节”)一种或多种所识别的关系)。在一些实施方案中,仅显示超过某一强度或权重的关系。如本公开通篇所详述的,生物活性改性剂(例如生物群集)可以被配置为使得在被引入到目标环境中时,一种或多种生物活性改性剂调节目标环境/生物环境/系统的至少一种性质(尤其是与测得元数据相关的一种或多种性质)。
根据本公开的一些实施方案,方法包括检测多个样品中多种微生物类型的存在,并确定每个样品中每种检出微生物类型的绝对细胞数量。可以测量每个样品中独特的第一标记物的数量及其量,独特的第一标记物是微生物株系的基因组。测量一种或多种独特的第二标记物的值或水平,独特的第二标记物指示特定微生物株系的代谢活性。基于测得值或水平,确定或定义每个样品的每种检出微生物株系的活性(例如,基于超过指定阈值的测得值或水平)。确定样品中每种检出活性微生物株系的加权或细胞调整值(加权或细胞调整值不是相对丰度)。在一些实现方式中,加权或细胞调整值是某株系相对于所有株系的所有绝对细胞计数的总和而言的绝对细胞计数。
分析每个样品(或样品集合)的每种检出活性微生物株系。该分析可以包括识别每种检出的具有某加权值的活性微生物株系与具有某加权值的每个其他活性微生物株系之间的关系及其强度,以及具有某加权值的每种活性微生物株系与一个或多个测得元数据之间的关系及其强度。
然后可以显示或以其他方式输出所识别的关系(在一些实施方案中,诸如加权值和强度的相关数据),并且可以将其用于生成合成群集。在一些实施方案中,显示或输出所识别的每个元数据的关系。在一些实施方案中,所显示的或输出的关系识别或被配置为有助于识别导致疾病的一种或多种微生物株系。在一些实施方案中,所显示的或输出的关系识别或被配置为有助于识别用于治疗疾病或病症的一种或多种微生物株系。
在一些实施方案中,仅显示或输出超过某一强度或权重(例如,超过指定阈值或基值)的关系。如本公开通篇所详述的,可以配置合成群集,使得在引入到目标环境中时,合成群集可以更改或改变目标环境的性质(尤其是与测得元数据相关的性质)。在一些实现方式中,上述方法可用于识别、合成和/或形成被配置为改变生物环境中的性质的生物活性改性剂,诸如活性微生物株系的合成群集,并且基于两个或更多个样品集合(每个样品集合具有多个环境参数),所述多个环境参数中的至少一个参数是在所述两个或更多个样品集合之间相似的共同环境参数,并且至少一个环境参数是在所述两个或更多个样品集合中的每一个之间不同的不同环境参数。在一些实现方式中,每个样品集合包括至少一个样品,其包含从生物样品来源获得的异质微生物群落。在一些实现方式中,至少一种活性微生物株系是一种或多种微生物类型的亚分类单元。
在本公开的一些实施方案中,所述一种或多种微生物类型是一种或多种细菌(例如,支原体、球菌、芽孢杆菌、立克次体、螺旋体)、真菌(例如,丝状真菌、酵母菌)、线虫、原生动物、古细菌、藻类、甲藻、病毒(例如,噬菌体)、类病毒和/或它们的组合。在一个实施方案中,所述一种或多种微生物株系是一种或多种细菌(例如,支原体、球菌、芽孢杆菌、立克次体、螺旋体)、真菌(例如,丝状真菌、酵母菌)、线虫、原生动物、古细菌、藻类、甲藻、病毒(例如,噬菌体)、类病毒和/或它们的组合。在进一步的实施方案中,所述一种或多种微生物株系是一种或多种真菌物种或真菌亚种。在进一步的实施方案中,所述一种或多种微生物株系是一种或多种细菌物种或细菌亚种。在更进一步的实施方案中,样品是瘤胃样品。在一些实施方案中,瘤胃样品来自牛。在更进一步的实施方案中,样品是胃肠样品。在一些实施方案中,胃肠样品来自猪或鸡。
在一些实施方案中,该方法包括测定样品中一种或多种活性微生物株系的绝对细胞计数,检测样品中一种或多种微生物类型的存在,以及确定样品中所述一种或多种微生物类型中的每一种的绝对数量。测量独特的第一标记物的数量以及每个独特的第一标记物的量或丰度。如本文所述,独特的第一标记物可以是独特的微生物株系的基因组。然后可以通过测量一种或多种独特的第二标记物的表达水平例如在蛋白质或rna水平上评估活性。根据该实施方案,独特的第二标记物可以与第一独特标记物相同或不同,并且是生物株系活性的标记物;例如,如果在一些实施方案中靶向特定的基因或区域,则第二和第一独特的标记物可以是相同的,并且其中如果生物是活跃转录的,则dna和rna序列可以是相同的。基于一种或多种独特的第二标记物的表达水平,确定哪一种或多种(如果有的话)微生物株系是有活性的。在一个实施方案中,如果微生物株系以阈值水平或高于阈值水平的百分比表达第二独特标记物,则认为其是有活性的。基于所述一种或多种活性微生物株系的所述一种或多种第一标记物的量和所述一种或多种微生物株系是其中的亚分类单元的微生物类型的绝对数量,来确定所述一种或多种活性微生物株系的绝对细胞计数。
在一个实施方案中,确定样品中所述一种或多种生物类型中的每一种的数量包括对样品或其部分进行核酸测序、离心、光学显微镜检查、荧光显微镜检查、染色、质谱分析、微流体分析、定量聚合酶链式反应(qpcr)或流式细胞术。
在另一个实施方案中,测量独特的第一标记物的数量及其量包括对来自样品的基因组dna进行高通量测序反应。在一个实施方案中,对独特的第一标记物的测量包括标记物特异性反应,例如,使用对独特的第一标记物具有特异性的引物。在另一个实施方案中,使用元基因组方法。
在一个实施方案中,测量一种或多种独特的第二标记物的表达水平包括对样品中的rna(例如,mirna、trna、rrna和/或mrna)进行表达分析。在进一步的实施方案中,基因表达分析包括测序反应。在另一个实施方案中,rna表达分析包括定量聚合酶链式反应(qpcr)、元转录组测序和/或转录组测序。
在一些实施方案中,测量样品中第二独特标记物的数量包括测量独特蛋白质标记物的数量。在一些实施方案中,测量样品中独特的第二标记物的数量包括测量独特代谢物标记物的数量。在一些实施方案中,测量样品中独特代谢物标记物的数量包括测量独特碳水化合物标记物的数量。在一些实施方案中,测量样品中独特代谢物标记物的数量包括测量独特脂质标记物的数量。在一些实施方案中,测量多个样品中所述一种或多种微生物株系的绝对细胞计数。在进一步的实施方案中,所述多个样品获自相同的环境或类似的环境。在一些实施方案中,在多个时间点获得所述多个样品。
在一些实施方案中,测量一种或多种独特的第二标记物的水平包括对样品或其部分进行质谱分析。在一些实施方案中,测量一种或多种独特的第二标记物的表达水平包括对样品或其部分进行元核糖体图谱分析(metaribosomeprofiling)和/或核糖体图谱分析。
在本公开的另一方面,确定了在多个样品中用于确定一种或多种活性微生物株系的绝对细胞计数的方法,并且绝对细胞计数水平与一种或多种元数据(例如,环境)参数相关联。在一个实施方案中,将绝对细胞计数水平与一种或多种元数据参数相关联包括共现测量(co-occurrencemeasurement)、互信息测量(mutualinformationmeasurement)、链接分析等。在一个实施方案中,所述一种或多种元数据参数是第二活性微生物株系的存在。因此,在该方法的一个实施方案中,使用绝对细胞计数值来确定微生物群落中所述一种或多种活性微生物株系与环境参数的共现。在另一个实施方案中,将所述一种或多种活性微生物株系的绝对细胞计数水平与诸如饲喂条件、ph、营养物或从中获得微生物群落的环境的温度之类的环境参数相关联。
在这方面,将一种或多种活性微生物株系的绝对细胞计数与一个或多个环境参数相关联。环境参数可以是样品本身的参数,例如,ph、温度、样品中蛋白质的量、群落中其他微生物的存在。在一个实施方案中,该参数是从中获得样品的宿主的特定基因组序列(例如,特定的基因突变)。另选地,环境参数是影响微生物群落特性变化的参数(即,其中微生物群落的“特性”由群落中微生物株系的类型和/或特定微生物株系的数量表征),或受微生物群落特性变化的影响的参数。例如,在一个实施方案中,环境参数是动物的食物摄入量或由泌乳反刍动物产生的乳汁量(或乳汁的蛋白质或脂肪含量)。在本文描述的一些实施方案中,环境参数被称为元数据参数。
在一个实施方案中,确定样品中一种或多种活性微生物株系的共现包括创建使用表示一个或多个环境参数和活性微生物株系关联的链接来填充的矩阵。
在一个实施方案中,确定一种或多种活性生物株系和元数据参数的共现包括用于测量网络内株系的连通性的网络和/或聚类分析方法,其中该网络是共有一个共同或类似环境参数的两个或更多个样品的集合体。在一些实施方案中,网络分析和/或网络分析方法包括图论、物种群落规则(speciescommunityrule)、特征向量(eigenvector)/模块性矩阵、分组策略(gambitofthegroup)和/或网络测量中的一个或多个。在一些实现方式中,网络测量包括观测矩阵、时间聚合网络、分层聚类分析、节点级度量和/或网络级度量中的一个或多个。在一些实施方案中,节点级度量包括度、强度、中介中心性、特征向量中心性、页面排名和/或范围中的一个或多个。在一些实施方案中,网络级度量包括密度、同质性/同配性和/或传递性中的一个或多个。
在一些实施方案中,网络分析包括链接分析、模块性分析、稳健性测量、中介性测量、连通性测量、传递性测量、中心性测量或其组合。在另一个实施方案中,聚类分析方法包括建立连通性模型、子空间模型、分布模型、密度模型或质心模型。在另一个实施方案中,网络分析包括通过链接挖掘和预测、集体分类(collectiveclassification)、基于链接的聚类、关系相似性或其组合的网络预测建模。在另一个实施方案中,网络分析包括在变量之间建立连接的互信息、最大信息系数计算或其他非参数方法。在另一个实施方案中,网络分析包括基于微分方程的群体建模。在另一个实施方案中,网络分析包括lotka-volterra建模。
基于该分析,可以显示或以其他方式输出株系关系,和/或识别包括在微生物群集中的一种或多种活性相关株系。
附图说明
图1示出了本公开的一些实施方案的示例高级过程流。
图1-1示出了本公开的一些实施方案的示例高级过程流。
图1-2示出了本公开的一些实施方案的示例高级过程流。
图1a示出了根据一些实施方案的用于筛选和分析来自复杂异质群落的微生物株系、预测其功能关系及相互作用以及基于此选择和合成微生物群集的示例高级过程流。
图1b示出了根据一些实施方案的用于确定一种或多种活性微生物株系的绝对细胞计数的一般过程流。
图1c示出了根据一些实施方案的用于微生物群落分析、类型/株系-元数据关系确定、显示和生物群集生成的过程流。
图1d示出了根据一些实施方案的所分析的株系和关系的示例性视觉输出。
图1e示出了根据一些实施方案的瘤胃细菌的mic得分分布和乳脂效率。
图1f示出了根据一些实施方案的瘤胃真菌的mic得分分布和乳脂效率。
图1g示出了根据一些实施方案的瘤胃细菌的mic得分分布和产乳效率。
图1h示出了根据一些实施方案的瘤胃真菌的mic得分分布和产乳效率。
图2a提供了整合本公开的一些实施方案的元基因组学(在图2b中详述)、元转录组学(在图2c中详述)、封闭的基因组和特征选择部件的概述流程。
图2d提供了根据本公开的一些实施方案的代谢组学的概述。
图2e提供了示出根据本公开的一些实施方案的多组学整合的流程图,而图2f提供了其实例的结果。
图2g示出了根据一些实施方案确定样品中的一种或多种活性微生物株系与一种或多种元数据(环境)参数的共现的一般过程流。
图3a是示出根据一些实施方案的示例性微生物相互作用分析和选择系统300的示意图,并且图3b是用于这种系统的示例过程流。参考图3a和图3b描述了用于确定天然微生物群落内的多维种间相互作用和依赖性、识别活性微生物以及选择多种活性微生物以形成将改变一个或多个指定参数和/或相关测量的微生物的群集、聚集体或其他合成分组的系统和过程。
图3c和图3d提供了示出本公开的一些方面的示例性数据。
图4示出了在实验过程中产生的乳脂磅数的非线性,以用于确定影响奶牛乳脂产生的瘤胃微生物群落成分。
图5示出了在过滤活性的情况下目标株系ascus_713的绝对细胞计数与产生的乳脂磅数(lbs)的相关性。
图6示出了在过滤活性的情况下目标株系ascus_7的绝对细胞计数和实验过程中产生的乳脂磅数(lbs)。
图7示出了在不过滤活性的情况下目标株系ascus_3038的相对丰度与产生的乳脂磅数(lbs)的相关性。
图8示出了给奶牛施用根据所公开的方法制备的微生物群集的现场试验结果;图8a示出了随时间产生的平均乳脂磅数;图8b示出了随时间产生的平均乳蛋白磅数;并且图8c示出了随时间产生的平均能量校正乳(ecm)磅数。
具体实施方式
微生物群落是许多不同类型的生态系统中的环境过程以及地球生物地球化学的核心,例如通过循环营养物和固定碳(falkowski等人(1998)science281,第237-240页,该文献全文出于所有目的以引用方式并入本文)。然而,由于群落复杂性和任何给定微生物群落的大多数成员缺乏可培养性,对这些过程的分子和生态细节以及影响因素仍然知之甚少。
微生物群落的定性和定量组成不同,每个微生物群落都是独特的,其组成取决于其所在的给定生态系统和/或环境。微生物群落成员的绝对细胞计数受该群落所处环境的变化以及微生物引起的生理和代谢变化(例如,细胞分裂、蛋白质表达等)的影响。群落内环境参数和/或一种活性微生物的量的变化可能对该群落的其他微生物以及发现该群落的生态系统和/或环境产生深远影响。为了理解、预测和应对这些微生物群落的变化,有必要识别样品中的活性微生物以及相应群落中活性微生物的数量。然而,到目前为止,对微生物群落成员的绝大多数研究都集中在特定微生物群落中微生物的比例上,而不是绝对细胞计数(segata等人(2013)molecularsystemsbiology9,第666页,全文出于所有目的以引用方式并入本文)。
尽管可以容易地确定微生物群落组成(例如,通过使用高通量测序方法),但是需要更深入地理解相应群落的聚集和维持方式。
微生物群落参与多种关键过程,诸如基本元素(例如碳、氧、氮、硫、磷和各种金属)的生物地球化学循环;并且相应群落的结构、相互作用和动力学对生物圈的存在至关重要(zhou等人(2015)mbio6(1):e02288-14.doi:10.1128/mbio.02288-14,全文出于所有目的以引用方式并入本文)。此类群落是高度异质的,并且几乎总是包括细菌、病毒、古细菌和其他微真核生物诸如真菌的复杂混合物。人体环境(诸如肠道和阴道)中的微生物群落异质性水平与诸如炎症性肠病和细菌性阴道病的疾病有关(nature(2012),第486卷,第207页,全文出于所有目的以引用方式并入本文)。然而,值得注意的是,即使是在健康个体的此类环境中,组织中存在的微生物也有着显著差异(nature(2012),第486卷,第207页)。
由于许多微生物可能是无法培养的或者难以培养/培养费用高昂,因此不依赖于培养的方法(诸如核酸测序)增进了对各种微生物群落的多样性的理解。小亚基核糖体rna(ssurrna或16srrna)基因的扩增和测序是部分地基于基因的普遍存在和相对一致的进化速率来研究群落中的微生物多样性的基本方法。高通量方法的进展已导向元基因组学分析,其中对微生物的整个基因组进行测序。此类方法不需要群落的先验知识,使得能够发现新的微生物株系。元基因组学、元转录组学、元蛋白质组学和代谢组学都能够探测群落以辨别结构和功能。
不仅能够对群落中的微生物进行编目,而且能够辨认哪些成员是有活性的、这些生物的数量以及微生物群落成员彼此之间的共现及与环境参数的共现(例如,群落中两种微生物响应群落环境的某些变化的共现),这样的能力将使得可以理解相应环境因素(例如,气候、存在的营养物、环境ph值)对微生物群落内微生物特性(及其相应数量)的重要性,以及某些群落成员对群落所处环境的重要性。本公开解决了这些和其他需求。
如本说明书中所使用的,除非上下文另有明确说明,否则单数形式“一个/种”和“该/所述”包括复数指代。因此,例如,术语“一种生物类型”旨在表示单种生物类型或多种生物类型。又如,术语“一个环境参数”可以表示单个环境参数或多个环境参数,使得词语“一个/种”不排除存在多于一个环境参数的可能性,除非上下文明确要求有且只有一个环境参数。
在本说明书通篇中对“一个实施方案”、“一个方面”或“一种实现方式”的引用表示结合该实施方案描述的特定特征、结构或特性包括在本公开的至少一个实施方案中。因此,在本说明书通篇中各个位置出现的短语“在一个实施方案中”不一定都指的是同一个实施方案。此外,特定特征、结构或特性可以在一个或多个实施方案中以任何合适的方式组合。
如本文所用,在特定实施方案中,当术语“约”或“大约”出现在数值之前时,表示该值加或减10%的范围。在提供值的范围的情况下,应当理解的是,在该范围的上限与下限之间的每个居间值(除非上下文另外明确指出,否则达到下限的单位的十分之一)以及该规定范围内的任何其他规定值或居间值都涵盖在本公开内。这些较小范围的上限和下限可以独立地包括在本公开也涵盖的较小范围内,受限于该规定范围内的任何明确排除的限值。在该规定范围包括一个或两个限值的情况下,排除所包括的那些限值之一或两者的范围也包括在本公开中。
如本文所用,“分离”、“分离的”、“分离的微生物”和类似术语旨在表示一种或多种微生物已经与在特定环境(例如土壤、水、动物组织)中与其相关的至少一种材料分开。因此,“分离的微生物”不存在于其天然存在的环境中;相反,通过本文所述的各种技术,微生物已从其自然环境中去除并被置于非天然存在的状态。因此,分离的株系可以作为例如生物学纯培养物存在,或者作为与可接受的载体结合的孢子(或其他形式的株系)存在。
如本文所用,“生物活性改性剂”是指一种组合物,诸如包含一种或多种活性微生物的微生物群集,其通过本公开的方法、系统和/或设备识别并且不自然存在于天然存在的环境中,和/或其比率、百分比和/或量与天然存在的不一致和/或不存在于自然界中。例如,可以从识别或产生的化合物/组合物、和/或一种或多种分离的微生物株系、以及合适的培养基或载体形成生物活性改性剂,诸如微生物群集(也称为合成群集或生物群集)或生物活性改性剂聚集体。生物活性改性剂可以应用或施用于目标,诸如目标环境、群体、个体、动物等。
在一些实施方案中,根据本公开的生物活性改性剂诸如微生物群集选自和/或基于活性相关单个微生物物种或物种的株系的集合、子集和/或分组。通过本公开的方法识别的关系和网络基于执行一项或多项共同的功能而被分组、相关和/或链接,或者可被描述为参与、或导向可识别参数诸如感兴趣的表型性状(例如反刍动物中产奶量增加)和/或与可识别参数相关联。在一些实现方式中,从中选择微生物群集和/或由此选择生物活性改性剂的组、和/或生物活性改性剂诸如微生物群集本身可以包括微生物的两个或更多个种、种的株系、或不同种的株系。在一些情况下,微生物可以共生地在组、生物活性改性剂和/或微生物群集中共存。
在本公开的某些方面,生物活性改性剂和/或微生物群集是或基于一种或多种分离的作为分离的且生物学纯的培养物存在的微生物。应当理解,特定微生物的分离的且生物学纯的培养物表示所述培养物基本上不含(在科学可解释的范围内)其他活生物,并且仅含有所讨论的单个微生物。培养物可含有不同浓度的所述微生物。本公开内容指出,分离的且生物学纯的微生物通常“必然不同于不太纯的或不纯的材料”。参见例如bergstrom,427f.2d1394,(ccpa1970)(讨论纯化的前列腺素),另参见bergy,596f.2d952(ccpa1979)(讨论纯化的微生物),另参见parke-davis&co.v.h.k.mulford&co.,189f.95(s.d.n.y.1911)(learnedhand,讨论纯化的肾上腺素),部分维持原判(aff’dinpart),部分撤销原判(rev’dinpart),196f.496(第二巡回上诉法院(2dcir.),1912),其中每篇文献均全文以引用方式并入本文。此外,在一些方面,本公开的实现方式可能需要对在所公开的微生物群集中使用的分离的且生物学纯的微生物培养物必须达到的浓度或纯度限制进行某些定量测量。在某些实施方案中,这些纯度值的存在是进一步的属性,其将通过本发明公开的方法识别的微生物与以天然状态存在的那些微生物区分开。参见例如merck&co.v.olinmathiesonchemicalcorp.,253f.2d156(第四巡回上诉法院,1958)(讨论由微生物产生的维生素b12的纯度限制),该文献以引用方式并入本文。
如本文所用,“载体”、“可接受的载体”或“药物载体”是指与生物活性改性剂诸如微生物群集一起使用或在其中使用的稀释剂、佐剂、赋形剂或媒介物。此类载体可以是无菌液体,诸如水和油,包括石油、动物油、植物油或合成来源的油;诸如花生油、大豆油、矿物油、芝麻油等。优选使用水或水溶液、盐水溶液、葡萄糖水溶液和甘油溶液作为载体,在一些实施方案中作为可注射溶液。另选地,载体可以是固体剂型载体,包括但不限于粘合剂(用于压制丸剂)、助流剂、包封剂、调味剂和着色剂中的一种或多种。可以关于预期的施用途径和标准药学实践来进行载体的选择。参见hardee和baggo(1998.developmentandformulationofveterinarydosageforms.第2版,crcpress.第504页);e.w.martin(1970.remington’spharmaceuticalsciences.第17版,mackpub.co.);以及blaser等人(美国公布us20110280840a1),每篇文献全文以引用方式明确地并入本文。
术语“微生物”和“微小生物”在本文中可互换使用,并且是指细菌、真核生物或古细菌域的任何微生物。微生物类型包括但不限于细菌(例如,支原体、球菌、芽孢杆菌、立克次体、螺旋体)、真菌(例如,丝状真菌、酵母菌)、线虫、原生动物、古细菌、藻类、甲藻、病毒(例如,噬菌体)、类病毒和/或它们的组合。生物株系是生物类型的亚分类单元,并且可以是例如特定微生物的种、亚种、亚型、遗传变体、致病变型或血清变型。
如本文所用的术语“标记物”或“独特标记物”是独特微生物类型、微生物株系或微生物株系活性的全基因组、全转录组、全蛋白质组和/或全代谢组的指示物。标记物可以在生物样品中测量,并且包括但不限于基于核酸的标记物(诸如核糖体rna基因)、基于肽或蛋白质的标记物、和/或代谢物或其他小分子标记物。
如本文所用的术语“代谢物”是代谢的中间体或产物。在一个实施方案中,代谢物是小分子。代谢物在以下方面具有多种功能,包括:燃料、结构、信号传导、对酶的刺激和抑制作用、作为酶的辅助因子、防御、以及与其他生物体的相互作用(如色素、气味剂和信息素)。初级代谢物直接参与正常生长、发育和繁殖。次级代谢物不直接参与这些过程,但通常具有重要的生态功能。代谢物的实例包括但不限于抗生素和色素,诸如树脂和萜烯等。一些抗生素使用初级代谢物作为前体,诸如由初级代谢物色氨酸产生的放线菌素。如本文所用,代谢物包括小的亲水性碳水化合物;大的疏水性脂质和复杂的天然化合物。
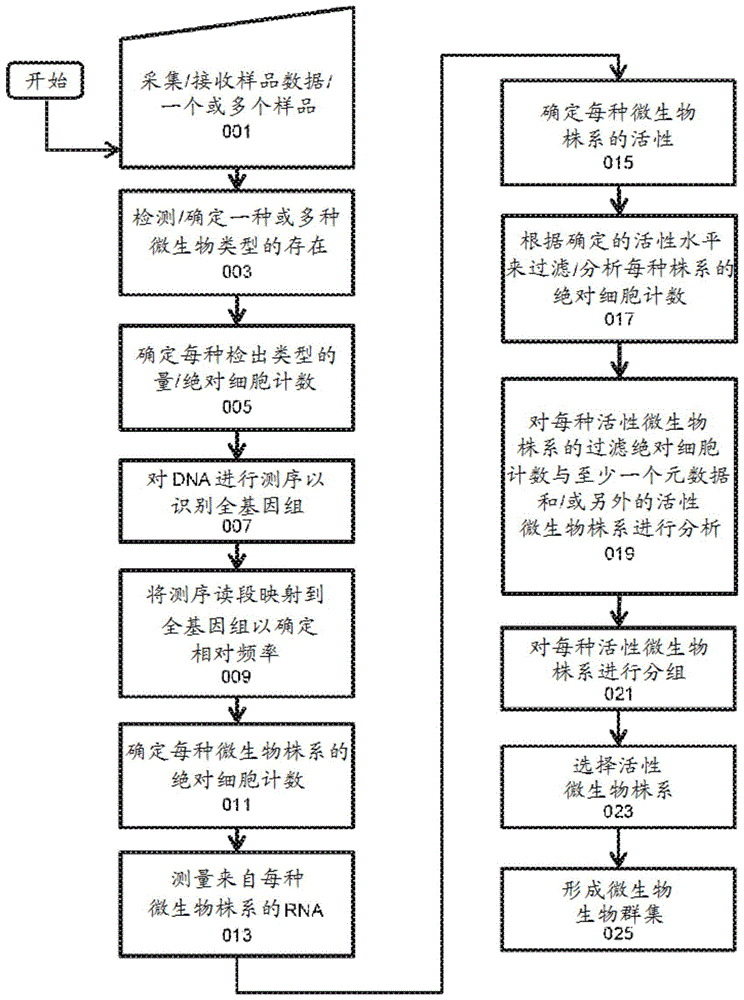
在本公开的一个方面,公开了用于从多个样品中识别活性微生物以及对识别的微生物与至少一个元数据进行分析的方法。如图1所示,方法可包括获得可共有至少一个共同环境参数的样品和/或样品数据(001),以及针对每个样品,检测每个样品中一种或多种微生物类型的存在(003)及其相应的量(005)。然后对每个样品中的dna进行测序以识别多种微生物株系的全基因组(007),并通过将测序读段映射到所识别的全基因组来确定样品中每种株系的相对频率(009)。每个样品中每种微生物株系的绝对细胞计数例如由每种微生物类型的量和每种株系的相对频率确定(011)。例如通过对每个样品中的cdna进行测序并将所测序的cdna映射到所识别的全基因组来测量每种微生物株系的rna(013)。基于映射到每个全基因组的cdna读段的量确定每个样品中每种微生物株系的活性(015)。根据所确定的活性来过滤(017)每种微生物株系的绝对细胞计数,以提供所述至少两个样品中每一个的活性微生物株系及其相应绝对细胞计数的集合。将所述至少两个样品中每一个的活性微生物株系的经过滤的绝对细胞计数用于与所述至少两个样品中每一个的其他株系和/或至少一个测得元数据的株系比较/分析中(019)。根据预测的功能和/或化学性质,将活性微生物株系分组成至少两组(021)。选择来自所述至少两组中的每一组的至少一种微生物株系(023),并将所选择的微生物株系与载体培养基组合以形成活性微生物的生物群集(025),该生物群集被配置为当被引入到目标生物环境中时改变与该目标生物环境的至少一个元数据相对应的性质。
本文详细讨论了可以利用的各种非限制性比较方法,并且以非限制性实例的方式,这些比较方法可以包括确定每个样品中活性微生物株系与至少一个测得元数据的共现。以非限制性实例的方式,确定每个样品中活性微生物株系和至少一个测得元数据的共现可包括创建使用表示元数据和微生物株系关系的链接以及活性微生物株系的绝对细胞计数填充的矩阵,以代表一个或多个异质微生物群落网络。本文详细讨论了可以利用的各种非限制性分组方法,并且以非限制性实例的方式,这些分组方法可包括基于非参数网络分析和聚类分析中的至少一种进行分组,所述非参数网络分析和聚类分析识别活性异质微生物群落网络内的每种活性微生物株系和测得元数据的连通性。
如图1-1所示,本公开的一些实施方案包括获得样品数据/一个或多个样品(031)(例如,共有至少一个共同环境参数的至少两个样品),以及针对每个样品,检测每个样品中一种或多种微生物类型的存在(033)及其相应的量/绝对细胞计数(035)。然后对每个样品中的dna进行测序以识别多种微生物株系的全基因组(037),并通过将测序读段映射到所识别的全基因组来确定样品中每种株系的相对频率(039)。每个样品中每种微生物株系的绝对细胞计数由每种微生物类型的量和每种株系的相对频率确定(041)。通过处理每个样品中的蛋白质(例如,通过质谱法)并将得自该处理的氨基酸序列映射到所识别的全基因组来测量每种微生物株系的蛋白质(043)。基于映射到每个全基因组的氨基酸序列的量确定每个样品中每种微生物株系的活性(045)。根据确定的活性来过滤每种微生物株系的绝对细胞计数(047),以提供所述至少两个样品中每一个的活性微生物株系及其相应绝对细胞计数的列表或集合。可以对所述至少两个样品中每一个的活性微生物株系的经过滤的绝对细胞计数与所述至少两个样品中每一个的其他活性微生物株系和/或至少一个测得元数据进行比较或分析(049),并且根据预测的功能和/或化学性质将活性微生物株系分组(051)或分类成至少两组。选择来自所述至少两组中的每一组的至少一种微生物株系(053),并将其与载体培养基组合以形成活性微生物的生物群集(055),该生物群集被配置为当被引入到目标生物环境中时改变与该目标生物环境的至少一个元数据相对应的性质。
如图1-2所示,本公开的一些实施方案包括获得一个或多个样品(061)(例如,可共有至少一个共同的环境参数的至少两个样品),以及针对每个样品,检测每个样品中一种或多种微生物类型的存在(063)及其相应的量(065)。然后对每个样品中的dna进行测序以识别多种微生物株系的全基因组(067),并通过将测序读段映射到所识别的全基因组来确定样品中每种株系的相对频率(069)。每个样品中每种微生物株系的绝对细胞计数由每种微生物类型的量和每种株系的相对频率确定(071)。通过对每个样品中的cdna进行测序并将所测序的cdna映射到所识别的全基因组来测量每种微生物株系的rna(073)。基于映射到每个全基因组的cdna读段的量确定每个样品中每种微生物株系的活性(075)。根据所确定的活性来过滤每种微生物株系的绝对细胞计数,以提供所述至少两个样品中每一个的活性微生物株系及其相应绝对细胞计数的集合(077)。对所述至少两个样品中每一个的活性微生物株系的经过滤的绝对细胞计数与所述至少两个样品中每一个的至少一个测得元数据进行比较(079)。例如,根据预测的功能和/或化学性质,将活性微生物株系分组成至少两组(081)。然后,对于每组,确定该组中至少两种活性微生物株系之间的遗传相似性(083),并基于此识别该组的至少一种感兴趣的功能(085)(例如,酶功能、抗微生物功能等)。这可以包括相同类型的株系和/或不同类型但在同一组内的株系。然后可以基于每组的所述至少一种识别的功能来识别一条或多条途径(087)。然后,可以亚克隆(089)(例如,通过细菌亚克隆)和/或以其他方式利用一条或多条识别的途径,例如,以用于工业和/或天然产品生产目的。另选地或另外地,可以基于所识别的至少一种感兴趣的功能而选择来自所述至少两组中的每一组的至少一种微生物株系和/或至少一种株系(091),并将其与载体培养基组合以形成生物群集(093),该生物群集被配置为当被引入到目标生物环境中时改变与该目标生物环境的至少一个元数据相对应和/或相关联的性质。另选地,或另外地,可以基于所识别的至少一种感兴趣的功能和/或途径来选择至少一种化合物和/或组合物(即,一种或多种生物活性改性剂组分,独立于一种或多种活性微生物株系和/或作为一种或多种活性微生物株系的补充)(095),并将其与载体培养基组合以形成生物活性改性剂(097),该生物活性改性剂被配置为当被引入到目标生物环境中时改变与该目标生物环境的至少一个元数据相对应和/或相关联的性质。
在本公开的一个方面,公开了一种用于识别多种微生物株系与一个或多个元数据和/或参数之间的关系的方法。如图1a中所示,从至少两个样品来源接收至少两个样品的样品和/或样品数据101,并且针对每个样品,确定一种或多种微生物类型的存在103。确定每个样品中所述一种或多种微生物类型中的每种检出微生物类型的绝对数量(细胞计数)105,并且基于全基因组和/或全转录组确定每个样品中独特的第一标记物的数量及其量107,每个独特的第一标记物是微生物株系的全基因组和/或全转录组的标记物。整合(例如,乘法或其他函数)每种微生物类型的绝对数量和第一标记物的数量(相对数量和/或相对百分比)以得到存在于每个样品中的每种微生物株系的绝对细胞计数109,并且基于超过指定阈值的每种微生物株系的至少一个独特的第二标记物的测量值,确定每个样品中每种微生物株系的活性水平111,如果微生物株系的至少一个独特的第二标记物的测量值超过对应阈值,则将该株系识别为有活性的。然后根据确定的活性来过滤每种微生物株系的绝对细胞计数,以提供所述至少两个样品中的每一个的活性微生物株系及其相应绝对细胞计数的集合或列表113。对所述至少两个样品中每一个的活性微生物株系的经过滤的绝对细胞计数的集合或列表与至少一个测得元数据或另外的活性微生物株系进行网络分析115,该网络分析以非限制性实例的方式包括确定每种活性微生物株系与每个其他活性微生物株系之间的最大信息系数得分,以及确定每种活性微生物株系与所述至少一个测得元数据或另外的活性微生物株系之间的最大信息系数得分。然后可以基于功能、预测的功能和/或化学性质对活性微生物株系进行分类117,并且基于该分类识别并输出多种活性微生物株系119。在一些实施方案中,该方法还包括从识别的多种微生物株系聚集生物活性改性剂121诸如活性微生物群集,该生物活性改性剂被配置为当被应用于目标时改变与至少一个测得元数据相对应和/或相关联的性质。该方法还可以包括基于所述输出的多种识别的活性微生物株系识别至少一种病原体(有关更多细节,参见实施例4)。在一些实施方案中,所述多种活性微生物株系可用于聚集活性微生物群集,该活性微生物群集被配置为当被应用于目标时处理所述至少一种识别的病原体和/或治疗与所述至少一种识别的病原体相关的症状。
在本公开的一个方面,提供了用于确定一个样品或多个样品中的一种或多种活性微生物株系的绝对细胞计数的方法,其中所述一种或多种活性微生物株系存在于样品中的微生物群落中。所述一种或多种微生物株系是一种或多种生物类型的亚分类单元(参见图1b中的方法1000)。针对每个样品,检测样品中一种或多种微生物类型的存在(1001)。确定样品中的所述一种或多种生物类型中每一种的绝对数量(1002)。测量独特的第一标记物的数量以及每个独特的第一标记物的量(1003)。如本文所述,独特的第一标记物是独特的微生物株系的标记物。然后通过测量一种或多种独特的第二标记物的表达水平在蛋白质和/或rna水平上评估活性(1004)。独特的第二标记物可以与第一独特标记物相同或不同,并且是生物株系活性的标记物。基于一种或多种独特的第二标记物的表达水平,确定哪种(如果有的话)微生物株系是有活性的(1005)。如果一种微生物株系以特定水平或高于阈值水平(例如,高于阈值水平至少约10%、至少约20%、至少约30%或至少约40%)表达第二独特标记物,则该微生物株系被认为是有活性的(1005)(应当理解,可以基于具体应用和/或实现方式来确定各种阈值,例如,阈值可以根据样品来源诸如具体物种、样品原位置、感兴趣的元数据、环境等而变化)。可以基于所述一种或多种活性微生物株系的所述一种或多种第一标记物的量和所述一种或多种微生物株系是其中的亚分类单元的生物类型的绝对数量,来确定所述一种或多种活性微生物株系的绝对细胞计数。
本公开的一些实施方案可以被配置用于分析微生物群落。如图1c所示,获得两个或更多个样品(和/或样品集合)的数据(1051),每个样品包括异质微生物群落,以及在每个样品中检测多种微生物类型(1053)。然后,确定每个样品中所述多种微生物类型中的每种检出微生物类型的绝对细胞数量(1055)(例如,通过facs或本文所讨论的其他方法)。测量每个样品中独特的第一标记物及其量(1057),每个独特的第一标记物是检出微生物类型的微生物株系的标记物。测量一种或多种独特的第二标记物的值(活性、浓度、表达等)(1059),独特的第二标记物指示检出的微生物类型的特定微生物株系的活性(例如,代谢活性),以及基于所述一个或多个独特的第二标记物的测得值(例如,基于超过指定设定阈值的值)确定每种检出的微生物株系的活性(1061)。确定每个样品中每种检出活性微生物株系的相应比率(1063),例如,基于相应的绝对细胞计数、值等。例如,在说明性实现方式中,对来自马粪样品的细胞进行染色并计数。然后,从每个样品中分离总核酸。将洗脱液分成两部分并酶促纯化以获得纯化的dna或纯化的rna。通过将rna酶促转化为cdna来稳定纯化的rna。制备总dna和cdna两者的illumina测序文库。测序后,将原始测序读段进行质量修整(qualitytrimmed)和合并,并识别微生物株系的总群体。将源自dna样品的测序文库映射回微生物株系的总群体,以识别每个样品中存在哪些株系,并对每个样品中的每个株系的读段数量进行定量。然后将定量的读段列表与绝对细胞计数数据整合以确定每种株系的绝对细胞数量。在整合细胞计数数据后,将来自cdna文库的读段映射回每个样品中的株系,以确定每个样品中的哪些株系是有活性的。从输出结果中去除无活性株系,以生成每个样品中每种检出活性微生物株系的相应比率的列表。
然后分析所述至少两个样品中的每种检出活性微生物株系(或其子集)以识别每种检出活性微生物株系与其他检出活性微生物株系之间的关系及其强度,以及每种检出活性微生物株系与至少一个测得元数据之间的关系及其强度(1065)。然后例如在图形显示器/界面上(例如,图1d)显示或以其他方式输出所识别的关系(1067),并且可以利用所述识别的关系来识别和/或生成生物活性改性剂(1069),诸如生物群集。在一些实施方案中,可以限制关系的显示/输出,使得仅显示超过某一强度或权重的关系(1066a、1066b)。
根据本公开的微生物群集可以选自活性的、相互联系的单个微生物物种或物种的株系的集合、子集和/或分组。通过本公开的方法识别的关系和网络基于执行一项或多项共同的功能而被分组和/或链接,或者可被描述为参与、或导向可识别参数诸如感兴趣的表型性状(例如反刍动物中产奶量增加)或与可识别参数相关联。在图1d中,使用louvain群落检测方法来识别与奶牛相关元数据参数相关联的组。每个节点代表特定的瘤胃微生物株系或元数据参数。节点之间的链接代表重要的关系。未连接的节点是无关的微生物。每个彩色“气泡”代表通过louvain分析检测到的组。该分组允许基于株系所分的组来预测株系的功能。
本公开的一些实施方案被配置成利用互信息来对驻留在动物胃肠道中的天然微生物株系对特定动物性状的重要性进行排名。计算所有微生物和所需动物性状的最大信息系数(mic)。关系的评分范围为0比1,其中1表示微生物株系与动物性状之间有紧密关系,0表示无关系。将基于该得分的截止值用于定义对于特定性状的改善有用和无用的微生物。图1e和图1f描绘了与乳脂效率有关系的瘤胃微生物株系的mic得分分布的实例。此处,曲线从指数形式变为线性形式的点(细菌得分~0.45-0.5,真菌得分~0.3)表示有用与无用微生物株系之间的截止值。图1g和图1h描绘了与产乳效率有关系的瘤胃微生物株系的mic得分分布的实例。曲线从指数形式变为线性形式的点(细菌得分~0.45-0.5,真菌得分~0.25)表示有用与无用微生物株系之间的截止值。
如图2g中所提供的,在本公开的另一方面,确定多个样品中一种或多种活性微生物的绝对细胞计数,并将绝对细胞计数与元数据(环境参数)相关联(2001-2008)。对多个样品进行一种或多种活性微生物株系的绝对细胞计数的分析,其中如果在所述多个样品的至少一个中的活性测量结果处于阈值水平或高于阈值水平,则认为所述一种或多种活性微生物株系是有活性的(2001-2006)。然后将所述一种或多种活性微生物株系的绝对细胞计数与特定实现方式和/或应用的元数据参数相关联(2008)。
在一个实施方案中,随着时间从相同的环境来源(例如,在一段时间内的同一种动物)采集多个样品。在另一个实施方案中,所述多个样品来自多个环境来源(例如,不同的动物)。在一个实施方案中,环境参数是第二活性微生物株系的绝对细胞计数。在进一步的实施方案中,将所述一种或多种活性微生物株系的绝对细胞计数值用于确定所述一种或多种活性微生物株系与微生物群落的第二活性微生物株系的共现。在进一步的实施方案中,将第二环境参数与所述一种或多种活性微生物株系的绝对细胞计数和/或第二环境株系的绝对细胞计数相关联。本公开通篇讨论了所公开实施方案的各方面。
重要的是,与本文提供的方法一起使用的样品可以是包括微生物群落的任何类型。例如,与本文提供的方法一起使用的样品包括但不限于动物样品(例如,哺乳动物、爬行动物、禽类)、土壤、空气、水(例如,海水、淡水、污水污泥)、沉积物、油、植物、农产品、植物、土壤(例如,根际土壤)和极端环境样品(例如,酸性矿山排水、热液系统)。在海水或淡水样品的情况下,样品可以来自水体表面,或任何深度的水体,例如深海样品。在一个实施方案中,水样是海洋、河流或湖泊样品。
在一个实施方案中,动物样品是体液。在另一个实施方案中,动物样品是组织样品。非限制性动物样品包括牙齿、汗液、指甲、皮肤、毛发、粪便、尿液、精液、粘液、唾液、胃肠道。动物样品可以是例如人、灵长类动物、牛、猪、犬、猫、啮齿动物(例如,小鼠或大鼠)或禽类样品。在一个实施方案中,禽类样品包括来自一只或多只鸡的样品。在另一个实施方案中,样品是人样品。人微生物群系包括在皮肤的表面和深层、乳腺、唾液、口腔粘膜、结膜和胃肠道中发现的微生物的集合体。微生物群系中发现的微生物包括细菌、真菌、原生动物、病毒和古细菌。身体的不同部分表现出不同的微生物多样性。微生物的量和类型可以表示个体的健康或患病状态。细菌类群的数量有数千种,病毒也可能非常多。身体上给定部位的细菌组成因人而异,这不仅在类型上,而且在丰度或量上也是如此。另外的应用包括用于食品,尤其是发酵食品和微生物食品,例如面包、奶酪、葡萄酒、啤酒、泡菜等。
在另一个实施方案中,样品是瘤胃样品。反刍动物诸如牛依靠不同的微生物群落来消化饲料。这些动物因具有改良的上消化道(网状瘤胃或瘤胃)而进化成能够食用低营养价值的饲料,这些饲料容纳在所述消化道中同时被厌氧微生物群落发酵。瘤胃微生物群落非常密集,每毫升中约有3×1010个微生物细胞。厌氧发酵微生物在瘤胃中占主导地位。瘤胃微生物群落包括生命的所有三个域的成员:细菌、古细菌和真核生物。瘤胃发酵产物相应的宿主需要它们以用于身体维持和生长以及产奶(vanhoutert(1993).anim.feedsci.technol.43,第189-225页;bauman等人(2011).annu.rev.nutr.31,第299-319页;各自的全文出于所有目的以引用方式并入)。此外,据报道,产奶量和组成与瘤胃微生物群落有关(sandri等人(2014).animal8,第572-579页;palmonari等人(2010).j.dairysci.93,第279-287页;各自的全文出于所有目的以引用方式并入)。在一个实施方案中,通过jewell等人(2015).appl.environ.microbiol.81,第4697-4710页中描述的方法采集瘤胃样品,该文献全文出于所有目的以引用方式并入本文。
在另一个实施方案中,样品是土壤样品(例如,根外土壤或根际土壤样品)。据估计,1克土壤含有数万种细菌类群,多达10亿个细菌细胞以及约2亿个真菌菌丝(wagg等人(2010).procnatl.acad.sci.usa111,第5266-5270页,该文献全文出于所有目的以引用方式并入)。细菌、放线菌、真菌、藻类、原生动物和病毒均在土壤中有发现。土壤微生物群落多样性涉及土壤微环境的结构和肥力、植物的营养物获取、植物多样性和生长,以及地上与地下群落之间的资源循环。因此,对土壤样品随着时间推移的微生物含量和活性微生物的共现(以及活性微生物的数量)进行评估可以了解与环境元数据参数(诸如营养物获取和/或植物多样性)相关联的微生物。
在一个实施方案中,土壤样品是根际土壤样品,即,直接受根分泌物和相关土壤微生物影响的狭窄土壤区域。根际土壤是群落密集的区域,在其中观察到升高的微生物活动且植物根系通过营养物质和生长因子的交换与土壤微生物相互作用(sanmiguel等人(2014),appl.microbiol.biotechnol.,doi10.1007/s00253-014-5545-6,该文献全文出于所有目的以引用方式并入)。当植物将许多化合物分泌到根际土壤中时,对根际土壤中生物类型的分析可用于确定在其中生长的植物的特征。
在另一个实施方案中,样品是海水或淡水样品。海水每毫升含有多达一百万个微生物,并且含有数千种微生物类型。在沿海水域,这些数字可能还要高出一个数量级,因为沿海水域会更多地产生大量的有机质和营养物。海洋微生物对以下方面至关重要:海洋生态系统的运作;所产生的二氧化碳与固定的二氧化碳之间的平衡的维持;通过海洋光养微生物诸如蓝藻(cyanobacteria)、硅藻以及微微型浮游植物和微型浮游植物在地球上产生超过50%的氧气;提供新型生物活性化合物和代谢途径;通过占据海洋食物网中的关键底层营养级,确保海产品的可持续供应。在海洋环境中发现的生物包括病毒、细菌、古细菌和一些真核生物。海洋病毒可能通过病毒裂解在控制海洋细菌群体中发挥重要作用。海洋细菌作为其他小型微生物的食物来源以及有机质的生产者是非常重要的。在整个海洋水层中发现的古细菌是远洋古细菌,它们的丰度与海洋细菌相当。
在另一个实施方案中,样品包括来自极端环境(即,具有对地球上大多数生命有害的条件的环境)的样品。在极端环境中茁壮成长的生物被称为极端微生物。虽然古细菌域包含众所周知的极端微生物的实例,但细菌域也可以具有这些微生物的代表。极端微生物包括:在ph值为3或更低水平下生长的嗜酸菌;在ph值为9或更高水平下生长的嗜碱菌;生长不需要氧气的厌氧菌,诸如spinoloricuscinzia;在深层地下中的充满地下水的岩石、裂缝、含水层和断层内的微观空间中生活的隐藏岩内生物;在浓度为约至少0.2m的盐中生长的嗜盐菌;在高温(约80-122℃)下茁壮成长的(诸如在热液系统中发现的)超嗜热菌;在寒冷沙漠中的岩石下生活的石下生物;从黄铁矿等还原矿物化合物中获取能量并且在地球化学循环中具有活性的无机自养生物,诸如亚硝化单胞菌(nitrosomonaseuropaea);耐受高浓度溶解重金属(诸如铜、镉、砷和锌)的金属耐受生物;在营养有限的环境中生长的寡营养菌;在高糖浓度的环境中生长的嗜高渗菌;在高压下茁壮成长的(诸如在海洋深处或地下深处发现的)嗜压微生物(或适压微生物);在约-15℃或更低的温度下存活、生长和/或繁殖的嗜冷菌/嗜寒微生物;耐高程度电离辐射的抗辐射生物;在45-122℃的温度下茁壮成长的嗜热菌;可以在极端干燥条件下生长的耐旱生物。多重嗜极生物是在多于一类极端环境的情况下能够成为极端微生物的生物,并且包括嗜热嗜酸菌(优选温度为70-80℃,ph介于2与3之间)。古细菌的泉古菌门包括嗜热嗜酸菌。
样品可包括来自一个或多个域的微生物。例如,在一个实施方案中,样品包含细菌和/或真菌(在本文中也称为细菌或真菌株系)的异质群。
在本文提供的用于确定样品中一种或多种微生物的存在和绝对细胞计数(例如从相同或不同环境中采集的和/或在多个时间点采集的多个样品中的一种或多种微生物的绝对细胞计数)的方法中,所述一种或多种微生物可以是任何类型。例如,所述一种或多种微生物可以来自细菌域、古细菌域、真核生物域或其组合。细菌和古细菌是原核生物,具有非常简单的细胞结构,没有内部细胞器。细菌可分为革兰氏阳性/无外膜、革兰氏阴性/有外膜和未分组的门。古细菌构成单细胞微生物的域或界。虽然外表上与细菌类似,但古细菌具有与真核生物更密切相关的基因和几种代谢途径,尤其是涉及转录和翻译的酶。古细菌生物化学的其他方面是独特的,诸如其细胞膜中存在醚脂质。古细菌分为四个公认的门:奇古菌门、曙古菌门、泉古菌门和初古菌门。
真核生物域包括真核生物,其由膜结合的细胞器(诸如细胞核)限定。原生动物是单细胞真核生物。所有多细胞生物都是真核生物,包括动物、植物和真菌。真核生物分为四个界:原生生物界、植物界、真菌界和动物界。然而,存在几种另选的分类。另一种分类将真核生物分为六个界:古虫界(各种鞭毛原生动物);变形虫界(叶状类变形虫(loboseamoeboid)和粘丝状真菌(slimefilamentousfungi));后鞭毛生物(动物、真菌、领鞭虫类);有孔虫界(有孔虫类、放射虫类和各种其他类变形虫原生动物);囊泡藻界(不等鞭毛类(褐藻、硅藻)、定鞭藻门、隐藻门(或淀粉鞭毛藻类)和囊泡虫类);泛植物/原始色素体植物(primoplantae)(陆生植物、绿藻、红藻和灰胞藻)。
在真核生物域内,真菌是在微生物群落中占主导地位的微生物。真菌包括酵母和丝状真菌等微生物以及常见的蘑菇。真菌细胞具有含有葡聚糖和几丁质的细胞壁,这是这些生物的独特特征。真菌形成一组拥有共同祖先的相关生物,命名为真菌门(eumycota)。据估计,真菌界有150万至500万个种类,其中约5%已被正式分类。大多数真菌的细胞生长为被称为菌丝的管状、细长状和丝状结构,这些结构可能含有多个细胞核。一些种类生长为通过出芽或二分裂来繁殖的单细胞酵母。真菌的主要门(有时称为分裂)主要根据其有性生殖结构的特征进行分类。目前,提出了七个门:微孢子门、壶菌门、芽枝霉门、新美鞭菌门、球囊菌门、子囊菌门和担子菌门。
通过本文描述的方法检测和定量的微生物也可以是病毒。病毒是一种小型致病因子,只在其他生物的活细胞内复制。病毒可以感染真核生物、细菌和古细菌域的所有类型的生命形式。病毒颗粒(称为病毒粒子)由两部分或三部分组成:(i)遗传物质,其可以是dna或rna;(ii)保护这些基因的蛋白质衣壳;以及在某些情况下(iii)当病毒在细胞外时包裹蛋白质衣壳的脂质膜。已确立了七个病毒目:有尾噬菌体目(caudovirales)、疱疹病毒目(herpesvirales)、线状病毒目(ligamenvirales)、单股反链病毒目(mononegavirales)、网巢病毒目(nidovirales)、小核糖核酸目(picornavirales)和芜菁黄花叶病毒目(tymovirales)。病毒基因组可以是单链(ss)或双链(ds)的rna或dna,并且可以使用或不使用逆转录酶(rt)。此外,ssrna病毒可以是有义的(+)或反义的(-)。该分类将病毒分为七组:i:dsdna病毒(诸如腺病毒、疱疹病毒、痘病毒);ii:(+)ssdna病毒(诸如细小病毒);iii:dsrna病毒(诸如呼肠孤病毒);iv:(+)ssrna病毒(诸如小核糖核酸病毒、披膜病毒);v:(-)ssrna病毒(诸如正粘病毒、弹状病毒);vi:在生命周期中具有dna中间体的(+)ssrna-rt病毒(诸如逆转录病毒);vii:dsdna-rt病毒(诸如嗜肝dna病毒)。
通过本文描述的方法检测和定量的微生物也可以是类病毒。类病毒是已知最小的传染性病原体,仅由环状单链rna的短链组成而没有蛋白质衣壳。它们主要是植物病原体,其中一些具有重要的经济意义。类病毒基因组非常小,含有约246至约467个核碱基。
根据本文提供的方法,处理样品以检测样品中一种或多种微生物类型的存在(图1b,1001;图2g,2001)。确定样品中一种或多种微生物类型的绝对数量(图1b,1002;图2g,2002)。确定所述一种或多种生物类型的存在和至少一种生物类型的绝对数量可以并行地或连续地进行。例如,在样品包含含有细菌(即,一种微生物类型)和真菌(即,第二种微生物类型)的微生物群落的情况下,在一个实施方案中,使用者检测样品中一种或两种生物类型的存在(图1b,1001;图2g,2001)。在进一步的实施方案中,使用者确定样品中至少一种生物类型的绝对数量—在该实例的情况下,确定样品中的细菌、真菌或其组合的数量(图1b,1002;图2g,2002)。
在一个实施方案中,对样品或其部分进行流式细胞术(fc)分析以检测一种或多种微生物类型的存在和/或数量(图1b,1001、1002;图2g,2001、2002)。在一个流式细胞术实施方案中,各个微生物细胞以至少约300*s-1、或至少约500*s-1、或至少约1000*s-1的速率通过照射区。但是,本领域普通技术人员将认识到,该速率可以根据所采用的仪器的类型而变化。电子门控的检测器测量表示光散射程度的脉冲的幅度。这些脉冲的幅度被电子地分类成“箱”(bin)或“通道”,从而允许显示具有某一定量性质(例如,细胞染色性质、直径、细胞膜)的细胞的数量相对于通道数量的直方图。这种分析允许确定每个“箱”中的细胞数量,在本文所述的实施方案中,所述“箱”是“微生物类型”箱,例如细菌、真菌、线虫、原生动物、古细菌、藻类、甲藻、病毒、类病毒等。
在一个实施方案中,将样品用一种或多种荧光染料染色,其中荧光染料对特定的微生物类型具有特异性,以便能够通过流式细胞仪或利用荧光的一些其他检测和定量方法(例如荧光显微术)检测。该方法可以提供样品中给定生物类型的细胞数和/或细胞体积的定量结果。在进一步的实施方案中,如本文所述,利用流式细胞术来确定生物类型的独特的第一标记物和/或独特的第二标记物的存在和量,例如酶表达、细胞表面蛋白表达等。还可以生成例如光散射相对于来自细胞膜染色的荧光(相对于来自蛋白质染色或dna染色的荧光)的双变量或三变量直方图或等高线图,并因此可以获得对整个群体的细胞中各种感兴趣特性的分布的认识。此类多参数流式细胞术数据的许多显示是常用的,并且适合与本文所述的方法一起使用。
在处理样品以检测一种或多种微生物类型的存在和数量的一个实施方案中,采用显微镜检测(图1b,1001、1002)。在一个实施方案中,显微镜检查是光学显微镜检查,其中将可见光和镜头系统用于放大小样品的图像。数字图像可以通过电荷耦合器件(ccd)相机来捕获。其他显微技术包括但不限于扫描电子显微镜检查和透射电子显微镜检查。根据本文提供的多个方面对微生物类型进行可视化和定量。
在本公开的另一个实施方案中,为了检测一种或多种微生物类型的存在和数量,对每个样品或其部分进行荧光显微镜检查。可以使用不同的荧光染料对样品中的细胞直接染色并使用落射荧光显微镜和上述流式细胞术对总细胞计数进行定量。用于定量微生物的有用染料包括但不限于吖啶橙(ao)、4,6-二氨基-2-苯基吲哚(dapi)和5-氰基-2,3二甲苯基氯化四唑(ctc)。活细胞可通过活力染色法估算,诸如
在另一个实施方案中,对每个样品或其部分进行拉曼显微光谱分析,以确定微生物类型的存在和至少一种微生物类型的绝对数量(图1b,1001-1002;图2g,2001-2002)。拉曼显微光谱是一种无损无标记技术,能够检测和测量单细胞拉曼光谱(scrs)。典型的scrs提供单个细胞的内在生化“指纹”。scrs包含生物分子(包括核酸、蛋白质、碳水化合物和脂质)的丰富信息,该信息使得能够表征不同的细胞种类、生理变化和细胞表型。拉曼显微镜检查不同细胞生物标记物的化学键对激光的散射。scrs是单个细胞中所有生物分子的光谱的汇总,表明细胞的表型谱。作为基因表达的结果,细胞表型通常反映基因型。因此,在相同的生长条件下,不同的微生物类型给出与其基因型中的差异相对应的不同scrs,并因此可以通过它们的拉曼光谱进行识别。
在另一个实施方案中,对样品或其部分进行离心以确定微生物类型的存在和至少一种微生物类型的数量(图1b,1001-1002;图2g,2001-2002)。该过程通过使用由离心机产生的离心力来沉积不均匀混合物。混合物的密度较大的组分远离离心机的轴线迁移,而混合物的密度较小的组分朝向轴线迁移。离心可以将样品分级分离成细胞质、膜和细胞外部分。它还可以用于确定感兴趣的生物分子的定位信息。另外,离心可用于分级分离总微生物群落dna。不同的原核生物组在其dna的鸟嘌呤加胞嘧啶(g+c)含量方面有所不同,因此基于g+c含量的密度梯度离心是区分生物类型和与每种类型相关的细胞数量的方法。该技术生成整个群落dna的分级分离谱,并根据g+c含量指示dna的丰度。将总群落dna物理分离成高度纯化的级分,每个级分代表不同的g+c含量,该含量可通过另外的分子技术进行分析,诸如变性梯度凝胶电泳(dgge)/扩增核糖体dna限制性分析(ardra)(参见本文的讨论),以评估总微生物群落多样性和一种或多种微生物类型的存在/量。
在另一个实施方案中,对样品或其部分进行染色以确定微生物类型的存在和至少一种微生物类型的数量(图1b,1001-1002;图2g,2001-2002)。染色剂和染料可用于可视化生物组织、细胞或细胞内的细胞器。染色可以与显微镜检查、流式细胞术或凝胶电泳结合使用,以可视化或标记对不同微生物类型来说是独特的细胞或生物分子。体内染色是对活组织染色的过程,而体外染色涉及对已从其生物背景中取出的细胞或结构染色。用于本文所述方法的具体染色技术的实例包括但不限于:用于确定细菌革兰氏状态的革兰氏染色法、用于识别内孢子存在的内孢子染色法、萋-尼(ziehl-neelsen)染色法、用于检查组织薄片的苏木精和伊红染色法、用于检查来自各种身体分泌物的细胞样品的巴氏染色法、碳水化合物的过碘酸-雪夫(schiff)染色法、采用三色染色方案来区分细胞与周围结缔组织的马森三色染色法(masson’strichome)、用于检查血液或骨髓样品的罗曼诺夫斯基(romanowsky)染色法(或包括瑞氏(wright's)染色法、哲纳尔氏(jenner)染色法、迈格林华(may-grunwald)染色法、雷氏曼(leishman)染色法和姬姆萨(giemsa)染色法的常见变型)、用于显示蛋白质和dna的银染色法、用于脂质的苏丹染色法和用于检测真正内孢子的conklin染色法。常见的生物染色剂包括用于细胞周期测定的吖啶橙;用于酸性粘蛋白的俾斯麦棕;用于糖原的胭脂红;用于细胞核的卡红明矾(carminealum);用于蛋白质的考马斯蓝;用于神经元胞质的酸性组分的甲酚紫;用于细胞壁的结晶紫;用于细胞核的dapi;用于细胞质材料、细胞膜、一些细胞外结构和红细胞的曙红;用于dna的溴化乙锭;用于胶原蛋白、平滑肌或线粒体的酸性品红;用于细胞核的苏木精;用于dna的赫斯特(hoechst)染色剂;用于淀粉的碘;用于吉曼尼兹(gimenez)染色技术中的细菌和用于孢子的孔雀石绿;用于染色质的甲基绿;用于动物细胞的亚甲蓝;用于尼斯尔(nissl)物质的中性红;用于细胞核的尼罗(nile)蓝;用于亲脂性实体的尼罗红;用于脂质的四氧化锇;用于荧光显微镜检查的罗丹明;用于细胞核的番红精。染色剂也用于透射电子显微镜检查以增强对比度,并包括磷钨酸、四氧化锇、四氧化钌、钼酸铵、碘化镉、碳酰肼、氯化铁、六胺、三氯化铟、硝酸镧、醋酸铅、柠檬酸铅、硝酸铅(ii)、高碘酸、磷钼酸、铁氰化钾、亚铁氰化钾、钌红、硝酸银、蛋白银、氯金酸钠、硝酸铊、氨基硫脲、乙酸铀酰、硝酸铀酰和硫酸氧钒。
在另一个实施方案中,对样品或其部分进行质谱分析(ms)以确定微生物类型的存在和至少一种微生物类型的数量(图1b,1001-1002;图2g,2001-2002)。如下所讨论,ms也可用于检测样品中一种或多种独特标记物的存在和表达(图1b,1003-1004;图2g,2003-2004)。例如,ms用于检测对微生物类型来说是独特的蛋白质和/或肽标记物的存在和量,从而提供对样品中相应微生物类型的数量的评估。定量可以在有稳定同位素标记或无标记的情况下进行。肽的从头测序也可以直接从ms/ms谱或序列标记(产生可以与数据库匹配的短标签)进行。ms还可以揭示蛋白质的翻译后修饰并识别代谢物。ms可与色谱和其他分离技术(诸如气相色谱、液相色谱、毛细管电泳、离子淌度)结合使用,以提高质量分辨率和测定效果。
在另一个实施方案中,对样品或其部分进行脂质分析,以确定微生物类型的存在和至少一种微生物类型的数量(图1b,1001-1002;图2g,2001-2002)。脂肪酸以细胞生物质的相对恒定的比例存在,并且特征性脂肪酸存在于可以区分群落内的微生物类型的微生物细胞中。在一个实施方案中,通过皂化提取脂肪酸,然后衍生化,得到相应的脂肪酸甲酯(fame),然后通过气相色谱进行分析。然后将一个实施方案中的fame谱与参考fame数据库进行比较,以通过多变量统计分析来识别脂肪酸及其对应的微生物特征。
在本文提供的方法的一些方面,测量样品或其部分(例如,样品等分试样)中独特的第一标记物的数量,以及每种独特的第一标记物的量(图1b,1003;图2g,2003)。独特的标记物是微生物株系的标记物,具体地讲,如本文所用,是株系的全基因组或转录组。应当理解,取决于探测和测量的独特标记物,不需要分析整个样品。例如,如果独特的标记物对细菌株系来说是独特的,则不需要分析样品的真菌部分。如上所述,在一些实施方案中,测量样品中一种或多种生物类型的绝对细胞计数包括按生物类型来分离样品,例如通过流式细胞术。
传统的微生物组研究侧重于16srrna扩增子测序以生成用于后续分析的微生物数据集,而本文公开了分析全基因功能谱、表达谱和代谢物谱的方法,这些方法提供微生物组的功能和代谢谱,从而明确地揭示微生物生物圈/微生物组与临床和/或环境参数之间的复杂联系。
图2a提供了整合元基因组学200a、元转录组学200b、封闭的基因组200c和特征选择200d的概述流程。对于元基因组学,如200a所示,使用来自混合微生物样品的dna制备文库。这包括混合的培养物和/或环境样品。对dna进行测序,并例如使用以下两种技术之一对数据进行分析:第一种是整体法,其中对来自一个样品或一组样品的所有序列一起进行分析而不依赖于它们的来源微生物;第二种是分箱方法(binnedapproach),其中将来自混合样品的序列分类到箱中(使用该组序列的独特性质),其中每个箱代表不同的微生物。然后,根据一些实施方案,可以在单个物种的情况下评估每个箱中的序列数据。对于元转录组学,如200b所示,使用来自混合微生物样品的rna制备文库。这包括混合的培养物或环境样品。通过去除其他rna来富集mrna,并且从纯化的mrna合成cdna。对cdna进行测序,并且如果没有参比基因组,则聚集数据,或者如果有参比,则不聚集数据。
对于封闭的基因组,如200c所示,在一些实施方案中,为了封闭基因组,从由一种生物组成的纯培养物、包含两种或更多种生物的混合培养物和/或混合的环境样品中提取dna。生成长和短读段序列数据。例如,仅使用长读段或使用长读段和短读段两者的混合方法来聚集数据。可以在评估基因组之前对混合样品进行分箱。
对于特征选择,如200d所示,根据实现方式和/或实施方案,特征可以指呈任何组合的所测量的任何事物,诸如株系、基因和/或代谢物等。可以将特征相关联(例如,使用元数据因子之间的mic得分或作为特征之间关系的邻接矩阵来关联)以产生关系列表,可以通过阈值对该列表进行过滤以确定重要性。可以对重要关系进行合并和/或分组,例如通过louvain群落检测和其他网络分析方法等。最后,对特征进行选择以作为可能的产物/目标/等进行测试。对上文的进一步讨论和详细阐述如下。
元基因组学
本公开的一些实施方案利用元基因组学来分析来自包括一种或多种微生物的一个或多个样品的基因功能。如图2b所示,在一些实施方案中,采集样品220a并将其储存在溶液(例如,苯酚乙醇混合物或其他终止溶液)中以防止储存的混杂效应。从每个样品中,提取微生物基因组dna220b,例如通过用超净试剂进行化学或动力学裂解。在一些实现方式中,例如,在污染高的情况下,可以使用空白样品,或者可以采用另外的处理来减少宿主污染。一些这样的方法可包括用fac或微流体技术去除宿主细胞,通过结合和去除cpd-甲基化区减少宿主核酸来富集微生物核酸等。进行测序220c。在一些情况下,使用长读段测序,例如,包括但不限于oxfordnanoporeminion或pacificbiosciencessequel。在其他实现方式中,在短读段测序仪(例如,包括但不限于illuminamiseq或hiseq)上对dna进行片段化或扩增和测序。在另一种实现方式中,结合使用长读段和片段化读段。每台测序仪上样品的数量可以根据这些样品中微生物组的预期复杂性而变化。此外,在一些实现方式中,对每个样品的平行样进行测序以防止测序偏差。在一些实现方式中,以非限制性实例的方式,通过基于转座的方法诸如illuminanextera(xt)或kappahyperplus来执行片段化。在测序后,例如通过序列比对清除所有数据中的宿主污染220d(参见例如schmieder和edwards2011,其全文出于所有目的以引用方式明确地并入本文)。在一些实施方案中,通过组装和注释对测序数据进行分析(222a-222e)。在其他实施方案中,在不组装的情况下进行功能谱分析(221a-221e)。在另一个实施方案中,将两种方法结合使用。在组装的情况下,通过全基因组组装将所有读段共同组装或单独组装(参见例如simpson和pop2015,其全文出于所有目的以引用方式明确地并入本文)。在一些实施方案中,通过基于kmer的德布鲁因图(debruijngraph)组装基因组(参见例如pevzner等人2001,其全文出于所有目的以引用方式明确地并入本文)。在其他实施方案中,将长读段单独组装或与片段化读段一起组装(参见例如sakakibara2011;bankevich等人2012;simpson等人2009,每篇文献全文出于所有目的以引用方式明确地并入本文)。在不脱离本公开的范围的情况下,可以基于特定实施方案和应用来利用和选择另外的组装方法。组装后,可以将所得的重叠群分箱成单独的基因组,这些单独的基因组代表通常称为箱的种类。有几种方法可以使用,但不限于k-mer频率(参见例如karlin等人1997;dick等人2009,其全文出于所有目的以引用方式明确地并入本文)、贝叶斯(bayes)分类器(参见例如rosen等人2008;strous等人2012;kelley和salzberg2010,其全文出于所有目的以引用方式明确地并入本文)、多样品(参见例如sharon等人2013;albertsen等人2013,其全文出于所有目的以引用方式明确地并入本文)和/或gc含量变化(korem等人2015,其全文出于所有目的以引用方式明确地并入本文)。然后分析箱的功能谱(参见例如howe等人2017,其全文出于所有目的以引用方式明确地并入本文)和/或分类学(参见例如segata等人2013,其全文出于所有目的以引用方式明确地并入本文)。在一些实施方案中,分箱是完全自动化的,并且在其他实施方案中结合有手动确认。在无组装的功能谱分析的实现方式中,使用一个或多个公共或专有数据库在功能上对读段进行注释(参见例如,brady和salzberg2009;huson等人2011,每篇文献全文出于所有目的以引用方式明确地并入本文)。用于群落分类的原始读段的分类学分类通过包括但不限于k-mer映射(参见例如wood和salzberg2014,其全文出于所有目的以引用方式明确地并入本文)、motu(参见例如sunagawa等人2013,其全文出于所有目的以引用方式明确地并入本文)和/或蛋白质标记物(参见例如truong等人2015,其全文出于所有目的以引用方式明确地并入本文)的多种方法中的任一种或多种来评估。在一些实施方案中,“箱”对应于株系的全基因组,并因此对应于第一独特标记物。因此,分箱从所有元基因组数据(其中代表所有株系)中拉取微生物的各个基因组。一旦获得了各个基因组,就可以将测序读段映射到基因组以确定该株系在该群落中的比例(微生物株系的独特的第一标记物的相对量),然后可以将该数量与绝对/总细胞计数数据整合以确定株系的绝对细胞计数/微生物株系的绝对细胞数量。在一些实施方案中,组装和无组装方法两者结合使用以将功能谱链接至分箱基因组和微生物群落中存在的株系的相对量。无论使用何种方法,主要输出可包括但不限于带注释的分箱基因组、功能或分类谱的矩阵、样品特征的矩阵以及与元数据因子相关的一些方法。箱相对量可以通过原始读段与各个分箱基因组的序列比对来确定(参见例如kang等人2015,其全文出于所有目的以引用方式明确地并入本文)。可以通过整合序列数据与细胞计数信息来确定箱绝对细胞计数。
元转录组学
本公开的一些实施方案利用元转录组学,即,对一种或多种微生物的一个或多个样品的基因表达的整体研究。如图2c所示,采集样品并将其储存在溶液(例如,苯酚乙醇混合物或其他终止溶液)中以防止储存的混杂效应。从每个样品中,提取所有微生物总rna,例如通过用超净试剂进行化学或动力学裂解(参见例如yuan等人2012;kennedy等人2014;probst等人2015;tanner等人1998;salter等人2014,每篇文献全文出于所有目的以引用方式明确地并入本文)。在一些实施方案中,例如,在污染高的情况下,可以使用空白样品,和/或可以采用另外的处理来减少宿主污染。此类方法可涉及用fac或微流体技术去除宿主细胞,或通过结合和去除多腺苷酸化mrna减少宿主核酸来富集微生物核酸。从总rna中,富集微生物mrna,例如,包括但不限于用于从缺少多聚腺苷酸尾的rna合成cdna的寡核苷酸(参见例如peano等人2013;sultan等人2014;wendisch等人2001;sharma等人2010,每篇文献全文出于所有目的以引用方式明确地并入本文)和/或通过特异性探针耗尽rrna(例如,sultan等人2014)。在富集mrna后,合成cdna,和/或将mrna片段化然后合成cdna(参见例如giannoukos等人2012,其全文出于所有目的以引用方式明确地并入本文)。在一些实施方案中,利用长读段cdna(来自mrna)测序,例如,包括但不限于oxfordnanoporeminion和/或pacificbiosciencessequel。在其他实施方案中,将mrna片段化,合成cdna并在短读段测序仪(例如但不限于illuminamiseq或hiseq)上进行测序。在另一个实施方案中,结合使用长读段和片段化读段。每台测序仪上样品的数量可以根据这些样品中微生物组的预期复杂性和特定实施方案和/或应用而变化。此外,在一些实施方案中,对每个样品的平行样进行测序以识别测序偏差(参见例如nelson等人2014;sinha等人2017;baym等人2015,每篇文献全文出于所有目的以引用方式明确地并入本文)。在一些实施方案中,可将具有已知量的mrna分子的标准品用于识别样品中每种mrna分子类型的数量。在测序后,从所有数据剪掉衔接子序列、低质量读段,并清除宿主污染和非mrna读段,例如通过序列比对(参见例如schmieder和edwards2011;krueger2015;bolger等人2014,每篇文献全文出于所有目的以引用方式明确地并入本文)。清除后,识别所有开放阅读框,将读段映射到数据库,归一化,然后计算mrna基因表达水平以及其他统计信息。在一些实施方案中,将mrna基因组装成重叠群并将其与先验参比基因组比对以识别特定株系内的活性基因(参见例如morgan和huttenhower2014;conesa等人2005;salzberg和schatz2011,每篇文献全文出于所有目的以引用方式明确地并入本文)。在一些实施方案中,将mrna基因组装成重叠群并在没有参比的情况下对其进行分析(参见例如grabherr等人,full-lengthtranscriptomeassemblyfromrna-seqdatawithoutareferencegenome.natbiotechnol.2011年5月15日;29(7):644-52.doi:10.1038/nbt.1883,其全文出于所有目的以引用方式明确地并入本文)。在一些实施方案中,对于真核微生物,可以分析选择性剪接和选择性mrna结构(参见例如grabherr等人2011)。在一些实施方案中,可以确定一个或多个样品之间的差异表达(anders等人“differentialexpressionanalysisforsequencecountdata.”genomebiology,11,第r106页.doi:10.1186/gb-2010-11-10-r106,
http://genomebiology.com/2010/11/10/r106/;以及trapnell等人,naturebiotechnology2010,每篇文献全文出于所有目的以引用方式明确地并入本文)。样品之间基因表达的差异可以与元数据一起用于后续分析。
代谢组学
本公开的一些实施方案可以利用代谢组学来分析每个细胞过程留下的特征性代谢物谱。如图2d所示,在一些实施方案中,采集样品并立即在4至-80摄氏度的温度下储存。在一些实现方式中,对“外显子组”或外部代谢物进行分析,而在其他实施方案中,对细胞内代谢物进行研究。在一些实施方案中,靶向代谢组学并测量特定代谢物的量。也可以不靶向代谢组学,并且目标代谢物的身份不是事先已知的。质谱法(ms)可用于测量分子和分子片段的质量。质荷比用作关于每个分子的信息。在一些实施方案中,在ms之前进行分离,以非限制性实例的方式,通过液相色谱和/或气相色谱进行分离。在其他实施方案中,使用直接进样而无需分离方法。结合质荷比,可以通过串联ms获得碎裂模式(参见例如siuzdak2006,其全文以引用方式明确地并入本文)。非靶向代谢组学(也称为全局代谢组学)可用于高通量测量以对样品代谢物进行新的表征。然而,在一些实施方案中,在非靶向代谢组学实现方式中,可以使用许多不同的设置(例如ph、溶剂、柱化学和/或电离)以检测不同的代谢物组合。此外,在一些实施方案中,所测量的一些或许多代谢物可以是新的并且可能不能在常用数据库中找到。根据本公开的实施方案,将靶向代谢组学用于聚焦已知代谢物,其中对所讨论的代谢物的浓度曲线进行绘制。根据这些定量曲线,可以对一种特定代谢物或一组代谢物进行定量测量。基于图像的靶向代谢组学诸如基质辅助激光解吸电离(maldi)(参见例如tanaka等人1988,其全文以引用方式明确地并入本文)、纳米结构成像质谱(nims)(参见例如northen等人2007;siuzdak等人2001,每篇文献全文以引用方式明确地并入本文)、解吸电喷雾电离质谱(desi)(参见例如wiseman等人2006,其全文以引用方式明确地并入本文)和/或二次离子质谱(sims)(参见例如norris2007,其全文以引用方式明确地并入本文)可以在组织样品中定位小或大代谢物。在数据采集之后,开始进行数据预处理,并识别、选择、过滤峰,检索、归一化和换算缺失值。在处理之后,根据实施方案、实现方式和/或生物学问题,应用一种或多种不同的多变量和单变量统计分析方法。在统计分析之后,使用已知的代谢物或标准品曲线的数据库来识别代谢物。最后,对样品的相对量或绝对量进行定量。样品之间代谢物浓度的差异可以与元数据一起用于后续分析。
封闭基因组和纳米孔测序
在一些实施方案中,采集并储存样品(例如,如上所讨论)以防止储存的混杂效应。在其他实施方案中,可能不需要储存溶液。从每个样品中,提取所有微生物总dna,例如通过用超净试剂进行化学或动力学裂解(如上所讨论)。在一些实施方案中,利用长读段测序(例如,如上所讨论),另外地或另选地,在短读段测序仪上对dna进行片段化和/或扩增和测序(例如,如上所讨论)。在一些实施方案中,如上所讨论,利用短读段和长读段方法。在一些实施方案中,评估dna序列的表观遗传修饰,例如甲基化(参见例如mcintyre等人2017;simpson等人2016,每篇文献全文出于所有目的以引用方式明确地并入本文)。将序列读段组装成不间断的连续基因组。在一些实施方案中,使用短读段组装软件,例如但不限于spades或velvet(参见例如earl等人2011)。在一些实施方案中,使用长读段组装软件,例如但不限于canu、miniasm或hinge(参见例如earl等人2011;koren等人2017;li2016;kamath等人2017,每篇文献全文出于所有目的以引用方式明确地并入本文)。在一些实施方案中,在组装之前对长读段进行纠错。在一些实施方案中,组装长读段并将短读段用于纠错(koren等人2012,每篇文献全文出于所有目的以引用方式明确地并入本文)。在一些实施方案中,两种方法都用于纠错。在一些实施方案中,共组装所述读段(wick等人2016,每篇文献全文出于所有目的以引用方式明确地并入本文)。
分选污染和富集某些组
如上所讨论,采集并储存样品。在一些实施方案中,随后将样品稀释至有效细胞密度以用于细胞分选。在一些实施方案中,处理样品以消除与感兴趣的细胞无关的外源分子。这可包括但不限于去除细胞外dna的dna酶、去除细胞外rna的rna酶、去除外源蛋白的蛋白酶。在一些实施方案中,基于大小对细胞进行分选。在其他实施方案中,通过散射分离细胞(参见例如buzatu等人2014,其全文据此以引用方式并入)。在一些实施方案中,用荧光染料处理细胞,并通过荧光分离细胞。在一些实施方案中,将荧光标记的核酸探针应用于细胞,然后通过荧光分离细胞。在一些实施方案中,将缀合的抗体应用于细胞,然后通过荧光分离细胞。在一些实施方案中,结合使用两种或更多种上述技术。在一些实施方案中,细胞分选是从样品中消除特定群体。在其他实施方案中,细胞分选是为了富集特定群体。在一些实施方案中,根据代谢活性和/或生理状态选择细胞。在一些实施方案中,将分选的细胞用于元基因组分析(参见上文的元基因组讨论)。在其他实施方案中,使混合的分选细胞群体在液体或固体培养基上生长。在其他实施方案中,从混合的样品中分选单个细胞以在液体或固体培养基上生长。在一些实施方案中,分选单个细胞并对其进行全基因组测序(wgs)。在一些实施方案中,分选单个细胞并对其进行转录组测序。在一些实施方案中,可以应用以上两种或更多种技术。
整合多组学数据集
在一些实施方案中,结合使用每种上述方法的不同组合。图2e示出了对样品或样品集合合使用的所有方法的实施方案的实现方式的概述,每个样品被均质化并且样品被分成每次测量的所需体积。将前述方法全用于来自每个子样品的微生物群体的组合或分离的微生物。元基因组学和元转录组学的整合从基因组组装开始,其中将两者都用于构建重叠群。此外,来自分离的微生物的最终分箱基因组或完整的基因组既包含功能谱也包含基因表达谱。在一些情况下,可将16srrna基因扩增子测序用于确定分类学分类和量/相对量。在其他情况下,可将分箱的基因组用于确定生物质总量/相对量,并且可将表达谱用于解析总活性。结合绝对细胞计数和活性谱,然后可以使用代谢物(相对或绝对)量来获得生物学知识并在后续分析中选择微生物株系。所获得的生物学知识的任何组合均可以用于但不限于生物标记物识别、微生物组状态识别、微生物组洞察计划和产品开发。
实验数据和实施例
实施例1a:多组学整合(图2f)
该研究的目的是产生马绞痛的生物标记物和可能的生物机制。对总共60名患者进行多次取样,其中30名患者被确定为患有绞痛。其他30名患者被确定为健康,没有其他诊断病情。
样品处理。从每个取样点采集粪便样品,并将其立即加入预装有稳定溶液的15ml锥形管中,并在4℃下储存。通过数次倒置将溶液混合并在之后立即储存在4℃下。将粪便样品以4,000rpm离心15分钟,倾析上清液,并等分0.5ml以使用
16srrna。按照
细胞染色和计数
将每个样品的小等分试样分离到新的1.7ml管中并称重。向每个样品中加入1ml无菌pbs,并在没有珠的情况下进行珠磨,以从纤维瘤胃内容物中分离细胞。然后将样品离心以去除大的碎片。将上清液的等份试样在pbs中稀释,然后过滤。将计数珠加入每根管(spherotechacfp-70-10)。然后在sonysh800细胞分选仪(sony,sanjose,ca,usa)上处理经染色的样品,并测定每克原始样品的真菌和细菌细胞数。
元基因组学。使用kapahyperprep文库制备方法(kapabiosystems,wilmington,ma,usa)制备用于鸟枪元基因组学测序的纯化dna。使用定量pcr(qpcr)和生物分析仪(agilenttechnologies,paloalto,ca,usa)对文库进行质量评估,随后使用hiseq(illumina,sandiego,ca,usa)进行测序。将所用数据使用trimgalore剪去衔接子序列并进行质量控制。通过与污染数据库的bowtie比对将样品的可能的人和小鼠污染过滤掉。例如,以非限制性实例的方式,通过metaphlan2的python2.7版(参见truong等人2015)对未组装读段的功能注释进行分析。使用带有元标志(metaflag)的spades(参见bankevich等人2012)组装读段。使用metabat(参见kang等人2015)进行分箱。使用phylophlan(参见segata等人2013)评估分类学分类。通过用bowtie2(参见例如langdon2015)对分箱基因与一般组装进行比对来确定相对箱样品量。通过将细胞计数数据与相对箱量整合来确定绝对细胞计数。
元转录组学。分离纯化的rna,并使用metabacteriaribozerorrna去除试剂盒(epicentre,madison,wi,usa)的改良版本去除rrna。使用纯化的mrna生成cdna,并使用kapahyperprep文库制备方法(kapabiosystems,wilmington,ma,usa)构建测序文库。使用定量pcr(qpcr)和生物分析仪(agilenttechnologies,paloalto,ca,usa)对文库进行质量评估,随后使用hiseq(illumina,sandiego,ca,usa)进行测序。将所用数据使用trimgalore剪去衔接子序列并进行质量控制。通过与污染数据库的bowtie比对将样品的可能的人和小鼠污染过滤掉(参见langmead等人2009)。使用短读段比对工具bowtie2(参见例如langdon2015)将所有获得的读段映射到来自鸟枪元基因组学的分箱草案基因组。还将deseq(参见例如anders和huber,2010)用于确认样品之间的差异表达谱。
代谢组学。将来自每个患者的倾析的上清液(50μl)在含有0.1%甲酸的乙腈:水:甲醇(2:2:1)中稀释(参见例如kamphorst和lewis2017)。使用synergyhydro-rp柱(100×2mm,2.5μm粒度)分离代谢物。将流速设定为0.200ml/min的恒定流速,将柱温保持在25℃。将自动进样器托盘保持在4℃,并将10μl样品进样至dionexultimate3000uplc系统(thermofisherscientic,waltham,ma)。采用已建立的方法(参见例如lu等人2010),使用电喷雾电离将样品引入exactiveplusorbitrapms(thermofisherscientic,waltham,ma)。
结果.通过这种综合方法,在每次测量中都形成了清晰的高水平聚类。在聚类之间的差异内,识别出基因表达和代谢物/分类谱中的微生物模式的生物标记物。此外,从体外测试的样品中分离所识别的基于微生物和代谢物的生物标记物(图2f)。这些提取产物在绞痛患者中进行了测试并逆转了绞痛的微生物病因。图2f示出了多组学测量的组合:(左上)16srrna谱,按otu划分样品;(中上)按株系的箱绝对细胞计数划分元基因组学样品;(左下)按基因表达谱划分元转录组学分箱的基因组;(中下)与基因表达相关的代谢组学代谢物量;(左)otu、基因表达和代谢物的网络。
实施例2a:细胞分选和封闭的全基因组测序
如上所讨论,可以从混合群落数据集组装微生物的各个全基因组,但是生成高质量的元基因组学数据并不简单。例如,测序深度要求可能非常高,并且先前的方法通常在经济上是不可行的(参见例如feehery等人2013,以引用方式并入本文),并且当最终目标是从非常复杂的和/或与宿主相关的微生物群落组装全基因组时更是如此。如本文所公开的,去除宿主细胞/dna和/或以其他方式降低样品的复杂性有利于/能够降低测序深度,从而实现期望的结果。下文详述根据本公开的一种这样的新方法的说明性实例,这里,使用流式细胞术从混合群落中分离和富集微生物,以允许以最小测序深度进行全基因组组装。
材料与方法
以下方法是概念验证,其描述和说明可以如何通过流式细胞术分选混合的微生物样品以降低复杂性和/或去除宿主污染。
样品制备
(1)将3种ascus株系、2种细菌(ascusb_5和ascusb_3138)和1种真菌(ascusf_15)培养至最大光密度。
(2)收获细胞并将其以相等的比率、相同的细胞密度和体积混合在一起。
(3)将混合细胞样品连续稀释:1:10、1:100、1:1000、1:10000。
(4)通过流式细胞术处理每个稀释的样品以识别用于分选的最佳浓度。
(5)用dna酶处理最佳稀释样以去除细胞外dna。
fac分选
将经dna酶处理的样品分选成4个不同的门(b、e、f、g0),使得每个门含有不同的微生物群体。每个门采集100万个事件,总共分选400万个事件。
文库制备和dna测序
(1)通过膜浓缩来自每个门的分选细胞,并提取基因组dna。
(2)使用kapabiosystemshyperplus试剂盒制备文库。
(3)然后将制备的文库加载到illuminamiseq上,并使用v21x300循环试剂盒进行测序。
数据分析
(1)对所有序列进行质量过滤和修剪。
(2)使用spades3.10(例如,参见nurk等人2013,其全文出于所有目的以引用方式明确地并入本文)组装修剪的序列。未使用元标志,因为不支持单端数据。
(3)使用metabat2(例如,参见kang等人2015,其全文出于所有目的以引用方式明确地并入本文)根据基因组对组装的序列进行分箱。
(4)使用bowtie2(例如,参见langmead和salzberg2012,其全文出于所有目的以引用方式明确地并入本文)对所得的箱与参比基因组进行比对以评估完整性。
结果
使用fac分选,fac分选能够成功降低样品的复杂性。可以根据门对不同的群体成员进行分选。过滤掉污染后,经分选的箱的组成如下:
还研究了每个门的读段以确定门内每种株系的百分比组成。每种分离物都存在参比基因组,从而允许使用一般序列读段比对来识别每个读段的来源基因组。下面是典型的bowtie2比对的实例。这里,通过将读段与ascusf_15参比基因组进行比对,从门b中识别出来自ascusf_15的读段。可以以更大的测序深度恢复更多的基因组。
●1197个读段;其中:
○1197个(100.00%)未配对;其中:
■728个(60.82%)比对0次
■186个(15.54%)只比对1次
■283个(23.64%)比对>1次
○39.18%总体比对率
为了进一步理解和详细说明本公开的方面,以下参考文献出于所有目以引用方式明确地并入本文:feehery等人,2013,“amethodforselectivelyenrichingmicrobialdnafromcontaminatingvertebratehostdna.”plosone8(10):e76096.;gilbert等人,2011,“microbialmetagenomics:beyondthegenome.”annualreviewofmarinescience3(1):347–71;kang等人,2015,“metabat,anefficienttoolforaccuratelyreconstructingsinglegenomesfromcomplexmicrobialcommunities.”peerj3(august):e1165;langmead等人,2012,“fastgapped-readalignmentwithbowtie2.”naturemethods9(4):357–59;nurk等人,2013,“assemblinggenomesandmini-metagenomesfromhighlychimericreads.”lecturenotesincomputerscience,158–70;
本文可以使对生物株系来说是独特的任何标记物。例如,标记物可包括但不限于小亚基核糖体rna基因(16s/18srdna)、大亚基核糖体rna基因(23s/25s/28srdna)、插入5.8s基因、细胞色素c氧化酶、β-微管蛋白、延伸因子、rna聚合酶和内部转录间隔区(its)。
核糖体rna基因(rdna),尤其是小亚基核糖体rna基因(即,在真核生物的情况下为18srrna基因(18srdna),在原核生物的情况下为16srrna(16srdna)),已成为用于评估微生物群落中的生物类型和株系的主要目标。然而,大亚基核糖体rna基因28srdna也已被作为目标。rdna适用于分类学识别,因为:(i)它们普遍存在于所有已知生物中;(ii)它们既具有保守区也具有可变区;(iii)它们存在可用于比较的指数级扩展的序列数据库。在样品的群落分析中,保守区用作对应的通用pcr和/或测序引物的退火位点,而可变区可用于系统发育分化。此外,细胞中高拷贝数的rdna有助于从环境样品中检测。
位于18srdna与28srdna之间的内部转录间隔区(its)也已被作为目标。its被转录但在组装核糖体之前被剪掉。its区由两个高度可变的间隔区its1和its2以及插入5.8s基因组成。该rdna操纵子在基因组中以多个拷贝出现。由于its区域不编码核糖体组分,因此它是高度可变的。
在一个实施方案中,独特的rna标记物可以是mrna标记物、sirna标记物或核糖体rna标记物。
编码蛋白质的功能基因在本文中也可用作独特的第一标记物。此类标记物包括但不限于:重组酶a基因家族(细菌reca、古细菌rada和radb、真核生物rad51和rad57、噬菌体uvsx);rna聚合酶β亚基(rpob)基因,其负责转录起始和延伸;伴侣蛋白。还识别了用于细菌加古细菌的候选标记基因:核糖体蛋白s2(rpsb)、核糖体蛋白s10(rpsj)、核糖体蛋白l1(rpla)、翻译延伸因子ef-2、翻译起始因子if-2、金属内肽酶、核糖体蛋白l22、ffh信号识别颗粒蛋白、核糖体蛋白l4/l1e(rpld)、核糖体蛋白l2(rplb)、核糖体蛋白s9(rpsi)、核糖体蛋白l3(rplc)、苯丙氨酰-trna合成酶β亚基、核糖体蛋白l14b/l23e(rpln)、核糖体蛋白s5、核糖体蛋白s19(rpss)、核糖体蛋白s7、核糖体蛋白l16/l10e(rplp)、核糖体蛋白s13(rpsm)、苯丙氨酰-trna合成酶α亚基、核糖体蛋白l15、核糖体蛋白l25/l23、核糖体蛋白l6(rplf)、核糖体蛋白l11(rplk)、核糖体蛋白l5(rple)、核糖体蛋白s12/s23、核糖体蛋白l29、核糖体蛋白s3(rpsc)、核糖体蛋白s11(rpsk)、核糖体蛋白l10、核糖体蛋白s8、trna假尿苷合酶b、核糖体蛋白l18p/l5e、核糖体蛋白s15p/s13e、胆色素原脱氨酶、核糖体蛋白s17、核糖体蛋白l13(rplm)、磷酸核糖甲酰甘氨脒环连接酶(rpse)、核糖核酸酶hii和核糖体蛋白l24。用于细菌的其他候选标记基因包括:转录延伸蛋白nusa(nusa)、rpobdna引导的rna聚合酶亚基β(rpob)、gtp结合蛋白enga、rpocdna引导的rna聚合酶亚基β'、pria引发体装配蛋白、转录-修复偶联因子、ctp合酶(pyrg)、secy前蛋白移位酶亚基secy、gtp结合蛋白obg/cgta、dna聚合酶i、rpsf30s核糖体蛋白s6、poadna引导的rna聚合酶亚基α、肽链释放因子1、rpli50s核糖体蛋白l9、多核糖核苷酸核苷酸转移酶、tsf延伸因子ts(tsf)、rplq50s核糖体蛋白l17、trna(鸟嘌呤-n(1)-)-甲基转移酶(rpls)、rply可能的50s核糖体蛋白l25、dna修复蛋白rada、葡萄糖抑制的分裂蛋白a、核糖体结合因子a、dna错配修复蛋白mutl、smpbssra结合蛋白(smpb)、n-乙酰氨基葡萄糖转移酶、s-腺苷甲基转移酶mraw、udp-n-乙酰基胞壁酰基丙氨酸--d-谷氨酸连接酶、rpls50s核糖体蛋白l19、rplt50s核糖体蛋白l20(rplt)、ruva霍利迪连结体(hollidayjunction)dna解旋酶、ruvb霍利迪连结体dna解旋酶b、sers丝氨酰-trna合成酶、rplu50s核糖体蛋白l21、rpsr30s核糖体蛋白s18、dna错配修复蛋白muts、rpst30s核糖体蛋白s20、dna修复蛋白recn、frr核糖体再循环因子(frr)、重组蛋白recr、未知功能蛋白upf0054、miaatrna异戊烯基转移酶、gtp结合蛋白ychf、染色体复制起始蛋白dnaa、脱磷酸辅酶a激酶、16srrna加工蛋白rimm、atp-锥域蛋白、1-脱氧-d-木酮糖5-磷酸还原异构酶、2c-甲基-d-赤藓糖醇2,4-环焦磷酸合成酶、脂肪酸/磷脂合成蛋白plsx、trna(ile)-赖氨酸合成酶、dnagdna引发酶(dnag)、ruvc霍利迪连结体解离酶、rpsp30s核糖体蛋白s16、重组酶areca、核黄素生物合成蛋白ribf、甘氨酰-trna合成酶β亚基、trmutrna(5-甲基氨基甲基-2-硫代尿苷)-甲基转移酶、rpmi50s核糖体蛋白l35、heme尿卟啉原脱羧酶、杆状决定蛋白、rpma50s核糖体蛋白l27(rpma)、肽基-trna水解酶、翻译起始因子if-3(infc)、udp-n-乙酰胞壁酰-三肽合成酶、rpmf50s核糖体蛋白l32、rpil50s核糖体蛋白l7/l12(rpil)、leus亮氨酰-trna合成酶、liganad依赖性dna连接酶、细胞分裂蛋白ftsa、gtp结合蛋白typa、atp依赖性clp蛋白酶、atp结合亚基clpx、dna复制和修复蛋白recf以及udp-n-乙酰烯醇丙酮酰葡糖胺还原酶。
根据本文所述的方法,磷脂脂肪酸(plfa)也可用作独特的第一标记物。由于plfa在微生物生长期间迅速合成,在储存分子中未发现并且在细胞死亡期间迅速降解,因此它提供了对当前活群体的准确普查。所有细胞都含有可被提取和酯化形成脂肪酸甲酯(fame)的脂肪酸(fa)。当使用气相色谱-质谱法分析fame时,得到的谱图构成样品中微生物的“指纹”。细菌和真核生物域中的生物的膜的化学组成由通过酯型键合连接到甘油的脂肪酸(磷脂脂肪酸(plfa))构成。相比之下,古细菌的膜脂质则由通过醚型键合与甘油连接的长支链烃(磷脂醚脂质(plel))构成。这是区分这三个域的最广泛使用的非遗传标准之一。在这种情况下,来源于微生物细胞膜的磷脂(其特征在于不同的酰基链)是优异的特征分子,因为这种脂质结构多样性可以与特定的微生物类群联系起来。
如本文所提供的,为了确定生物株系是否有活性,测量一种或多种独特的第二标记物的表达水平,第二标记物可以与第一标记物相同或不同(图1b,1004;图2g,2004)。上面描述了独特的第一标记物。独特的第二标记物是微生物活性的标记物。例如,在一个实施方案中,上述任何第一标记物的mrna或蛋白质表达被认为是用于本公开目的的独特的第二标记物。
在一个实施方案中,如果第二标记物的表达水平高于阈值水平(例如,对照水平)或处于阈值水平,则认为微生物是有活性的(图1b,1005;图2g,2005)。在一个实施方案中,如果第二标记物的表达水平与在一些实施方案中作为对照水平的阈值水平相比改变至少约5%、至少约10%、至少约15%、至少约20%、至少约25%或至少约30%,则确定活性。
在一个实施方案中,在蛋白质、rna或代谢物水平上测量第二独特的标记物。独特的第二标记物与第一独特的标记物相同或不同。
如上文所提供,可以根据本文描述的方法检测独特的第一标记物和独特的第二标记物的数量。此外,对独特的第一标记物的检测和定量根据本领域普通技术人员已知的方法进行(图1b,1003-1004,图2g,2003-2004)。
在一个实施方案中,核酸测序(例如,gdna、cdna、rrna、mrna)用于确定独特的第一标记物和/或独特的第二标记物的绝对细胞计数。测序平台包括但不限于桑格(sanger)测序和可从roche/454lifesciences、illumina/solexa、pacificbiosciences、iontorrent和nanopore获得的高通量测序方法。测序可以是特定dna或rna序列的扩增子测序或整个元基因组/转录组鸟枪法测序。
传统的桑格测序(sanger等人(1977)dnasequencingwithchain-terminatinginhibitors.procnatl.acad.sci.usa,74,第5463–5467页,其全文以引用方式并入本文)依赖于在体外dna复制期间通过dna聚合酶选择性地掺入链终止双脱氧核苷酸,并且适合与本文所述的方法一起使用。
在另一个实施方案中,对样品或其部分进行核酸提取,用合适的引物扩增感兴趣的dna(诸如rrna基因),并使用测序载体构建克隆文库。然后通过桑格测序对所选的克隆进行测序,并检索感兴趣的dna的核苷酸序列,从而允许计算样品中独特微生物株系的数量。
来自roche/454lifesciences的454焦磷酸测序产生长读段并且可以用于本文所描述的方法(margulies等人(2005)nature,437,第376-380页;美国专利no.6,274,320、6,258,568、6,210,891,每篇文献全文出于所有目的以引用方式并入本文)。待测序的核酸(例如,扩增子或雾化的基因组/元基因组dna)具有通过pcr或通过连接固定在任一端的特异性衔接子。将具有衔接子的dna固定到悬浮在油包水乳液中的微珠上(理想情况下,一个珠粒将具有一个dna片段)。然后执行乳液pcr步骤以制备每个dna片段的多个拷贝,得到一组珠粒,其中每个珠粒含有相同dna片段的许多克隆拷贝。然后将每个珠粒放入光纤芯片的孔中,该光纤芯片还含有边合成边测序反应所必需的酶。添加碱基(诸如a、c、g或t)引发焦磷酸盐释放,其产生闪光,对所述闪光进行记录以推断每个孔中的dna片段的序列。每次可以运行约100万个读段,读段长度可达1,000个碱基。可以进行双端测序,其产生成对的读段,每个读段始于给定dna片段的一端。可以产生分子条形码并将其置于多重反应中的衔接子序列与感兴趣序列之间,从而允许将每个序列按照生物信息分配给样品。
illumina/solexa测序产生的平均读段长度为约25个碱基对(bp)至约300bp(bennett等人(2005)pharmacogenomics,6:373-382;lange等人(2014).bmcgenomics15,第63页;fadrosh等人(2014)microbiome2,第6页;caporaso等人(2012)ismej,6,第1621–1624页;bentley等人(2008)accuratewholehumangenomesequencingusingreversibleterminatorchemistry.nature,456:53–59)。该测序技术也是边合成边测序,但采用可逆染料终止子和附接有一批寡核苷酸的流动池。待测序的dna片段在任一端均具有特异性衔接子,并在填充有与片段末端杂交的特定寡核苷酸的流动池中冲刷。然后复制每个片段以产生相同片段的聚类。可逆染料终止子核苷酸接着在流动池中冲刷并给予其时间以便附接。洗掉过量的核苷酸,对流动池成像,并且可以去除可逆终止子,使得该过程可以重复,并且可以在随后的循环中继续添加核苷酸。可以实现每个读段的长度为300个碱基的双端读段。illumina平台可以在单次运行中以双端方式生成40亿个片段,每个读段有125个碱基。条形码也可用于样品多路复用,但需使用索引引物(indexingprimer)。
solid(寡核苷酸连接和检测测序,lifetechnologies)法是“边连接边测序”方法,并且可以与本文描述的方法一起用于检测第一标记物和/或第二标记物的存在和量(图1b,1003-1004;图2g,2003-2004)(peckham等人,solidtmsequencingand2-baseencoding.sandiego,ca:americansocietyofhumangenetics,2007;mitra等人(2013)analysisoftheintestinalmicrobiotausingsolid16srrnagenesequencingandsolidshotgunsequencing.bmcgenomics,14(增刊5):s16;mardis(2008)next-generationdnasequencingmethods.annurevgenomicshumgenet,9:387–402;每篇文献全文以引用方式并入本文)。dna片段的文库从待测序的样品制备,并用于制备克隆珠粒群,其中在每个磁珠的表面上仅存在一种片段。附接在磁珠上的片段将具有通用p1衔接子序列,使得每个片段的起始序列都是已知且相同的。引物与文库模板内的p1衔接子序列杂交。一组共四个荧光标记的二碱基探针竞争与测序引物的连接。通过在每个连接反应中询问每个第一和第二碱基来实现二碱基探针的特异性。以决定最终读段长度的循环数进行多个连接、检测和切割循环。solid平台每次运行可产生多达30亿个读段,读段长度为75个碱基。双端测序是可用的并且可以在本文中使用,但是该对中的第二读段的长度仅为35个碱基。样品的多路复用可以通过类似于illumina使用的系统进行,其中采用单独的索引运行。
iontorrent系统(如454测序)适用于本文所述的方法,以检测第一标记物和/或第二标记物的存在和量(图1b,1003-1004;图2g,2003-2004)。它使用含有珠粒的微孔板,珠粒上将附接dna片段。然而,在检测碱基掺入的方式上,它与所有其他系统均不同。当碱基加入到正在生长的dna链中时,释放出质子,这会略微改变周围的ph值。能够灵敏检测ph的微检测器与板上的孔相关联,并且记录何时发生这些变化。将不同的碱基(a、c、g、t)依次通过孔洗涤,从而可以推断出来自每个孔的序列。ionproton平台每次运行可产生多达5000万个读段,读段长度为200个碱基。personalgenomemachine平台具有达400个碱基的更长读段。双向测序是可用的。通过标准在线分子条形码测序可以实现多路复用。
pacificbiosciences(pacbio)smrt测序使用单分子实时测序方法,并且在一个实施方案中,与本文所述的方法一起用于检测第一标记物和/或第二标记物的存在和量(图1b,1003-1004;图2g,2003-2004)。pacbio测序系统不涉及扩增步骤,这使其与其他主要的下一代测序系统区别开来。在一个实施方案中,在包含许多零模波导(zmw)检测器的芯片上执行测序。将dna聚合酶连到zmw检测器上,并在合成dna链时对磷酸化连接染料标记的核苷酸掺入进行实时成像。pacbio系统每次运行产生长度非常长(平均约4,600个碱基)且数量非常大(约47,000个)的读段。典型的“双端”方法不与pacbio一起使用,因为读段通常足够长,使得通过ccs可以多次覆盖片段而无需独立地从每个末端进行测序。用pacbio进行多路复用不涉及独立的读段,而是遵循标准“在线”条形码模型。
在第一独特标记物是its基因组区域的一个实施方案中,使用自动核糖体基因间隔分析(arisa)来确定样品中微生物株系的数量和特性(图1b,1003,图2g,2003)(ranjard等人(2003).environmentalmicrobiology5,第1111-1120页,全文出于所有目的以引用方式并入)。its区域在长度和核苷酸序列上均具有显著的异质性。使用荧光标记的正向引物和自动dna测序仪可以实现高分离度和高通量。在每个样品中包含内标为确定一般片段大小提供了准确性。
在另一个实施方案中,将pcr扩增的rdna片段的片段长度多态性(rflp)(也称为扩增的核糖体dna限制性分析(ardra))用于表征样品中独特的第一标记物及其量(图1b,1003,图2g,2003)(有关另外的细节,参见massol-deya等人(1995),mol.microb.ecol.manual.3.3.2,第1-18页,其全文出于所有目的以引用方式并入本文)。使用通用引物通过pcr产生rdna片段,用限制酶消化,在琼脂糖或丙烯酰胺凝胶中电泳,并用溴化乙锭或硝酸银染色。
用于检测独特的第一标记物的存在和丰度的一种指纹技术是单链构象多态性(sscp)(参见lee等人(1996).applenvironmicrobiol62,第3112-3120页;scheinert等人(1996).j.microbiol.methods26,第103-117页;schwieger和tebbe(1998).appl.environ.microbiol.64,第4870-4876页,每篇文献全文以引用方式并入本文)。在此技术中,对使用16srrna基因特异性引物获得的dna片段(诸如pcr产物)进行变性,并且直接在非变性凝胶上进行电泳。分离基于影响电泳淌度的单链dna的大小和折叠构象的差异来进行。可通过多种策略防止电泳期间dna链的再退火,包括在pcr中使用一种磷酸化引物,接着使用λ外切核酸酶对磷酸化链进行特异性消化,并使用一种生物素化引物来实施对变性后的一条单链的磁分离。在一个实施方案中,为了评估给定微生物群落中的优势群体的身份,切除条带并且进行测序,或者可将sscp模式与特异性探针杂交。电泳条件(诸如凝胶基质、温度和向凝胶添加甘油)可能影响分离。
除基于测序的方法之外,用于定量第二标记物的表达(例如,基因、蛋白质表达)的其他方法也适于与本文提供的方法一起用于确定一种或多种第二标记物的表达水平(图1b,1004;图2g,2004)。例如,定量rt-pcr、微阵列分析、线性扩增技术如基于核酸序列的扩增(nasba)都适合与本文所述的方法一起使用,并且可以根据本领域普通技术人员已知的方法进行。
在另一个实施方案中,对样品或其部分进行定量聚合酶链式反应(pcr)以检测第一标记物和/或第二标记物的存在和量(图1b,1003-1004;图2g,2003-2004)。通过将转录的核糖体和/或信使rna(rrna和mrna)逆转录成互补dna(cdna),然后进行pcr(rt-pcr)来测量特异性微生物株系活性。
在另一个实施方案中,对样品或其部分进行基于pcr的指纹技术以检测第一标记物和/或第二标记物的存在和量(图1b,1003-1004;图2g,2003-2004)。可以基于核苷酸组成通过电泳来分离pcr产物。不同dna分子之间的序列变异影响解链行为,因此具有不同序列的分子将在凝胶中的不同位置停止迁移。因此,电泳谱可通过不同条带或峰的位置和相对强度来限定,并且可被转换为数值数据以用于多样性指数计算。也可从凝胶上切下条带,然后测序以揭示群落成员的系统发育关系。电泳方法可包括但不限于:变性梯度凝胶电泳(dgge)、温度梯度凝胶电泳(tgge)、单链构象多态性(sscp)、限制性片段长度多态性分析(rflp)或扩增核糖体dna限制性分析(ardra)、末端限制性片段长度多态性分析(t-rflp)、自动核糖体基因间隔区分析(arisa)、随机扩增多态性dna(rapd)、dna扩增指纹分析(daf)和bb-peg电泳。
在另一个实施方案中,使样品或其部分接受基于芯片的平台如微阵列或微流体处理,以确定独特的第一标记物的量和/或独特的第二标记物的存在/量(图1b,1003-1004;图2g,2003-2004)。从样品中的总dna扩增pcr产物,并直接与固定到微阵列上的已知分子探针杂交。在荧光标记的pcr扩增子与探针杂交后,通过使用共聚焦激光扫描显微镜检查对阳性信号进行评分。微阵列技术允许通过复制而快速评估样品,这在微生物群落分析中具有显著优势。微阵列上的杂交信号强度可以与目标生物的量成正比。通用高密度16s微阵列(例如phylochip)含有约30,000个靶向几种培养的微生物物种和“候选菌门”(candidatedivision)的16srrna基因探针。这些探针靶向所有121个划定的原核生物目,并允许同时检测8,741个细菌和古细菌类群。用于分析微生物群落的另一种微阵列是功能基因阵列(fga)。与phylochp不同,fga主要设计用来检测细菌的特定代谢组。因此,fga不仅揭示群落结构,而且还阐明原位群落代谢潜力。fga含有来自具有已知生物功能的基因的探针,因此它们可用于将微生物群落组成与生态系统功能联系起来。称为geochip的fga含有>24,000个探针,这些探针来自各种生物地球化学、生态学和环境过程(如氨氧化、甲烷氧化和固氮)中涉及到的所有已知代谢基因。
在一个实施方案中,蛋白质表达测定法与本文所述的方法一起用于测定一种或多种第二标记物的表达水平(图1b,1004;图2g,2004)。例如,在一个实施方案中,利用质谱法或免疫测定法如酶联免疫吸附测定法(elisa)来定量一种或多种独特的第二标记物的表达水平,其中所述一种或多种独特的第二标记物是蛋白质。
在一个实施方案中,使样品或其部分接受溴脱氧尿苷(brdu)掺入以确定第二独特标记物的水平(图1b,1004;图2g,2004)。brdu是胸苷的合成核苷类似物,可以掺入正在复制的细胞的新合成dna中。然后可以使用brdu特异性抗体来检测碱基类似物。因此,brdu掺入可识别正在活跃复制它们的dna的细胞,这是根据本文所述的方法的一个实施方案的微生物活性量度。brdu掺入可与fish组合使用以提供所靶向的细胞的特性和活性。
在一个实施方案中,对样品或其部分进行结合fish的显微放射自显影术(mar)以确定第二独特标记物的水平(图1b,1004;图2g,2004)。mar-fish基于以下步骤:将放射性底物掺入细胞中,使用放射自显影术检测活性细胞以及使用fish识别细胞。用显微镜以单细胞分辨率进行活细胞的检测和识别。mar-fish提供关于以下方面的信息:总细胞,探针靶向的细胞,和掺入有给定放射性标记物质的细胞的百分比。该方法实现对所靶向的微生物的原位功能的评估,并且是研究微生物的体内生理学的有效方法。结合mar-fish开发用于定量细胞特异性底物摄取的技术被称为定量mar(qmar)。
在一个实施方案中,对样品或其部分进行结合fish的稳定同位素拉曼光谱分析(拉曼-fish)以确定第二独特标记物的水平(图1b,1004;图2g,2004)。该技术结合了稳定同位素探测、拉曼光谱分析和fish以将代谢过程与特定生物联系起来。被细胞结合的稳定同位素的比例影响光散射,从而导致标记的细胞组分(包括蛋白质和mrna组分)发生可测量的峰移位。拉曼光谱分析可用于识别细胞是否合成化合物,包括但不限于:油(如烷烃)、脂质(如三酰基甘油(tag))、特异性蛋白质(如血红素蛋白、金属蛋白)、细胞色素(如p450、细胞色素c)、叶绿素、发色团(如用于光捕获类胡萝卜素和视紫红质的色素)、有机聚合物(如聚羟基烷酸酯(pha)、聚羟基丁酸酯(phb))、藿烷类化合物、类固醇、淀粉、硫化物、硫酸盐和次级代谢物(如维生素b12)。
在一个实施方案中,对样品或其部分进行dna/rna稳定同位素探测(sip)以确定第二独特标记物的水平(图1b,1004;图2g,2004)。sip使得能够确定与特定代谢途径相关联的微生物多样性,并且一直以来通常用于研究碳和氮化合物利用中涉及到的微生物。用稳定同位素(例如13c或15n)标记感兴趣的底物并将其添加到样品中。只有能够代谢底物的微生物才能将底物掺入它们的细胞中。随后,可以通过密度梯度离心分离13c-dna和15n-dna并将其用于元基因组分析。基于rna的sip可以是用于sip研究的响应性生物标记物,因为rna本身是细胞活性的反映。
在一个实施方案中,使样品或其部分接受同位素阵列处理以确定第二独特标记物的水平(图1b,1004;图2g,2004)。同位素阵列允许以高通量方式对活性微生物群落进行功能和系统发育筛选。该技术使用用于监测底物摄取谱的sip和用于确定活性微生物群落的分类学特性的微阵列技术的组合。将样品与14c标记的底物一起温育,该底物在生长过程中掺入微生物生物质中。将14c标记的rrna与未标记的rrna分离,然后用荧色物进行标记。将荧光标记的rrna杂交到系统发育微阵列上,然后扫描放射性和荧光信号。因此,该技术允许同时研究微生物群落组成以及复杂微生物群落的代谢活性微生物的特异性底物消耗。
在一个实施方案中,对样品或其部分进行代谢组学测定以确定第二独特标记物的水平(图1b,1004;图2g,2004)。代谢组学研究代谢组,代谢组代表在生物细胞、组织、器官或生物中所有代谢物(即细胞过程的终产物)的集合体。该方法可用于监测微生物的存在和/或微生物介导的过程,因为它允许将特定代谢物谱与不同的微生物相关联。可使用诸如气相色谱法-质谱法(gc-ms)的技术获得与微生物活性相关联的细胞内和细胞外代谢物的谱。代谢组学样品的复杂混合物可通过诸如气相色谱法、高效液相色谱法和毛细管电泳的技术来分离。代谢物的检测可以通过质谱、核磁共振(nmr)光谱、离子淌度光谱、电化学检测(与hplc结合)和放射性标记(当与薄层色谱法组合时)进行。
根据本文描述的实施方案,确定样品中一种或多种活性微生物株系的存在和相应数量(图1b,1006;图2g,2006)。例如,对通过测定第一标记物的数量和存在而获得的株系特性信息进行分析以确定存在多少独特的第一标记物,从而代表独特的微生物株系(例如,通过计算测序测定中序列读段的数量)。在一个实施方案中,该值可表示为第一标记物的总序列读段的百分比,以给出特定微生物类型的独特微生物株系的百分比。在进一步的实施方案中,将该百分比乘以微生物类型的数量(在步骤1002或2002中获得,参见图1b和图2g),得出样品中和给定体积中所述一种或多种微生物株系的绝对细胞计数。
如上所述,如果第二独特标记物表达水平处于阈值水平,高于阈值,例如比对照水平高至少约5%、至少约10%、至少约20%或至少约30%,那么所述一种或多种微生物株系则被视为有活性的。
在本公开的另一个方面,在多个样品中确定用于确定一种或多种微生物株系的绝对细胞计数的方法(图2g,具体参见2007)。要将微生物株系分类为有活性的,其仅需在一个样品中是有活性的即可。样品可在多个时间点取自相同的来源,或者可来自不同的环境来源(例如,不同的动物)。
在一个实施方案中,使用样品的绝对细胞计数值将所述一种或多种活性微生物株系与环境参数相关联(图2g,2008)。在一个实施方案中,环境参数是第二活性微生物株系的存在。在一个实施方案中,通过网络分析和/或图论来确定株系和参数的共现,从而将所述一种或多种活性微生物株系与环境参数相关联。
在一个实施方案中,确定一种或多种活性微生物株系与环境参数的共现包括网络和/或聚类分析方法,以测量网络内的多种株系或一种株系与环境参数的连通性,其中所述网络是共有共同或相似的环境参数的两个或多个样品的集合体。本文提供并讨论了独立性测量的实例,并且通过配置以下教导内容和方法可以理解另外的细节:blomqvist"onameasureofdependencebetweentworandomvariables"theannalsofmathematicalstatistics(1950):593-600;hollander等人"nonparametricstatisticalmethods-wileyseriesinprobabilityandstatisticstextsandreferencessection"(1999);和/或blum等人"distributionfreetestsofindependencebasedonthesampledistributionfunction"theannalsofmathematicalstatistics(1961):485-498;上述公布中每个的全文出于所有目的以引用方式明确地并入本文。
在另一个实施方案中,使用包括皮尔逊(pearson)相关、斯皮尔曼(spearman)相关、肯德尔(kendall)相关、典型相关分析、似然比检验(例如,通过调整wilks,s.s.“ontheindependenceofksetsofnormallydistributedstatisticalvariables”econometrica,第3卷第3期,1935年7月,第309-326页中详述的教导内容和方法,该文献全文出于所有目的以引用方式明确地并入本文)和典型相关分析在内的相关方法建立变量之间的连通性。根据所比较的变量的数量,可以在适当时使用这些方法的多变量扩展、最大相关(参见例如alfrédrényi"onmeasuresofdependence"actamathematicahungarica10.3-4(1959):441-451,该文献全文以引用方式明确地并入本文)或两者(mac)。一些实施方案利用最大相关分析和/或被配置用于发现多维模式的其他多变量相关测量(例如,通过调整2014年中国北京第31届机器学习国际大会会议录(proceedingsofthe31stinternationalconferenceonmachinelearning)中的nguyen等人“multivariatemaximalcorrelationanalysis”的方法和教导内容,该文献全文出于所有目的以引用方式明确地并入本文)。在一些实施方案中,网络度量和分析(例如由farine等人在“constructing,conductingandinterpretinganimalsocialnetworkanalysis”journalofanimalecology,2015,84,第1144-1163页,doi:10.1111/1365-2656.12418中所讨论的(其全文出于所有目的以引用方式明确地并入本文))可以用于和配置用于本公开。
在一些实施方案中,网络分析包括在变量之间建立连通性的非参数方法(例如,通过调整以下文献中详述的教导内容和方法:taskinen等人"multivariatenonparametrictestsofindependence."journaloftheamericanstatisticalassociation100.471(2005):916-925;以及gieser等人“anonparametrictestofindependencebetweentwovectors.”journaloftheamericanstatisticalassociation,第92卷,第438期,1977年6月,第561-567页;每篇文献的全文出于所有目的以引用方式明确地并入本文),其包括互信息最大信息系数、最大信息熵(mie;例如,通过调整zhangya-hong等人"detectingmultivariablecorrelationwithmaximalinformationentropy[j]”journalofelectronics&informationtechnology,2015-01(37(1):123-129)的教导内容和方法,该文献全文出于所有目的以引用方式明确地并入本文)、核典型相关分析(kcca;例如,通过调整bach等人“kernelindependentcomponentanalysis”journalofmachinelearningresearch3(2002)1-48中详述的教导内容和方法,该文献全文出于所有目的以引用方式明确地并入本文)、交替条件期望或反向拟合算法(ace;例如,通过调整breiman等人"estimatingoptimaltransformationsformultipleregressionandcorrelation:rejoinder."journaloftheamericanstatisticalassociation80,第391期,(1985):614-19,doi:10.2307/2288477中详述的教导内容和方法,该文献全文出于所有目的以引用方式明确地并入本文)、距离相关性度量(dcor;例如,通过调整szekely等人“measuringandtestingdependencebycorrelationofdistances”theannalsofstatistics,2007,第35卷,第6期,2769-2794,doi:10.1214/009053607000000505中详述的教导内容和方法,该文献全文出于所有目的以引用方式明确地并入本文)、布朗(brownian)距离协方差(dcov;例如,通过调整szekely等人“browniandistancecovariance”theannalsofappliedstatistics,2009,第3卷,第4期,1236-1265,doi:10.1214/09-aoas312中详述的教导内容和方法,该文献全文出于所有目的以引用方式明确地并入本文)、希尔伯特-施密特(hilbert-schmidt)独立性准则(hsci/chsi;例如,通过调整gretton等人“akernaltwo-sampletest”journalofmachinelearningresearch13(2012)723-773和poczos等人“copula-basedkerneldependencymeasures”carnegiemellowuniversity,researchshowcase@cmu,第29届机器学习国际大会会议录中详述的教导内容和方法,每篇文献全文出于所有目的以引用方式明确地并入本文)、随机化依赖性系数(rdc;例如,通过调整lopez-paz等人“therandomizeddependencecoefficient”advancesinneuralinformationprocessingsystems(2013)中详述的教导内容和方法,该文献全文出于所有目的以引用方式明确地并入本文)。在一些实施方案中,这些方法中的一个或多个可以与装袋(bagging)或提升(boosting)方法或k最近邻估计量结合(例如,通过调整以下文献中详述的教导内容和方法:breiman,“arcingclassifiers”theannalsofstatistics,1998,第26卷,第3期,801-849;liu,“modifiedbaggingofmaximalinformationcoefficientforgenome-wideidentification”int.j.dataminingandbioinformatics,第14卷,第3期,2016,第229-257页;和/或gao等人“efficientestimationofmutualinformationforstronglydependentvariables”proceedingsofthe18thinternationalconferenceonartificialintelligenceandstatistics(aistats),2015,sandiego,ca,jmlr:w&cp第38卷;每篇文献全文出于所有目的以引用方式明确地并入本文)。
在一些实施方案中,网络分析包括节点级分析,其包括度、强度、中介中心性、特征向量中心性、页面排名和范围。在另一个实施方案中,网络分析包括网络级度量,其包括密度、同质性或同配性、传递性、链接分析、模块性分析、稳健性测量、中介性测量、连通性测量、传递性测量、中心性测量或其组合。在其他实施方案中,将物种群落规则(参见例如connor等人“theassemblyofspeciescommunities:chanceorcompetition?”ecology,第60卷,第6期(1979年12月),第1132-1140页,该文献全文出于所有目的以引用方式并入本文)应用于网络,其可以包括利用分组策略假设(例如,通过调整franks等人“samplinganimalassociationnetworkswiththegambitofthegroup”behavecolsociobiol(2010)64:493,doi:10.1007/x00265-0098-0865-8中详述的教导内容和方法,该文献全文出于所有目的以引用方式明确地并入本文)。在一些实施方案中,可以使用特征向量/模块性矩阵分析方法,例如,通过配置markejnewman在“findingcommunitystructureinnetworksusingtheeigenvectorsofmatrices”physicalreviewe74.3(2006):036104中讨论的教导内容和方法,该文献全文出于所有目的以引用方式明确地并入本文。
在一些实施方案中,利用时间聚合网络或时间有序网络。在另一个实施方案中,聚类分析方法包括使用图中群落检测和/或使用群落检测算法(例如,以非限制性实例的方式,louvain、bron-kerbosch、girvan-newman、clauset-newman-moore、pons-latapy和wakita-tsurumi算法)建立或构建观察矩阵、连通性模型、子空间模型、分布模型、密度模型或质心模型。
在一些实施方案中,聚类分析方法是基于模块性优化的启发式方法。在进一步的实施方案中,聚类分析方法是louvain方法(参见例如以下文献中描述的方法:blondel等人(2008)fastunfoldingofcommunitiesinlargenetworks.journalofstatisticalmechanics:theoryandexperiment,第2008卷,2008年10月,该文献全文出于所有目以引用方式并入本文,并且其可以适用于本文公开的方法)。
在其他实施方案中,网络分析包括通过链接挖掘和预测、集体分类、基于链接的聚类、分层聚类分析、关系相似性或其组合的网络预测建模。在另一个实施方案中,网络分析包括基于微分方程的群体建模。在另一个实施方案中,网络分析包括lotka-volterra建模。
在一些实施方案中,将所述一种或多种活性微生物株系与样品中的环境参数相关联(例如,确定共现)包括创建使用表示环境参数和微生物株系关联的链接来填充的矩阵。
在一些实施方案中,编译在步骤2007获得的多个样品数据(例如,针对两个或更多个样品,这些样品可以在两个或更多个时间点采集,其中每个时间点对应于单个样品)。在进一步的实施方案中,将每个样品中的所述一种或多种微生物株系中的每一种的细胞数量存储在关联矩阵中(在一些实施方案中,其可以是量矩阵)。在一个实施方案中,使用通过关联(例如,量)数据加权的规则挖掘方法,将关联矩阵用来识别特定时间点样品中的活性微生物株系之间的关联。在一个实施方案中应用过滤器以删除不重要的规则。
在一些实施方案中,将一种或多种、或两种或更多种活性微生物株系的绝对细胞计数与一个或多个环境参数相关联(图2g,2008),例如通过确定共现。环境参数可基于待分析的一个或多个样品来选择并且不被本文所述的方法限制。环境参数可以是样品本身的参数,例如,ph、温度、样品中的蛋白质的量。另选地,环境参数是影响微生物群落特性变化的参数(即,其中微生物群落的“特性”由群落中微生物株系的类型和/或特定微生物株系的数量表征),或受微生物群落特性变化的影响的参数。例如,在一个实施方案中,环境参数是动物的食物摄入量或由泌乳反刍动物产生的乳汁量(或乳汁的蛋白质或脂肪含量)。在一个实施方案中,环境参数是存在于相同样品中的微生物群落中第二微生物株系的存在、活性和/或量。在本文描述的一些实施方案中,环境参数被称为元数据参数,反之亦然。
元数据参数的其他实例包括但不限于来自获取样品的宿主的遗传信息(例如,dna突变信息)、样品ph、样品温度、特定蛋白质或mrna的表达、营养条件(例如,周围环境/生态系统的一种或多种营养物质的水平和/或特性)、对于疾病的易感性或抗性、疾病的发作或进展、样品对于毒素的易感性或抗性、异生物质化合物(药物)的效力、天然产物的生物合成或其组合。
例如,根据一个实施方案,根据图2g的方法(即,在2001-2007处)针对多个样品计算微生物株系数量变化。编译一种或多种活性株系(例如,根据步骤2006最初被识别为有活性的一种或多种株系)随时间的株系数量变化,并且记录变化的方向性(即,负值代表减少,正值代表增加)。将细胞随时间的数量表示为网络,其中微生物株系表示节点,量加权规则表示边缘。利用马尔可夫(markovchain)链和随机游走来确定节点之间的连通性并限定聚类。在一个实施方案中,使用元数据过滤聚类以便识别与合乎需要的元数据相关联的聚类(图2g,2008)。
在进一步的实施方案中,通过整合随时间的细胞数变化和存在于目标聚类中的株系根据重要性对微生物株系进行排名,其中最大细胞数变化排名最高。
在一个实施方案中,将网络和/或聚类分析方法用于测量网络内的所述一种或多种株系的连通性,其中所述网络是共有共同或相似的环境参数的两个或更多个样品的集合体。在一个实施方案中,网络分析包括链接分析、模块性分析、稳健性测量、中介性测量、连通性测量、传递性测量、中心性测量或其组合。在另一个实施方案中,网络分析包括通过链接挖掘和预测、社会网络理论、集体分类、基于链接的聚类、关系相似性或其组合的网络预测建模。在另一个实施方案中,网络分析包括在变量之间建立连接的互信息、最大信息系数计算或其他非参数方法。在另一个实施方案中,网络分析包括基于微分方程的群体建模。在又一个实施方案中,网络分析包括lotka-volterra建模。
聚类分析方法包括建立连通性模型、子空间模型、分布模型、密度模型或质心模型。
例如执行图2g的方法步骤2008的基于网络和聚类的分析可通过处理器、部件和/或模块来执行。如本文所用,部件和/或模块可以是例如任何组件、指令和/或成套的操作性耦合的电气部件,并且可以包括例如存储器、处理器、电迹线、光学连接器、软件(在硬件中执行)和/或类似物。
图3a是示出根据一个实施方案的微生物分析、筛选和选择平台和系统300的示意图。根据本公开的平台可包括确定天然微生物群落内的多维种间相互作用和依赖性的系统和过程,并且参照图3a描述一个实例。图3a是架构图并因此省略了某些方面以使描述更清楚,但这些方面当在本公开的上下文中理解时对于技术人员而言应是显而易见的。
如图3a所示,微生物筛选和选择平台和系统300可包括一个或多个处理器310、数据库319、存储器320、通信接口390、被配置为与用户输入装置396和外围装置397(包括但不限于数据采集和分析装置诸如fac、选择/温育/配制装置、和/或额外的数据库/数据源、远程数据采集装置(例如,可采集元数据环境数据诸如样品特征、温度、天气等的装置,包括运行应用程序以采集此类信息的移动智能电话以及其他移动或固定装置))交互的输入/输出接口、被配置为在通信网络392(例如,lan、wan和/或因特网)上接收数据并向客户端393b(其可包括用户界面和/或显示器,诸如图形显示器)和用户393a传送数据的网络接口、数据采集部件330、绝对计数部件335、样品相关部件340、活性部件345、网络分析部件350以及株系选择/途径识别/微生物群集生成部件355。在一些实施方案中,微生物筛选系统300可以是单一物理装置。在其他实施方案中,微生物筛选系统300可包括多个物理装置(例如,通过网络操作性地耦合),其中的每一个可包括图3a所示的一个或多个部件和/或模块。
微生物筛选系统300中的每个部件或模块可操作性地耦合到每个其余的部件和/或模块。微生物筛选系统300中的每个部件和/或模块可以是能够执行与该部件和/或模块相关联的一种或多种特定功能的硬件和/或软件(在硬件中存储和/或执行)的任何组合。
存储器320可以是例如随机存取存储器(ram)(例如,动态ram、静态ram)、闪存存储器、移动式存储器、硬盘驱动器、数据库等等。在一些实施方案中,存储器320可包括例如数据库(例如,如在319中的)、过程、应用程序、虚拟机和/或一些其他软件部件、程序和/或模块(在硬件中存储和/或执行)或被配置来执行用于微生物筛选和群集生成的微生物筛选过程和/或一种或多种相关方法(例如,通过数据采集部件330、绝对计数部件335、样品相关部件340、活性部件345、网络分析部件350、株系选择/途径识别/微生物群集生成部件355(和/或相似的模块))的硬件部件/模块。在此类实施方案中,执行微生物筛选和/或群集生成过程和/或相关方法的指令可存储在存储器320中并且在处理器310处执行。在一些实施方案中,通过数据采集部件330采集的数据可存储在数据库319中和/或存储器320中。
处理器310可被配置为控制例如通信接口390的操作,将数据编写入存储器320和从该存储器读取数据,以及执行存储在存储器320中的指令。处理器310还可以被配置为执行和/或控制例如数据采集部件330、绝对计数部件335、样品相关部件340、活性部件和网络分析部件350的操作,如本文进一步详细描述的。在一些实施方案中,在一个或多个处理器310的控制下并且基于存储在存储器320中的方法或过程,数据采集部件330、绝对计数部件335、样品相关部件340、活性部件345、网络分析部件350以及株系选择/途径识别/群集生成部件355可被配置为执行微生物筛选、选择和合成群集生成过程,如本文进一步详细描述的。
通信接口390可包括和/或被配置为管理微生物筛选系统300的一个或多个端口(例如,通过一个或多个输入输出接口395)。在一些情况下,例如,通信接口390(例如,网络接口卡(nic))可包括一个或多个线卡,其中的每一个可包括(操作性地)耦合到装置(例如,外围装置397和/或用户输入装置396)的一个或多个端口。通信接口390中包括的端口可以是可主动与耦合的装置通信或在网络392上通信(例如,与终端用户装置393b、主机装置、服务器等通信)的任何实体。在一些实施方案中,这样的端口不一定需要是硬件端口,而可以是虚拟端口或由软件定义的端口。通信网络392可以是能够传送信息(例如,数据和/或信号)的任何网络或网络组合,并且可以包括例如电话网络、以太网网络、光纤网络、无线网络和/或蜂窝网络。通信可以通过网络进行,例如wi-fi或无线局域网(“wlan”)连接、无线广域网(“wwan”)连接和/或蜂窝连接。网络连接可以是有线连接,例如以太网连接、数字用户线(“dsl”)连接、宽带同轴连接和/或光纤连接。例如,微生物筛选系统300可以是被配置为通过网络392被一个或多个计算装置393b访问的主机装置。以这种方式,计算装置可通过网络392向微生物筛选系统300提供信息和/或从该微生物筛选系统接收信息。此类信息可以是例如供微生物筛选系统300用来采集、相关、确定、分析和/或生成有活性的、经网络分析的微生物群集的信息,如本文进一步详细描述的。相似地,计算装置可被配置为检索和/或请求来自微生物筛选系统300的确定的信息。
在一些实施方案中,通信接口390可包括和/或被配置成包括输入/输出接口395。输入/输出接口可接受、连通和/或连接到用户输入装置、外围装置、加密处理器装置和/或类似装置。在一些情况下,一种输出装置可以是视频显示器,其可包括例如具有从视频接口接收信号的接口(例如,数字视频接口(dvi)电路和电缆)的阴极射线管(crt)或液晶显示器(lcd)、led或基于等离子体的监视器。在此类实施方案中,通信接口390可被配置成尤其用于接收数据和/或信息以及发送微生物筛选修改、命令和/或指令。
数据采集部件330可以是下述任何硬件和/或软件部件和/或模块(存储在诸如存储器320的存储器中和/或在诸如处理器310的硬件中执行),其被配置为采集、处理和/或归一化数据以用于通过绝对计数部件335、样品相关部件340、活性部件345、网络分析部件350和/或株系选择/途径识别/群集生成部件355实施的对天然微生物群落内的多维种间相互作用和依赖性的分析。在一些实施方案中,数据采集部件330可被配置为确定给定体积的样品中的一种或多种活性生物株系的绝对细胞计数。基于一种或多种活性微生物株系的绝对细胞计数,数据采集部件330可使用标记物序列来识别绝对细胞计数数据集内的活性株系。数据采集部件330可在一段时间内连续采集数据以表示样品内的微生物群体的动态学。数据采集部件330可编译时间数据并将量矩阵中的每种活性生物株系的细胞数存储在诸如存储器320的存储器中。
样品相关部件340和网络分析部件350可被配置为一起确定天然微生物群落内的多维种间相互作用和依赖性。样品相关部件340可以是下述任何硬件和/或软件部件(存储在诸如存储器320的存储器中和/或在诸如处理器310的硬件中执行),其被配置为将元数据参数(环境参数,例如通过共现)与一种或多种活性微生物株系的存在相关联。在一些实施方案中,样品相关部件340可将一种或多种活性生物株系与一个或多个环境参数相关联。
网络分析部件350可以是下述任何硬件和/或软件部件(存储在诸如存储器320的存储器中和/或在诸如处理器310的硬件中执行),其被配置为确定样品中的一种或多种活性微生物株系与环境(元数据)参数的共现。在一些实施方案中,基于通过数据采集部件330采集的数据以及通过样品相关部件340确定的所述一种或多种活性微生物株系与一个或多个环境参数之间的相关性,网络分析部件350可创建使用表示环境参数和微生物株系关联的链接、所述一种或多种活性微生物株系的绝对细胞计数以及所述一种多种独特的第二标记物的表达水平来填充的矩阵,以代表异质微生物株系群体的一个或多个网络。例如,网络分析可使用关联(量和/或丰度)矩阵来使用以量数据加权的规则挖掘方法来识别样品中的活性微生物株系与元数据参数之间的关联(例如,两种或更多种活性微生物株系的关联)。在一些实施方案中,网络分析部件350可应用过滤器来选择和/或删除规则。网络分析部件350可计算活性株系随时间的细胞数变化,记录变化的方向性(即,负值代表减少,正值代表增加)。网络分析部件350可将矩阵表示为网络,其中微生物株系表示节点,量加权规则表示边缘。网络分析部件350可利用杠杆马尔可夫链和随机游走来确定节点之间的连通性并限定聚类。在一些实施方案中,网络分析部件350可使用元数据来过滤聚类,以便识别与合乎需要的元数据相关联的聚类。在一些实施方案中,网络分析部件350可通过整合随时间的细胞数变化和存在于目标聚类中的株系对目标微生物株系进行排名,其中最大细胞数变化排名最高。
在一些实施方案中,网络分析包括链接分析、模块性分析、稳健性测量、中介性测量、连通性测量、传递性测量、中心性测量或其组合。在另一个实施方案中,可以使用包括建立连通性模型、子空间模型、分布模型、密度模型或质心模型的聚类分析方法。在另一个实施方案中,网络分析包括通过链接挖掘和预测、集体分类、基于链接的聚类、关系相似性或其组合的网络预测建模。在另一个实施方案中,网络分析包括在变量之间建立连接的互信息、最大信息系数计算或其他非参数方法。在另一个实施方案中,网络分析包括基于微分方程的群体建模。在另一个实施方案中,网络分析包括lotka-volterra建模。
图3b示出了根据本公开的一个实施方案的示例性逻辑流。首先,采集和/或接收多个样品和/或样品集合3001。应当理解,如本文所用,“样品”可以指一个或多个样品、样品集合、多个样品(例如,来自特定群体),使得当讨论两个或更多个不同的样品时,为了便于理解,每个样品可包括多个子样品(例如,当讨论第一样品和第二样品时,第一样品可包括从第一群体采集的2个、3个、4个、5个或更多个子样品,第二样品可包括从第二群体或另选地从第一群体但是在不同时间点(诸如采集第一子样品之后的一周或一个月)采集的2个、3个、4个、5个或更多个子样品)。当采集子样品时,可监测并存储每个子样品的各个采集指标和参数,包括环境参数、定性和/或定量观察结果、群体成员身份(例如,因此,当从相同的群体在两个或更多个不同的时间采集样品时,子样品通过身份配对,所以将在时间1来自动物1的子样品与在时间2从该相同动物采集的子样品相关联,依次类推)。
针对每个样品、样品集合和/或子样品,基于目标生物类型对细胞进行染色3002,对每个样品/子样品或其部分称重并顺序地稀释3003,且进行处理3004以确定每个样品/子样品中的每种微生物类型的细胞数。在一种示例性实现方式中,可使用细胞分选仪对来自样品(诸如来自环境样品)的各个细菌和真菌细胞计数。作为本公开的一部分,对特定染料显色以实现对根据传统方法先前不可计数的微生物的计数。遵循本公开的方法,使用特定染料对细胞壁(例如,针对细菌和/或真菌)进行染色,并且可使用激光基于细胞特征从较大群体对目标细胞的离散群体进行计数。在一个特定实例中,制备环境样品并稀释到等渗缓冲液中且使用染料进行染色:(a)对于细菌,可使用以下染料染色-dna:sybr绿,呼吸作用:5-氰基-2,3-二甲苯基氯化四唑和/或ctc,细胞壁:孔雀石绿和/或结晶紫;(b)对于真菌,可使用以下染料染色-细胞壁:calcofluor白、刚果红、台盼蓝、直接黄96、直接黄11、直接黑19、直接橙10、直接红23、直接红81、直接绿1、直接紫51、麦胚凝集素-wga、活性黄2、活性黄42、活性黑5、活性橙16、活性红23、活性绿19和/或活性紫5。
在本公开的开发中,有利地发现虽然直接染料和活性染料通常与纤维素基材料(即,棉、亚麻和粘胶人造丝)的染色相关联,但是它们分别由于β-(1→4)-连接的n-乙酰葡萄糖胺链以及β-(1→4)-连接的d-葡糖胺和n-乙酰基-d-葡糖胺链的存在也可用来对甲壳质和壳聚糖染色。当这些亚基装配成链时,形成与纤维素链非常相似的扁平纤维状结构。直接染料通过染料与纤维分子之间的范德华力附着到甲壳质和/或壳聚糖分子上。两者之间的表面积接触越大,相互作用越强。另一方面,活性染料与甲壳质和/或壳聚糖形成共价键。
将每个染色的样品加载到fac上3004以用于计数。可将样品运行通过具有产生单个液滴(例如,约1/10微升(0.1μl))的料流的特定尺寸喷嘴(例如,100μm,根据实现方式和应用而选择)的微流体芯片。这些变量(喷嘴尺寸、液滴形成)可针对每种目标微生物类型进行优化。理想地,包封在每个液滴中的是一个细胞或“事件”,并且当每个液滴被激光击中时,染色的任何物质被激发并发射不同波长的光。fac以光学方式检测每个发射,并且可将它们绘制为事件(例如,在2d图上绘制)。典型的图由针对事件大小(通过“正向散射”确定)的一条轴和针对荧光强度的另一条轴组成。可在这些图上的离散群体周围画“门”,并且可对这些门中的事件进行计数。
图3c示出了来自使用直接黄染色的真菌的示例数据;包括酵母单一培养物3005a(阳性对照,左)、大肠杆菌3005b(阴性对照,中)和环境样品3005c(实验,右)。在该图中,“后向散射”(bsc-a)测量事件的复杂性,而fitc测量来自直接黄的荧光发射的强度。每个点表示一个事件,并且事件的密度通过从绿至红的颜色变化指示。门b指示其中预期将发现靶向事件(在这种情况下是使用直接黄染色的真菌)的一般区域。
返回到图3b,以从一个或多个来源采集3001的两个或更多个样品(包括从各个动物或单一地理位置随时间采集的样品;从在地理学、品种、表现、饮食、疾病等方面不同的两个或更多个组采集的样品;从经历生理学扰动或事件的一个或多个组采集的样品;等等)开始,可使用流式细胞术(包括染色3002)分析样品以建立绝对计数,如上所讨论的。对样品进行称重并顺序地稀释3003,且使用fac进行处理3004。然后处理来自fac的输出以确定每个样品中的所需生物类型的绝对数量3005。以下代码段示出根据一个实施方案的用于此类处理的示例性方法:
从每个样品分离总核酸3006。将核酸样品洗脱液分成两个部分(通常,两个等量部分),并对每个部分进行酶促纯化以获得纯化的dna3006a或纯化的rna3006b。将纯化的rna通过酶促转化为cdna3006c来稳定化。使用pcr针对纯化的dna和纯化的cdna两者制备测序文库(例如,illumina测序文库)以附接适当的条形码和衔接子区域,并且扩增对于测量所需生物类型适当的标记物区域3007。可评估并定量文库质量,然后可集中所有的文库并进行测序。
对原始测序读段进行质量修整和合并3008。对处理的读段进行去重和聚类以生成存在于多个样品中的所有独特的株系的集合或列表3009。该集合或列表可用于存在于多个样品中的每种株系的分类学识别3010。可识别来源于dna样品的测序文库,并且将来自所识别的dna文库的测序读段映射回去重株系的集合或列表,以便识别每个样品中存在哪些株系,并且对每个样品中的每种株系的读段数量进行定量3011。然后将定量的读段列表与目标微生物类型的绝对细胞计数整合,以便确定每种株系的绝对数量或细胞计数3013。以下代码段示出根据一个实施方案的用于此类处理的示例性方法:
#userdefinedvariables
#
#input=quantifiedcountoutputfroinsequenceanalysis
#count=calculatedabsolutecellcountoforganismtype
#taxonomy=predictedtaxonomyofeachstrain
#
readabsolutecellcountfileascounts
readtaxonomyfileastax
ncols=len(counts)
num_samples=ncols/2
tax_level=[]
tax_level.append(unique(taxonomy[′kingdom′].values.ravel()))
tax_level.append(unique(taxonomy[′phylum′].values.ravel()))
tax_level.append(unique(taxonomy[′class′].values.ravel()))
tax_level.append(unique(taxonomy[′order′].values.ravel()))
tax_level.append(unique(taxonomy[′family′]values.ravel()))
tax_level.append(unique(taxonomy[′genus′].values.ravel()))
tax_level.append(unique(taxonomy[′species′].values.ravel()))
tax_counts=merge(left=counts,right=tax)
#specieslevelanalysis
tax_counts.to_csv(′species.txt′)
#onlypulldnasamples
data_mule=loadcsv(′species.txt′,usecols=xrange(2,ncols,2))
data_mule_normalized=data_mule/sum(data_mule)
data_mule_with_counts=data_mule_normalized*counts
repeatforeverytaxonomiclevel
识别来源于cdna样品的测序文库3014。然后将来自所识别的cdna文库测序读段映射回去重株系的列表,以便确定每个样品中哪些株系是有活性的。如果读段的数量低于指定的或指明的阈值3015,则所述株系被认为或识别为无活性的并且从随后的分析去除3015a。如果读段的数量超过阈值3015,则所述株系被认为或识别为有活性的并且保留在分析中3015b。然后将无活性株系从输出中过滤3013,以生成每个样品的活性株系和相应绝对数量/细胞计数的列表3016。以下代码段示出根据一个实施方案的用于此类处理的示例性方法:
针对每个样品(样品集合、子样品等)识别、检索和/或采集定性和定量元数据(例如,环境参数等)3017并将其存储在数据库(例如,319)中3018。根据应用/实现方式,可识别适当的元数据并查询数据库,以拉取所分析的每个样品的识别的和/或相关的元数据3019。然后将元数据的子集与活性株系及其相应绝对数量/细胞计数的集合或列表合并以通过样品矩阵创建大物种和元数据3020。
然后计算株系与元数据之间3021a以及株系之间3021b的最大信息系数(mic)。集中结果以创建所有关系及其对应mic得分的集合或列表3022。如果关系得分低于给定阈值3023,则该关系被认为/识别为是不相关的3023b。如果关系高于给定阈值3023,则该关系被认为/识别为是相关的3023a,且进一步接受网络分析3024。以下代码段示出根据一个实施方案的用于此类分析的示例性方法:
基于网络分析的输出,选择活性株系3025以用于制备含有所选株系的产物(例如,群集、聚集体和/或其他合成分组)。网络分析的输出还可用于告知对株系的选择以用于进一步的产物组成测试。
上面针对分析和确定讨论了阈值的使用。根据实现方式和应用,阈值可以:(1)凭经验确定(例如,基于分布水平,设定在去除指定的或显著部分的低水平读段的数量下的截止值);(2)是任何非零值;(3)基于百分比/百分位数;(4)仅是归一化的第二标记物(即,活性)读段大于归一化的第一标记物(细胞计数)读段的株系;(5)是活性与量或细胞计数之间的log2倍变化;(6)对于整个样品(和/或样品集合),归一化的第二标记物(活性)读段大于平均第二标记物(活性)读段;和/或除统计学阈值(即,显著性检验)之外的上述任何量级阈值。以下实例提供根据一个实施方案关于基于dna的第一标记物测量结果的基于rna的第二标记物测量结果的分布的阈值分割细节。
采集一只雄性cobb500的小肠内容物并根据本公开对其进行分析。简而言之,使用fac确定样品中细菌细胞的总数(例如,3004)。从固定的小肠样品分离总核酸(例如,3006)。制备dna(第一标记物)和cdna(第二标记物)测序文库(例如,3007),并将它们加载到illuminamiseq上。对来自每个文库的原始测序读段进行质量过滤、去重、聚类和定量(例如,3008)。将来自基于dna的和基于cdna的文库两者的定量株系列表与细胞计数数据整合,以建立样品内每种株系的绝对细胞数量(例如,3013)。虽然cdna不一定是株系量的直接量度(即,高度活性株系可具有相同rna分子的许多拷贝),但是在该实例中将基于cdna的文库与细胞计数数据整合以维持用于dna文库的相同归一化程序。
在分析之后,在基于cdna的文库中识别出702种株系(46种独特的),并且在基于dna的文库中识别出1140种株系。如果使用0作为活性阈值(即,保留任何非零值),则该样品内具有基于dna的第一标记物的株系中的57%也与基于cdna的第二标记物相关联。这些株系被识别为/认为是微生物群落的活性部分,并且仅这些株系继续进行随后的分析。如果使阈值更加严格并且仅第二标记物值超过第一标记物值的株系被认为是有活性的,则仅289个株系(25%)达到阈值。达到该阈值的株系对应于高于图3d中的dna(第一标记物)线的那些株系。
本公开包括多种方法,这些方法识别影响彼此以及一个或多个参数或元数据的多种活性微生物株系,并选择所识别的微生物以用在微生物群集中,该微生物群集包括在执行或影响共同的功能方面相联系的或可被描述为参与或引起可识别参数(诸如感兴趣的表型性状(例如,反刍动物的产奶量增加))或与可识别参数相关联的各个微生物物种或物种株系的微生物群落的选择子集。本公开还包括实施和/或有利于所述方法的各种系统和设备。
在一些实施方案中,该方法包括:获得共有至少一个共同特征(诸如样品地理位置、样品类型、样品来源、样品来源个体、样品目标动物、样品时间、品种、饮食、温度等)并且具有至少一个不同特征(诸如样品地理位置/时间位置、样品类型、样品来源、样品来源个体、样品目标动物、样品时间、品种、饮食、温度等,不同于共同特征)的至少两个样品。针对每个样品,检测一种或多种微生物类型的存在,确定每个样品中所述一种或多种微生物类型中的每种检出微生物类型的数量;以及测量每个样品中独特的第一标记物的数量及其量,每种独特的第一标记物是微生物株系的标记物。然后将每种微生物类型的数量和第一标记物的数量整合,以得到存在于每个样品中的每种微生物株系的绝对细胞计数;基于指定阈值测量每种微生物株系的至少一个独特的第二标记物,以确定每个样品中所述微生物株系的活性水平;根据确定的活性来过滤绝对细胞计数,以提供所述至少两个样品中的每一个的活性微生物株系及其相应绝对细胞计数的集合或列表;对所述至少两个样品中的每一个的活性微生物株系的经过滤的绝对细胞计数与彼此以及与所述至少两个样品中的每一个的至少一个测得元数据进行比较,并且基于预测的功能和/或化学性质将活性微生物株系归类为至少2组、至少3组、至少4组、至少5组、至少6组、至少7组、至少8组、至少9组、至少10组、至少15组、至少20组、至少25组、至少50组、至少75组或至少100组中的一种。例如,该比较可以是识别相应微生物株系之间和每种微生物株系与元数据之间和/或元数据与微生物株系之间的联系的网络分析。至少一种微生物可选自至少两组,并组合形成被配置为改变与所述至少一个元数据相对应的性质(例如,目标中的性质,诸如奶牛或奶牛群体的产奶量)的微生物群集。形成该群集可包括分离所述微生物株系或每种微生物株系,基于所述分析选择先前分离的微生物,和/或基于所述分析温育/生长特定的微生物株系,以及组合所述株系(包括基于应用,以特定的量/计数和/或比率和/或培养基/载体)以形成微生物群集。该群集可包括适当的培养基、载体和/或药学载体,其使得能够以该群集中的微生物可影响受者(例如,增加产奶量)的方式递送所述微生物。
测量独特的第一标记物的数量可包括测量每个样品中独特的基因组dna标记物的数量、测量每个样品中独特的rna标记物的数量、测量每个样品中独特的蛋白标记物的数量和/或测量每个样品中独特的代谢物标记物的数量(包括测量每个样品中独特的脂质标记物的数量和/或测量每个样品中独特的碳水化合物标记物的数量)。
在一些实施方案中,测量独特的第一标记物的数量及其量包括对来自每个样品的基因组dna进行高通量测序反应,和/或对来自每个样品的基因组dna进行元基因组测序。独特的第一标记物可包括mrna标记、sirna标记物和/或核糖体rna标记物中的至少一种。独特的第一标记物可另外或另选地包括σ因子、转录因子、核苷相关蛋白和/或代谢酶中的至少一种。
在一些实施方案中,测量所述至少一个独特的第二标记物包括测量每个样品中所述至少一个独特的第二标记物的表达水平,并且可包括对样品中的mrna进行基因表达分析。基因表达分析可包括测序反应、定量聚合酶链式反应(qpcr)、元转录组测序和/或转录组测序。
在一些实施方案中,测量所述至少一个独特的第二标记物的表达水平包括对每个样品或其部分进行质谱分析和/或对每个样品或其部分进行元核糖体图谱分析或核糖体图谱分析。所述一种或多种微生物类型包括细菌、古细菌、真菌、原生动物、植物、其他真核生物、病毒、类病毒或它们的组合,并且所述一种或多种微生物株系包括一种或多种细菌株系、古细菌株系、真菌株系、原生动物株系、植物株系、其他真核生物株、病毒株系、类病毒株系或它们的组合。所述一种或多种微生物株系可以是一种或多种真菌种或亚种,和/或所述一种或多种微生物株系可以是一种或多种细菌种或亚种。
在一些实施方案中,确定每个样品中的所述一种或多种微生物类型中的每一种的数量包括对每个样品或其部分进行测序、离心、光学显微镜检查、荧光显微镜检查、染色、质谱分析、微流体分析、定量聚合酶链式反应(qpcr)、凝胶电泳和/或流式细胞术。
独特的第一标记物可包括系统发育标记物,其包括5s核糖体亚基基因、16s核糖体亚基基因、23s核糖体亚基基因、5.8s核糖体亚基基因、18s核糖体亚基基因、28s核糖体亚基基因、细胞色素c氧化酶亚基基因、β-微管蛋白基因、延伸因子基因、rna聚合酶亚基基因、内部转录间隔区(its)或它们的组合。测量独特的标记物的数量及其量可包括对来自每个样品的基因组dna进行高通量测序反应,对基因组dna进行基因组测序,和/或对基因组dna进行扩增子测序。
在一些实施方案中,所述至少一个不同的特征包括:采集时间,在该采集时间采集所述至少两个样品中的每一个,使得第一样品的采集时间不同于第二样品的采集时间;采集位置(地理位置差异和/或各个样品目标/动物采集差异),在该采集位置处采集所述至少两个样品中的每一个,使得第一样品的采集位置不同于第二样品的采集位置。所述至少一个共同特征可包括样品来源类型,使得第一样品的样品来源类型与第二样品的样品来源类型相同。样品来源类型可以是动物类型、器官类型、土壤类型、水类型、沉积物类型、油类型、植物类型、农产品类型、根外土壤类型、根际土壤类型、植物部分类型等中的一种。在一些实施方案中,所述至少一个共同特征包括所述至少两个样品中的每一个均是胃肠样品,在一些实现方式中,所述胃肠样品可以是瘤胃样品。在一些实现方式中,相反,本文提供的共同/不同特征可以是某些样品之间的不同/共同特征。在一些实施方案中,所述至少一个共同特征包括动物样品来源类型,每个样品具有另外的共同特征,使得每个样品是组织样品、血液样品、牙齿样品、汗液样品、指甲样品、皮肤样品、毛发样品、粪便样品、尿液样品、精液样品、粘液样品、唾液样品、肌肉样品、脑样品或器官样品。
在一些实施方案中,上述方法可以进一步包括基于所述至少一个测得元数据从目标获得至少一个另外的样品,其中来自所述目标的所述至少一个另外的样品与所述至少两个样品共有至少一个共同的特征。然后,对于来自所述目标的所述至少一个另外的样品,检测一种或多种微生物类型的存在,确定所述一种或多种微生物类型中的每种检出微生物类型的数量,测量独特的第一标记物的数量及其量,整合每种微生物类型的数量和第一标记物的数量以得到存在的每种微生物株系的绝对细胞计数,测量每种微生物株系的至少一个独特的第二标记物以确定该微生物株系的活性水平,根据确定的活性来过滤绝对细胞计数以提供来自所述目标的所述至少一个另外的样品的活性微生物株系及其相应绝对细胞计数的集合或列表。在此类实施方案中,从所述至少两组选择所述至少一种微生物株系是基于来自所述目标的所述至少一个另外的样品的活性微生物株系及其相应绝对细胞计数的集合或列表,使得形成的群集被配置为改变目标的与所述至少一个元数据相对应的性质。例如,使用此实现方式,可从取自荷斯坦奶牛的样品和取自泽西(jersey)奶牛或水牛的目标样品中识别微生物群集,其中该分析识别来自初始样品和目标样品的相同或相似微生物株系之间的相同、基本上相似或相似的网络关系。
在一些实施方案中,对所述至少两个样品中的每一个的活性微生物株系的经过滤的绝对细胞计数与所述至少两个样品中的每一个的至少一个测得元数据或另外的活性微生物株系进行比较包括确定每个样品中所述一种或多种活性微生物株系与所述至少一个测得元数据或另外的活性微生物株系的共现。所述至少一个测得元数据可包括一个或多个参数,其中所述一个或多个参数是样品ph、样品温度、脂肪丰度、蛋白质丰度、碳水化合物丰度、矿物质丰度、维生素丰度、天然产物丰度、指定化合物丰度、样品来源的体重、样品来源的进食量、样品来源的重量增加、样品来源的饲料效率、一种或多种病原体的存在或不存在、样品来源的物理特征或测量结果、样品来源的生产特征中的至少一个或它们的组合。参数还可包括由样品来源产生的奶中的乳清蛋白丰度、酪蛋白丰度和/或脂肪丰度。
在一些实施方案中,确定每个样品中所述一种或多种活性微生物株系和所述至少一个测得元数据或另外的活性微生物株系的共现可以包括创建使用表示两个或更多个样品集合中元数据和微生物株系关联的链接、所述一种或多种活性微生物株系的绝对细胞计数以及所述一种或多种独特的第二标记物的测量结果填充的矩阵,以代表一个或多个异质微生物群落的一个或多个网络。确定所述一种或多种活性微生物株系和所述至少一个测得元数据或另外的活性微生物株系的共现并对活性微生物株系进行分类可包括网络分析和/或聚类分析,以测量网络内每种微生物株系的连通性,该网络表示共有共同特征、测得元数据和/或相关环境参数的至少两个样品的集合体。网络分析和/或聚类分析可以包括链接分析、模块性分析、稳健性测量、中介性测量、连通性测量、传递性测量、中心性测量或其组合。聚类分析可以包括构建连通性模型、子空间模型、分布模型、密度模型和/或质心模型。在一些实现方式中,网络分析可以包括通过链接挖掘和预测、集体分类、基于链接的聚类、关系相似性或其组合的网络预测建模。网络分析可以包括基于微分方程的群体建模和/或lotka-volterra建模。分析可以是启发式方法。在一些实施方案中,分析可以是louvain方法。网络分析可以包括用于在变量之间建立连通性的非参数方法,和/或用于建立连通性的变量之间的互信息和/或最大信息系数计算。
对于一些实施方案,一种基于两个或更多个样品集合形成活性微生物株系的群集的方法,该群集被配置为改变环境中的性质或特征,这两个或更多个样品集合在其之间共有至少一个共同的或相关的环境参数并且在其之间具有至少一个不同的环境参数,每个样品集合包含至少一个含有异质微生物群落的样品,其中所述一个或多个微生物株系是一种或多种生物类型的亚分类单元,该方法包括:检测每个样品中多种微生物类型的存在;确定每个样品中每种检出微生物类型的绝对细胞数量;以及测量每个样品中独特的第一标记物的数量及其量,其中独特的第一标记物是微生物株系的标记物。然后,在蛋白质或rna水平上,测量一种或多种独特的第二标记物的表达水平,其中独特的第二标记物是微生物株系的活性的标记物;基于超过指定阈值的所述一种或多种独特的第二标记物的表达水平,确定每个样品的检出微生物株系的活性;基于所述一种或多种第一标记物的量和所述一种或多种微生物株系是其中的亚分类单元的微生物类型的绝对细胞数量计算每个样品中每种检出活性微生物株系的绝对细胞计数,其中所述一种或多种活性微生物株系表达高于该指定阈值的第二独特的标记物。然后基于最大信息系数网络分析确定样品中活性微生物株系与至少一个环境参数的共现,以测量网络内每种微生物株系的连通性,其中该网络是所述至少两个或更多个样品集合与至少一个共同或相关环境参数的集合体。基于网络分析从所述一种或多种活性微生物株系选择多种活性微生物株系,并由选择的多种活性微生物株系形成活性微生物株系的群集,该活性微生物株系的群集被配置为当被引入到某环境中时选择性地改变该环境的性质或特征。对于一些实现方式,第一样品集合的至少一个共同或相关环境因素的至少一个测得指标不同于第二样品集合的至少一个共同或相关环境因素的测得指标。例如,如果样品/样品集合来自奶牛,则第一样品集合可以来自以草饲料饲喂的奶牛,而第二样品集合可以来自以玉米饲料饲喂的奶牛。虽然一个样品集合可以是单个样品,但是它也可以另选地是多个样品,并且样品集合内每个样品的至少一个共同或相关环境因素的测得指标基本相似(例如,一个组中的样品全部从以草饲料饲喂的畜群中采集),并且一个样品集合的平均测得指标与来自另一个样品集合的平均测得指标不同(第一样品集合来自以草饲料饲喂的畜群,第二样品集合来自以玉米饲料饲喂的畜群)。在分析中可能存在其他纳入考虑的不同和相似之处,诸如不同品种、不同饮食、不同表现、不同年龄、不同饲料添加剂、不同生长阶段、不同生理特征、不同健康状况、不同海拔、不同环境温度、不同季节、不同抗生素等。虽然在一些实施方案中,每个样品集合包括多个样品,并且第一样品集合从第一群体采集,第二样品集合从第二群体采集,但在另外的或另选的实施方案中,每个样品集合包括多个样品,并且第一样品集合在第一时间从第一群体采集,第二样品集合在不同于第一时间的第二时间从第一群体采集。例如,第一样品集合可以在畜群正在进食草料的第一时间从畜群采集,第二样品集合可以在第二时间(例如,2个月后)采集,其中该畜群在被采集第一样品后立即切换为用玉米饲料饲喂。在此类实施方案中,可以采集样品并对群体进行分析,和/或该分析可以包括对个体动物的特定参考,以便可以识别在该时间段内发生在个体动物上的变化,以及提供更精细的数据粒度级别。在一些实施方案中,一种基于两个或更多个样品(或样品集合,每个集合包括至少一个样品)形成活性微生物株系的合成群集的方法,该合成群集被配置为改变生物环境中的性质,这两个或更多个样品各自具有多个环境参数(和/或元数据),所述多个环境参数中的至少一个参数是在所述两个或更多个样品或样品集合之间相似的共同环境参数,并且至少一个环境参数是在所述两个或更多个样品或样品集合中的每一个之间不同的不同环境参数,每个样品集合包括至少一个包含获自生物样品来源的异质微生物群落的样品,所述活性微生物株系中的至少一种是一种或多种生物类型的亚分类单元,该方法包括:检测每个样品中多种微生物类型的存在;确定每个样品中每种检出微生物类型的绝对细胞数量;测量每个样品中独特的第一标记物的数量及其量,独特的第一标记物是微生物株系的标记物;测量一种或多种独特的第二标记物的水平(例如,表达水平),其中独特的第二标记物是微生物株系活性的标记物;基于超过指定阈值的所述一种或多种独特的第二标记物的水平(例如,表达水平)确定每个样品的每种检出微生物株系的活性,以识别一种或多种活性微生物株系;由所述一种或多种独特的第一标记物中的每一种的量(相对量、比例数量、比例量、百分比量等)和所述一种或多种微生物株系是其中的亚分类单元的相应和对应微生物类型的绝对细胞数量来计算每个样品中每种检出活性微生物株系的绝对细胞计数(其中该计算是数学函数,诸如乘法、点运算和/或其他运算),所述一种或多种活性微生物株系具有或表达高于该指定阈值的一种或多种独特的第二标记物;分析所述两个或更多个样品集合的活性微生物株系,该分析包括对所述两个或更多个样品集合中的每一个的每种活性微生物株系、所述至少一个共同环境参数以及所述至少一个不同环境参数进行非参数网络分析,该非参数网络分析包括确定每种活性微生物株系与每种其他活性微生物株系之间的最大信息系数得分以及确定每种活性微生物株系与所述至少一个不同环境参数之间的最大信息系数得分;基于所述非参数网络分析从所述一种或多种活性微生物株系中选择多种活性微生物株系;以及形成包含选择的多种活性微生物株系和微生物载体培养基的活性微生物株系的合成群集,所述活性微生物株系的合成群集被配置为当被引入到生物环境中时选择性地改变该生物环境的性质。根据实施方案或实现方式,所述至少两个样品或样品集合可包括3个样品、4个样品、5个样品、6个样品、7个样品、8个样品、9个样品、10个样品、11个样品、12个样品、13个样品、14个样品、15个样品、16个样品、17个样品、18个样品、19个样品、20个样品、21个样品、22个样品、23个样品、24个样品、25个样品、26个样品、27个样品、28个样品、29个样品、30个样品、35个样品、40个样品、45个样品、50个样品、60个样品、70个样品、80个样品、90个样品、100个样品、150个样品、200个样品、300个样品、400个样品、500个样品、600个样品等。根据实施方案或实现方式、样品的总数可以为小于5、5至10、10至15、15至20、20至30、30至40、40至50、50至60、60至70、70至80、80至90、90至100、小于100、大于100、小于200、大于200、小于300、大于300、小于400、大于400、小于500、大于500、小于1000、大于1000、小于5000、小于10000、小于20000等。
在一些实施方案中,至少一种共同或相关环境因素包括营养物信息、饮食信息、动物特征、感染信息、健康状况等。
所述至少一个测得指标可以包括样品ph、样品温度、脂肪丰度、蛋白质丰度、碳水化合物丰度、矿物质丰度、维生素丰度、天然产物丰度、特定化合物丰度、样品来源的体重、样品来源的进食量、样品来源的重量增加、样品来源的饲料效率、一种或多种病原体的存在或不存在、样品来源的物理特征或测量、样品来源的生产特征、由样品来源产生的奶中的乳清蛋白丰度、由样品来源产生的奶中的酪蛋白丰度和/或由样品来源产生的奶中的脂肪丰度或它们的组合。
根据实施方案,测量每个样品中独特的第一标记物的数量可以包括测量独特的基因组dna标记物的数量、测量独特的rna标记物的数量和/或测量独特的蛋白质标记物的数量。所述多种微生物类型可包括一种或多种细菌、古细菌、真菌、原生动物、植物、其他真核生物、病毒、类病毒或它们的组合。
在一些实施方案中,确定每个样品中每种微生物类型的绝对数量包括对样品或其部分进行测序、离心、光学显微镜检查、荧光显微镜检查、染色、质谱分析、微流体分析、定量聚合酶链式反应(qpcr)、凝胶电泳和/或流式细胞术。在一些实施方案中,一种或多种活性微生物株系是选自一种或多种细菌、古细菌、真菌、原生动物、植物、其他真核生物、病毒、类病毒或它们的组合的一种或多种微生物类型的亚分类单元。在一些实施方案中,一种或多种活性微生物株系是一种或多种细菌株系、古细菌株系、真菌株系、原生动物株系、植物株系、其他真核生物株系、病毒株系、类病毒株系或它们的组合。在一些实施方案中,一种或多种活性微生物株系是一种或多种细菌种或亚种。在一些实施方案中,一种或多种活性微生物株系是一种或多种真菌种或亚种。
在一些实施方案中,至少一个独特的第一标记物包括系统发育标记物,其包括5s核糖体亚基基因、16s核糖体亚基基因、23s核糖体亚基基因、5.8s核糖体亚基基因、18s核糖体亚基基因、28s核糖体亚基基因、细胞色素c氧化酶亚基基因、β-微管蛋白基因、延伸因子基因、rna聚合酶亚基基因、内部转录间隔区(its)或它们的组合。
在一些实施方案中,测量独特的第一标记物的数量及其量包括对来自每个样品的基因组dna进行高通量测序反应,和/或对来自每个样品的基因组dna进行元基因组测序。在一些实现方式中,独特的第一标记物可包括mrna标记物、sirna标记物和/或核糖体rna标记物。在一些实现方式中,独特的第一标记物可包括σ因子、转录因子、核苷相关蛋白、代谢酶或它们的组合。
在一些实施方案中,测量一种或多种独特的第二标记物的表达水平包括对每个样品中的mrna进行基因表达分析,并且在一些实现方式中,基因表达分析包括测序反应。在一些实现方式中,基因表达分析包括定量聚合酶链式反应(qpcr)、元转录组测序和/或转录组测序。
在一些实施方案中,测量一种或多种独特的第二标记物的表达水平包括对每个样品或其部分进行质谱分析、元核糖体图谱分析和/或核糖体图谱分析。
在一些实施方案中,测量所述至少一种或多种独特的第二标记物的表达水平包括对每个样品或其部分进行元核糖体图谱分析或核糖体图谱分析(ribo-seq)(参见例如ingolia,n.t.、s.ghaemmaghami、j.r.newman和j.s.weissman,2009,“genome-wideanalysisinvivooftranslationwithnucleotideresolutionusingribosomeprofiling”science324:218-223;ingolia,n.t.,2014,“ribosomeprofiling:newviewsoftranslation,fromsinglecodonstogenomescale”nat.rev.genet.15:205-213;每篇文献全文出于所有目的以引用方式并入)。ribo-seq是一种分子技术,可用于在基因组规模上确定体内蛋白质合成。该方法直接测量哪些转录物由于与mrna结合并相互作用而通过足迹核糖体(footprintingribosome)进行主动翻译。然后处理结合的mrna区域并进行高通量测序反应。已证明ribo-seq与定量蛋白质组学有很强的相关性(参见例如li、g.w.、d.burkhardt、c.gross和j.s.weissman.2014“quantifyingabsoluteproteinsynthesisratesrevealsprinciplesunderlyingallocationofcellularresources”cell157:624-635,该文献全文以引用方式明确地并入本文)。
本公开的一些实施方案利用元蛋白质组学来分析和/或评估酶促过程留下的蛋白质谱。可利用质谱法(ms)来测量蛋白质和蛋白质片段的质量。可以使用蛋白酶(例如lysc和胰蛋白酶)对蛋白质的混合物进行蛋白质水解消化,以生成蛋白质片段的复杂混合物。在一些实施方案中,需要在ms之前对样品进行多次分离和分级分离,包括液相色谱或气相色谱、一维凝胶电泳、二维凝胶电泳、等电点聚焦、强阳离子交换分离、反相色谱分离等。在其他实施方案中,使用直接进样而无需分离方法。结合质荷比,可以通过串联ms获得碎裂模式。在数据采集之后,开始进行数据预处理,并识别、选择、过滤峰,检索、归一化和换算缺失值。在处理之后,根据特定实施方案和/或实现方式/用途,应用许多不同的多变量和单变量统计分析方法。在统计分析之后,使用已知的蛋白质或标准品曲线的数据库来识别蛋白质。最后,对样品的相对量或绝对量进行定量。样品之间蛋白质浓度的差异可以与元数据一起用于后续分析。
在一些实施方案中,靶向蛋白质组学并测量特定蛋白质的量。也可以不靶向蛋白质组学,其中目标蛋白质的身份不是事先已知的。非靶向蛋白质组学可用于高通量测量以对样品蛋白质进行新的表征。在非靶向蛋白质组学中,可以使用许多不同的设置(例如ph、溶剂、柱化学和电离)以检测不同的蛋白质组合。靶向蛋白质组学侧重于已知蛋白质,其中对所讨论的蛋白质的浓度曲线进行绘制。根据这些定量曲线,可以对一种特定蛋白质或一组蛋白质进行定量测量。还可以使用数据库搜索算法(包括但不限于sequest、mascat、myrimatch、omssa或x!tandem)来识别蛋白质。
蛋白质组学可以与稳定同位素探针技术一起进行,以便促进和/或定量分析,以及确定微生物株系与活跃翻译的蛋白质谱之间的时间关系。蛋白质组学还可以与源自相同样品或类似样品的分离株系的全基因组、元基因组学、元转录组学或核糖体图谱分析数据集整合,以识别活跃产生该蛋白质的株系。当以这种方式利用时,蛋白质组学提供了各个株系活性的指示物。蛋白质组分析也可以独立于其他元级数据集进行,以提供鸟枪分析,从而深入了解微生物群落的功能。在一些情况下,利用结晶化来识别翻译后修饰。
根据实施方案,可以使用另外的或另选的方法来获得蛋白质组学数据集,包括基于图像的靶向代谢组学,例如基质辅助激光解吸电离(maldi)、纳米结构成像质谱(nims)、解吸电喷雾电离质谱(desi)、二次离子质谱(sims)或基质辅助激光解吸电离-飞行时间质谱(maldi-tofms)。
样品的来源类型可以是动物、土壤、空气、盐水、淡水、污水污泥、沉积物、油、植物、农产品、食品样品(特别是发酵食品和微生物食品,例如面包、奶酪、葡萄酒、啤酒、泡菜等)、根外土壤、根际土壤、植物部分、蔬菜、极端环境中的一种或它们的组合。在一些实现方式中,每个样品是消化道和/或瘤胃样品。在一些实现方式中,样品可以是组织样品、血液样品、牙齿样品、汗液样品、指甲样品、皮肤样品、毛发样品、粪便样品、尿液样品、精液样品、粘液样品、唾液样品、肌肉样品、脑样品、组织样品和/或器官样品。
根据实现方式,本公开的微生物群集可包括两种或更多种基本上纯的微生物或微生物株系、所需微生物/微生物株系的混合物,并且还可以包括可施用到目标(例如以用于恢复动物的微生物区)的任何另外的组分。根据本公开制备的微生物群集可与药剂一起施用以允许微生物在目标环境(例如,动物的胃肠道,其中该群集被配置成抵抗低ph并且在胃肠环境中生长)中存活。在一些实施方案中,微生物群集可包括增加一种或多种所需微生物或微生物株系的数量和/或活性的一种或多种药剂,所述株系在包括在所述群集中的微生物/株系中存在或不存在。此类药剂的非限制性实例包括低聚果糖(例如,果寡糖、菊糖、菊糖型果聚糖)、低聚半乳糖、氨基酸、醇及其混合物(参见ramirez-farias等人2008.br.j.nutr.4:1-10以及pool-zobel和sauer2007.j.nutr.137:2580-2584和增刊,其中每篇文献全文出于所有目的以引用的方式并入本文)。
通过本公开的方法识别的微生物株系可在包括在群集中之前进行培养/生长。培养基可用于这种生长,并且可包括适于支持微生物生长的任何培养基,以非限制性实例的方式,其包括天然的或人工的,包括胃泌素补充琼脂、lb培养基、血清和/或组织培养凝胶。应当理解,培养基可单独使用或与一种或多种其他培养基组合使用。它还可在添加或不添加外源营养物的情况下使用。培养基可被改进或富含另外的化合物或组分,例如可有助于微生物和/或其株系的特定组的相互作用和/或选择的组分。例如,可存在抗生素(诸如青霉素)或消毒剂(例如,季铵盐和氧化剂)和/或可修改物理条件(诸如盐度、营养物(例如有机和无机矿物质(诸如磷、含氮盐、氨、钾和微量营养物,诸如钴和镁))、ph和/或温度)。
如上所讨论,系统和设备可根据本公开配置,并且在一些实施方案中可包括处理器和存储器,该存储器存储可由处理器读取/发布的指令来执行所述方法。在一个实施方案中,系统和/或设备被配置来执行所述方法。还公开了所述方法的处理器实现方式,如参考图3a所讨论的。例如,处理器实施的方法可包括:从共有至少一个共同特征并且具有至少一个不同特征的至少两个样品接收样品数据;针对每个样品,确定每个样品中的一种或多种微生物类型的存在;确定每个样品中的所述一种或多种微生物类型中的每种检出微生物类型的细胞数量;确定每个样品中独特的第一标记物的数量及其量,每个独特的第一标记物是微生物株系的标记物;通过一个或多个处理器整合每种微生物类型的数量和第一标记物的数量,以得到存在于每个样品中的每种微生物株系的绝对细胞计数;基于超过指定阈值的每种微生物株系的至少一个独特的第二标记物的测量值,确定每个样品中每种微生物株系的活性水平,如果微生物株系的至少一个独特的第二标记物的测量值超过对应阈值,则该株系被识别为有活性的;根据确定的活性来过滤每种微生物株系的所述绝对细胞计数,以提供所述至少两个样品中的每一个的活性微生物株系及其相应绝对细胞计数的列表;通过一个或多个处理器对所述至少两个样品中的每一个的活性微生物株系的经过滤的绝对计数与所述至少两个样品中的每一个的至少一个测得元数据或另外的活性微生物株系进行分析,并且基于功能、预测功能和/或化学性质对活性微生物株系进行分类;基于所述分类识别多种活性微生物株系;以及输出所识别的多种活性微生物株系以用于聚集被配置成当应用于目标时改变目标的与至少一个测得元数据相对应的性质的活性微生物群集。在一些实施方案中,输出可用于合成的和/或转基因的微生物和微生物株系的生成、合成、评估和/或测试。一些实施方案可包括存储用于执行所述方法和/或有利于所述方法的执行的指令的处理器可读非暂态计算机可读介质。在一些实施方案中,根据本公开的分析和筛选方法、设备和系统可用于识别产生问题的微生物和株系,诸如病原体,如在以下实施例4中所讨论的。在此类情形下,已知的症状元数据(诸如病变得分)将用于样品的网络分析。
预期本文所述的系统和方法可通过软件(存储在存储器中和/或在硬件上执行)、硬件或其组合来执行。硬件部件和/或模块可包括例如通用处理器、现场可编程门阵列(fpga)和/或专用集成电路(asic)。软件部件和/或模块(在硬件上执行)可以多种软件语言(例如,计算机代码)表达,包括unix实用程序、c、c++、javatm、javascript(例如,ecmascript6)、ruby、sql、
本文所述的一些实施方案涉及具有非暂态计算机可读介质(也可称为非暂态处理器可读介质或存储器)的装置,该介质上具有用于执行各种计算机实施的操作的指令或计算机代码。计算机可读介质(或处理器可读介质)在某种意义上是非暂态的,即它本身不包括暂态传播信号(例如,在传输介质诸如空间或电缆上运载信息的传播电磁波)。介质和计算机代码(也可以称为代码)可以是为一个或多个特定目的而设计和构造的那些。非暂态计算机可读介质的实例包括但不限于:磁存储介质,诸如硬盘、软盘和磁带;光存储介质,诸如压缩盘/数字视频光盘(cd/dvd)、光盘只读存储器(cd-rom)和全息装置;磁光存储介质,诸如光盘;载波信号处理部件和/或模块;以及专门配置为存储和执行程序代码的硬件装置,诸如专用集成电路(asic)、可编程逻辑装置(pld)、只读存储器(rom)和随机存取存储器(ram)装置。本文描述的其他实施方案涉及计算机程序产品,其可以包括例如本文所讨论的指令和/或计算机代码。
虽然上文已经描述了图3a的各种实施方案,但是应当理解,它们仅以实例的方式而不是限制的方式来呈现。在上述方法和步骤指示某些事件以某一顺序发生的情况下,可以更改某些步骤的顺序。另外,某些步骤可以在可能的情况下在并行过程中同时执行,以及如上所述按顺序执行。尽管已经将各种实施方案描述为具有特定的特征和/或组成部分的组合,但是其他实施方案也可以具有来自本文描述的任何实施方案的任何特征和/或组成部分的任何组合或子组合。此外,尽管将各种实施方案描述为具有与特定计算装置相关联的特定实体,但是在其他实施方案中,不同的实体可以与其他和/或不同的计算装置相关联。
实验数据和实施例
通过参考以下实验数据和实施例进一步说明本发明。然而,应当注意,这些实验数据和实施例(如上述实施方案)是说明性的,不应被解释为以任何方式限制本公开的范围。
实施例1
参考图2g提供的步骤。
2000:将来自牛瘤胃样品的细胞剪切掉基质。这可以通过以下方式来完成:将样品通过超声处理或涡旋而充分共混或混合,然后进行差速离心以从细胞中除去基质。离心可包括使用nycodenz或percoll的梯度离心步骤。
2001:使用针对特定生物类型的荧光染料对生物进行染色。将流式细胞术用于基于染色性质和大小区分不同的群体。
2002:样品中生物的绝对数量通过例如流式细胞术确定。该步骤给出关于给定体积中有多少生物类型(诸如细菌、古细菌、真菌、病毒或原生生物)的信息。
2003:获得牛瘤胃样品,并通过珠磨直接裂解附着到基质上的细胞。纯化总核酸。用rna酶处理纯化的总核酸以获得纯化的基因组dna(gdna)。使用标准dna文库构建方法(例如,片段化-连接-扩增-测序方法、同时进行片段化和连接文库(可能需要扩增)的基于转座酶的方法、自由扩增方法(例如,片段化-连接-测序)等)从样品的gdna部分构建全元基因组ngs测序文库。合并样品并使用illuminahiseq对合并的样品进行多重测序。
2004:将附着到基质上的来自牛瘤胃样品的细胞通过珠磨直接裂解。使用基于柱的方法纯化总核酸。用dna酶处理纯化的总核酸以获得纯化的rna。从总rna中去除核糖体rna以产生纯化的mrna。使用标准dutp方法从样品的mrna部分构建全转录组ngs测序文库(例如,参见astrand-specificlibrarypreparationprotocolforrnasequencing;doi:10.1016/b978-0-12-385118-5.00005-0,其全文出于所有目的据此以引用方式并入)。合并样品并使用illuminahiseq对合并的样品进行多重测序。
2005:通过删除低质量碱基对和截短的读段以及宿主污染来处理测序输出(fastq文件)。使用定制的生物信息学管道来组装和分析基于dna的数据集。将得到的元基因组学分析分箱以识别各个株系的基因组,并对每个基因组进行注释以确定每种株系的代谢能力。然后将元转录组学读段映射到各个基因组以识别活性株系。
2006:使用在先前步骤(2005)中获得的株系身份数据,确定映射到每种株系的元基因组学读段的数量并将其表示为总读段数的百分比。将该百分比乘以细胞计数(2002)以计算样品和给定体积中每种生物类型的绝对细胞计数。使用基于mrna的数据集中存在的映射序列以及适当的阈值,在绝对细胞计数数据集内识别活性株系。从分析中删除不符合阈值的株系。
2007:重复2003-2006以建立代表多个牛瘤胃内微生物群体动态的时间进程。编译时间数据并以量或丰度矩阵存储每个样品的每种活性生物株系的细胞数和元数据。使用通过量数据加权的规则挖掘方法,将量矩阵用于识别特定时间点样品中的活性株系之间的关联。应用过滤器以删除无关紧要的规则。
2008:计算活性株系随时间的细胞数变化,记录变化的方向性(即,负值代表减少,正值代表增加)。将矩阵表示为网络,其中生物株系代表节点,量加权规则代表边缘。利用马尔可夫链和随机游走来确定节点之间的连通性并限定聚类。使用元数据过滤聚类以识别与期望的元数据(环境参数)相关联的聚类。通过整合随时间的细胞数变化和存在于目标聚类中的株系来对目标生物株系进行排名,其中最大细胞数变化排名最高。
实施例2
实验设计及材料和方法
目的:确定影响奶牛乳脂产量的瘤胃微生物群落成分。
动物:将八头瘤胃插管的泌乳荷斯坦奶牛安置在单独的拴养牛栏中以用于实验。每天喂食奶牛两次,每天挤奶两次,并持续为其提供淡水。在一头奶牛(奶牛1)由于在实验之前因流产引起的并发症而导致第一次饮食乳脂降低(milkfatdepression)之后,将其从研究中去除。
实验设计和处理:实验采用交叉设计,分为2组,1个实验周期。实验周期持续38天:10天的协变量期/洗脱期,28天的数据采集和取样期。数据采集期包括10天的饮食乳脂降低(mfd)和18天的恢复。在第一个实验周期后,所有奶牛在第2个周期开始前经历10天的洗脱期。
饮食mfd用低纤维(29%ndf)的总混合日粮(tmr)诱导,该总混合日粮具有高淀粉降解性(70%可降解)和高多不饱和脂肪酸水平(pufa,3.7%)。恢复阶段包括两种淀粉可降解性不同的饮食。给四头奶牛随机分配高纤维(37%ndf)、低pufa(2.6%)和高淀粉可降解性(70%可降解)的恢复饮食。其余的四头奶牛饲喂高纤维(37%ndf)、低pufa(2.6%)但低淀粉降解率(35%)的恢复饮食。
在10天协变量和10天洗脱期期间,给奶牛饲喂高纤维、低pufa和低淀粉降解性的饮食。
样品和测量:在整个协变量、洗脱和样品采集期中,每天测量每只动物的产奶量、干物质摄入量和饲料效率。测量tmr样品的营养组成。在采集期期间,每3天采集并分析奶样。分析样品的乳组分浓度(乳脂、乳蛋白、乳糖、乳尿素氮、体细胞计数和固体)和脂肪酸组成。
采集瘤胃样品并在采集期期间每3天分析微生物群落的组成和活性。在饮食mfd的第0天、第7天和第10天,在饲喂后0、2、4、6、8、10、12、14、16、18、20和22小时对瘤胃进行集中取样。类似地,在恢复期期间的第16天和第28天,在饲喂后0、2、4、6、8、10、12、14、16、18、20和22小时对瘤胃进行集中取样。分析瘤胃内容物的ph、乙酸盐浓度、丁酸盐浓度、丙酸盐浓度、异酸浓度以及长链和cla异构体浓度。
瘤胃样品制备和测序:采集后,将瘤胃样品在摇摆式离心机(swingbucketcentrifuge)中以4,000rpm在4℃下离心20分钟。倾析出上清液,将每个瘤胃内容物样品的等分试样(1-2mg)加入预装有0.1mm玻璃珠的无菌1.7ml管中。采集第二份等分试样并储存在空的无菌1.7ml管中以用于细胞计数。
将具有玻璃珠的瘤胃样品(第1个等分试样)用珠磨法均质化以裂解微生物。从每个样品中提取并纯化dna和rna,并准备用于在illuminahiseq上测序。使用双端化学对样品测序,其中在文库的每一端测序250个碱基对。将空管中的瘤胃样品(第2个等分试样)染色并使其通过流式细胞仪以对每个样品中的每种微生物类型的细胞数量进行定量。
测序读段处理和数据分析:对来自元基因组学样品的测序读段进行质量修整和处理,以识别瘤胃中存在的细菌种类的全基因组。将计数数据集和元转录组学数据集与测序读段整合以确定瘤胃微生物群落内活性微生物物种的绝对细胞数量。使用互信息,将随着时间推移的奶牛的产奶特性(包括所产奶的磅数)与实验过程中每个样品内的活性微生物的分布相关联。计算所产生的乳脂的磅数与每种活性微生物的绝对细胞计数之间的最大信息系数(mic)得分。根据mic得分对微生物排名,并选择mic得分最高的微生物作为与所产奶的磅数最相关的目标物种。
通过从测序分析中省略适当的数据集来测试用例以确定计数数据、活性数据以及计数和活性对最终输出结果的影响。为了评估使用线性相关而不是mic对目标选择的影响,还计算了与所有微生物的相对丰度和活性微生物的绝对细胞计数相比所产生的乳脂的磅数的皮尔逊系数。
结果和讨论
相对丰度与绝对细胞计数
针对包括细胞计数数据(绝对细胞计数,表2)的数据集以及不包括细胞计数数据(相对丰度,表1)的数据集,基于mic得分识别了前15个目标物种。在该分析中未使用活性数据以隔离细胞计数数据对最终目标选择的影响。最终发现,两个数据集之间的前8个目标是相同的。在剩余的7个中,5个株系以不同的顺序存在于两个列表中。尽管这5个株系的排名存在差异,但每个株系的计算出的mic得分在两个列表之间是相同的。存在于绝对细胞计数列表但不存在于相对丰度列表上的两个株系ascus_111和ascus_288分别在相对丰度列表上排第91位和第16位。存在于相对丰度列表但不存在于绝对细胞计数列表上的两个株系ascus_102和ascus_252分别在绝对细胞计数列表上排第50位和第19位。这4个株系在每个列表上确实具有不同的mic得分,因此解释了它们的排名变化以及随后对列表中其他株系的影响。
表1:未经活性过滤使用相对丰度得到的前15个目标株系
表2:未经活性过滤使用绝对细胞计数得到的前15个目标株系
细胞计数数据的整合并不总是影响分配给每个株系的最终mic得分。这可能归因于这样的事实:虽然微生物群体确实在38天的实验过程中每天都在瘤胃内变动,但其变动范围总是在每毫升107-108个细胞内。群体数量的大幅变动无疑会对最终mic得分产生更广泛的影响。
非活性物种与活性物种
为了基于活性数据评估过滤株系的影响,从具有(表3)和不具有(表1)活性数据的利用相对丰度的数据集以及具有(表4)和不具有(表2)活性数据的利用绝对细胞计数的数据集中识别目标物种。
对于相对丰度的情况,在应用活性数据之前,ascus_126、ascus_1366、ascus_1780、ascus_299、ascus_1139、ascus_127、ascus_341和ascus_252被认为是目标株系。在整合活性数据后,这八个株系(最初的前15个目标株系中的53%)的排名降至15以后。对于绝对细胞计数的情况,观察到类似的趋势。在整合活性数据集后,ascus_126、ascus_1366、ascus_1780、ascus_299、ascus_1139、ascus_127和ascus_341(最初的前15个目标株系中的46%)的排名降至15以后。
活性数据集对目标排名和选择的影响比细胞计数数据集严重得多。将这些数据集整合在一起时,如果发现样品无活性,则基本上将其更改为“0”,并且不将其视为分析的一部分。因此,样品内点的分布在整合后会有巨大改变或偏斜,这反过来又会极大地影响最终mic得分,从而影响目标微生物的排序。
表3:经过活性过滤使用相对丰度得到的前15个目标株系
表4:经过活性过滤使用绝对细胞计数得到的前15个目标株系
相对丰度和无活性与绝对细胞计数和有活性
最终,此处定义的方法利用细胞计数数据和活性数据两者来识别与相关元数据特征高度相关的微生物。在使用两种方法选择的前15个目标株系中(表4、表1),两个列表上均有的株系仅发现7个。八个株系(53%)是绝对细胞计数和活性列表所特有的。两个列表上的前3个目标的株系和排名都匹配。但是,三个中的两个在两个列表上没有相同的mic得分,表明它们受到活性数据集整合的影响,但这种影响不足以扰乱它们的排序。
线性相关与非参数方法
计算所产生的乳脂的磅数与每个样品中的活性微生物的绝对细胞数量之间的皮尔逊系数和mic得分(表5)。根据mic(表5a)或皮尔逊系数(表5b)对株系进行排名,以选择与乳脂产量最相关的目标株系。在每种情况下均报告mic得分和皮尔逊系数。在两个列表上均发现的株系有六个,这意味着使用mic方法识别了九个(60%)独特株系。列表之间的株系排序不匹配—每种方法识别的前3个目标株系也是独特的。
与皮尔逊系数一样,在0到1的范围内报告mic得分,1表明两个变量之间的关系非常紧密。此处,前15个目标的mic得分在0.97到0.74之间。然而,相关性测试用例的皮尔逊系数在0.53到0.45之间,远低于互信息测试用例。这种不一致可能是由于每种分析方法固有的差异导致的。虽然相关性是衡量线周围的点的分散性的线性估计,但互信息利用概率分布并测量两个分布之间的相似性。在实验过程中,所产生的乳脂的磅数非线性地变化(图4)。该特定函数可以通过互信息而不是相关性来更好地表示和逼近。为了研究这一点,绘制了使用相关性和互信息分别识别的排名最高的目标株系ascus_713(图5)和ascus_7(图6),以确定每种方法预测株系与乳脂之间的关系时表现如何。如果两个变量表现出强相关性,则当将它们相对于彼此绘制时,它们由点几乎不分散开的一条线表示。在图5中,ascus_713与乳脂弱相关,如由点的广泛分布所示。互信息再次衡量两种点分布的相似程度。当绘制ascus_7与乳脂的关系时(图6),很明显两种点分布非常相似。
作为整体的本发明的方法与传统方法
分析微生物群落的传统方法依赖于使用相对丰度数据而不结合活性信息,并且最终以微生物物种与元数据的简单相关性给出结论(参见例如美国专利no.9,206,680,其全文出于所有目的以引用方式并入本文)。在此,我们展示了每个数据集的并入如何逐步影响最终的目标列表。当整体应用时,与常规方法相比,本文所述的方法选择出完全不同的目标集合(表5a和表5c)。绘制使用常规方法选择的排名最高的目标株系ascus_3038与乳脂的关系图,以显示相关性的强度(图7)。与前面的实施例一样,ascus_3038也表现出与乳脂的弱相关性。
表5:使用互信息或相关性得到的前15个目标株系
表5a.经过活性过滤使用绝对细胞计数的mic
表5b.经过活性过滤使用绝对细胞计数的相关性
表5c.未经活性过滤使用相对丰度的相关性
实施例3
增加奶牛的总乳脂、乳蛋白和能量校正乳(ecm)
实施例3示出了一种具体实现方式,目的是增加由泌乳反刍动物产生的乳脂和乳蛋白的总量,以及计算的ecm。如本文所用,ecm表示基于乳体积、乳脂和乳蛋白计算的乳中能量的量。ecm将奶组分调整为3.5%的脂肪和3.2%的蛋白质,从而均衡动物表现并允许在各个动物和畜群水平上比较随时间的产量。与本公开相关的用于计算ecm的等式是:
ecm=(0.327x奶磅数)+(12.95x脂肪磅数)+(7.2x蛋白质磅数)
应用本文提出的方法,利用所公开的方法识别活性相关的微生物/微生物株系并从中生成微生物群集,证明了由泌乳反刍动物产生的乳脂和乳蛋白的总量增加。这些增加是在不需要进一步添加激素的情况下实现的。
在该实施例中,向泌乳中期的荷斯坦奶牛施用根据上述公开内容识别和产生的包含两种分离微生物ascusb_x和ascusf_y的微生物群集五周的时间。将奶牛随机分成2组,每组8头,其中一组是接受没有微生物群集的缓冲液的对照组。第二组(即实验组)每天施用一次包含ascusb_x和ascusf_y的微生物群集,持续五周。每头奶牛圈养在单独的围栏中,且自由获取饲料和水。饮食是高产乳饮食。对奶牛进行随意饲喂并在当天结束时对饲料称重,且将前一天的拒食饲料称重并丢弃。使用得自salterbrecknell(fairmont,mn)的ps-2000称进行称重。
给奶牛插管,使插管延伸到奶牛的瘤胃中。在施用对照剂量或实验剂量之前,向奶牛进一步提供至少10天的插管后恢复期。
向对照组的施用由20ml中性缓冲盐水组成,而向实验组的施用由悬浮在20ml中性缓冲盐水中的约109个细胞组成。对照组每天接受一次20ml盐水,而实验组接受还包含所述微生物群集的109个微生物细胞的20ml盐水。
在第0、7、14、21和35天对每头奶牛的瘤胃进行取样,其中第0天是微生物施用前一天。注意,实验和对照施用在当天对瘤胃取样后进行。从第0天开始,每日对瘤胃取样,将得自hannainstruments(woonsocket,ri)的ph计插入采集的瘤胃液中进行记录。瘤胃取样包括通过插管从瘤胃的中心、背部、腹部、前部和后部取样的颗粒和流体,并将五个样品全部合并到含有1.5ml终止液(95%乙醇,5%苯酚)的15ml锥形瓶中。还在每个取样日采集粪便样品,其中使用触诊套管从直肠采集粪便。在每次取样时对奶牛称重。
将粪便样品置于2盎司小瓶中,冷冻保存,并分析以确定表观中性洗涤纤维(ndf)消化率、表观淀粉消化率和表观蛋白质消化率的值。瘤胃取样包括对瘤胃的流体和颗粒部分两者进行取样,将其每一者储存在15ml锥形管中。用10%终止液(5%苯酚/95%乙醇混合物)固定细胞,将其保持在4℃下,置于冰上运送至ascusbiosciences(sandiego,california)。
每天测量两次产奶量,一次在早上,一次在晚上。每天测量两次奶组成(%脂肪和%蛋白质等),一次在早上,一次在晚上。在tularedairyherdimprovementassociation(dhia)(tulare,california),用近红外光谱进一步分析奶样的蛋白质脂肪、固体、奶尿素氮(mun)和体细胞计数(scc)。每天测定一次各奶牛的进食量和瘤胃ph。
在适应期的最后一天采集总混合日粮(tmr)的样品,然后连续每周采集一次。采用四分法进行取样,其中将样品储存在真空密封袋中,将其运送到cumberlandvalleyanalyticalservices(hagerstown,md)并用nir1软件包分析。缓冲液和/或微生物生物群集的最后一天施用是在第35天,但是其他所有测量和取样继续如前所述进行直至第46天。
图8a表明与仅施用缓冲液的奶牛相比,基于所公开的方法接受微生物群集的奶牛表现出乳脂平均产量增加20.9%。图8b表明与仅施用缓冲液的奶牛相比,施用微生物群集的奶牛表现出乳蛋白平均产量增加20.7%。图8c表明施用微生物群集的奶牛表现出能量校正乳的平均产量增加19.4%。在停止施用微生物群集后,图8a-c中所见的增加变得不太明显,如垂直线与数据点相交所示。
实施例4
检测作为肉鸡病变形成的病原体的产气荚膜梭菌(clostridiumperfringen)
用不同水平的产气荚膜梭菌挑战160只雄性cobb500(表6a)。将它们饲养21天后杀死,并对病变进行评分,以量化坏死性肠炎的进展和产气荚膜梭菌的影响。
表6a
实验设计
将鸡圈养在木地板围栏中的环境受控设施内(~4'×4'减去2.25平方英尺的饲槽空间),其提供[~0.69平方英尺/鸡]的地面空间和鸡密度、温度、照明、饲槽和水。将鸡放入铺有适当厚度木刨花的干净围栏中,为鸡提供舒适的环境。如果在研究过程中木刨花变得太潮湿而不能为测试鸡创造舒适条件,则向围栏中添加另外的木刨花。通过白炽灯照明,并如下使用商业照明程序。
表6b
对于所有处理组而言,鸡的环境条件(即鸡密度、温度、照明、饲槽和储水空间)均相似。为了防止鸡迁移和细菌在围栏间扩散,每个围栏都有一个在围栏之间的固体(塑料)分隔器,高度约24英寸。
疫苗接种和治疗药物:
在孵化场为鸡接种马立克氏病(mareks)疫苗。在收到鸡时(研究第0天),通过喷雾施用为鸡接种新城病(newcastle)和传染性支气管炎疫苗。疫苗制造商、批号和失效期的文档与最终报告一起提供。
水:
在整个研究期间,通过每个围栏一个plasson饮水器随意提供水。饮水器按需要每天检查两次并清理以保证对鸡的清洁且恒定的水供应。
饲料:
在整个研究期间,每个围栏通过一根悬挂的直径为~17英寸的管式饲槽随意提供饲料。在大约前4天,将鸡饲槽托盘放在每个围栏中。根据实验设计,在收到鸡时(第0天)使鸡接受它们相应的处理饮食。称重并记录从第0天到研究结束向围栏中添加和从中去除的饲料。
每日观察:
每天至少观察两次测试设施、围栏和鸡以了解一般鸡群状况、照明、水、饲料、通风和意外事件。如果在每日两次观察中的任何一次观察到异常情况或异常行为,则对其进行记载,并将该记载包括在研究记录中。每天记录一次测试设施的最低-最高温度。
围栏卡片:
每个围栏附有2张卡片。一张卡片标识围栏编号,第二张卡片表示处理编号。
动物处理:
将动物保持在理想的宜居条件下。以减少伤害和不必要压力的方式处理动物。严格执行人道措施。
兽医护理、干预和安乐死:
根据现场sop,将发展与测试程序不相关的临床上显著的并发疾病的鸡按照研究者或指定人员的判断从研究中去除并实施安乐死。另外,在现场兽医或合格技术人员授权后,也对濒死的或受伤的鸡实施安乐死。记载任何退出的原因。如果动物死亡,或因为人道原因而去除并实施安乐死,则将其记录在围栏的死亡表上,并实施尸检且记录以记载去除的原因。
如果研究者认为安乐死是必要的,则通过颈脱位法对动物实施安乐死。
死亡和淘汰:
从研究第0天开始,对发现已死亡或被去除并处死的任何鸡进行称重和尸检。将不能取得饲料或水的淘汰鸡处死、称重和记载。在围栏死亡记录上记录重量和可能的死因以及尸检发现。
体重和进食量:
在大约第14天和第21天,按照围栏和个体,对鸡称重。在研究第14天和第21天对每个围栏中剩余的饲料进行称重并记录。计算第14-21天期间的进食量。
增重和饲料转化率:
对每个称重日按照围栏和个体得出的平均鸡重量进行汇总。在研究第21天(即,第0-21天)使用围栏的总饲料消耗量除以存活鸡的总重量来计算平均饲料转化率。使用围栏中的总饲料消耗量除以存活鸡的总重量和死亡或从该围栏去除的鸡的重量来计算调整饲料转化率。
产气荚膜梭菌挑战
施用方法:
通过饲料施用本研究中的产气荚膜梭菌(cl-15,α型,α和β2毒素)培养物。使用来自每个围栏的饲槽的饲料与培养物混合。在将培养物放置在围栏中之前,将处理饲料从鸡中去除,保持约4-8小时。对于每个围栏的鸡,将浓度为约2.0–9.0x108cfu/ml的基于研究设计的固定量的肉汤培养物与饲槽托盘中的固定量的饲料(~25g/鸡)混合,并且所有挑战的围栏均以相同的方式处理。大多数培养物-饲料在1-2小时内被消耗。为了使所有处理中的鸡均以相似的方式处理,未挑战的组也在与挑战组相同的时间段内去除饲料。
梭菌挑战:
将产气荚膜梭菌培养物(cl-15)在~37℃下在含有淀粉的液体硫乙醇酸盐培养基中生长~5小时。cl-15是来自科罗拉多州的肉鸡爆发的产气荚膜梭菌的野外株系(fieldstrain)。每天制备并使用新鲜肉汤培养物。对于每个围栏的鸡,将固定量的过夜肉汤培养物与饲槽托盘中的固定量的处理饲料混合(参见施用)。将饲料的量、培养接种物的体积和定量以及给药天数记载在最终报告中,并且所有围栏将以相同的方式处理。鸡接受产气荚膜梭菌培养物一天(研究第17天)。
采集的数据:
-用于根据本公开使用ascus平台方法分析的肠内容物。
-在大约第14天和第21天按照围栏和个体得到的鸡重量以及按围栏得到的饲料效率。
-从第0天至研究结束向每个围栏添加的和从中去除的饲料量。
-死亡:第0天至研究结束的性别、重量和可能的死因。
-去除的鸡:第0天至研究结束的淘汰原因、性别和重量。
-设施和鸡的每日观察、每日设施温度。
-大约第21天的5只鸡/围栏的病变得分
病变评分:
在上一次产气荚膜梭菌培养物施用后四天,从每个围栏随机选择五只鸡(抓到哪只是哪只)、处死并且针对坏死性肠炎对肠病变进行评分。病变评分如下:
-0=正常:没有ne病变,小肠具有正常弹性(打开后卷回正常位置)
-1=轻度:小肠壁薄而松弛(打开时保持平坦并且打开后不卷回正常位置);过量粘液覆盖粘膜
-2=中度:可观察到的小肠壁发红和肿胀;肠膜的轻微溃疡和坏死;过量粘液
-3=重度:小肠膜的大面积坏死和溃疡;大量出血;粘膜上存在纤维蛋白层和坏死碎片(土耳其式毛巾外观)
-4=死亡或濒死:可能在24小时内死亡并且具有2或更大ne病变得分的鸡
结果
使用以上所公开的方法(例如,如参考图1a、图1b和图2以及在整个说明书中所讨论的)以及常规相关方法(如上文所讨论的)分析结果。针对每只鸡的小肠内容物确定株系水平上的微生物丰度和活性,并参考以下两种不同的鸡特征分析这些特性:个体病变得分和围栏的平均病变得分。
在个体病变得分分析中使用37只鸡—虽然对40只鸡进行评分,但是仅37只具有足够的用于分析的肠物质。将相同的测序读段和相同的测序分析管道用于本公开的ascus方法和常规方法两者。然而,ascus方法也整合活性信息以及每个样品的细胞计数信息,如早前详述的。
使用ascus互信息方法来对37只肉鸡的活性株系的量与个体病变得分之间的关系进行评分。针对常规方法计算37只肉鸡的株系与个体病变得分之间的皮尔逊相关性。通过针对从样品池识别的生物列表进行的全局比对搜索来确认致病株系产气荚膜梭菌。然后根据每种分析方法的输出确定该特定株系的排名。ascus方法将实验中施用的产气荚膜梭菌确定为与个体病变得分关联的第一位株系。常规方法将该株系确定为与个体病变得分关联的第26位株系。
在平均病变得分分析中使用102只鸡。如在先前用例中的,将相同的测序读段和相同的测序分析管道用于ascus方法和常规方法两者。同样,ascus方法也整合活性信息以及每个样品的细胞计数信息。
使用ascus互信息方法来对活性株系的量与每个围栏的平均病变得分之间的关系进行评分。针对常规方法计算株系与每个围栏的平均病变得分之间的皮尔逊相关性。通过针对从样品池识别的生物列表进行的全局比对搜索来确认致病株系产气荚膜梭菌。然后根据每种分析方法的输出确定该特定株系的排名。ascus方法将实验中施用的产气荚膜梭菌识别为与围栏的平均病变得分关联的第4位株系。常规方法将产气荚膜梭菌识别为与围栏的平均病变得分关联的第15位株系。由于产气荚膜梭菌感染的可变水平被大量/平均测量所掩盖,因此围栏的平均病变得分是不如个体病变得分准确的测量。当将个体病变得分分析与平均围栏病变得分分析进行比较时,预期排名将下降。
采集的元数据提供如下
表7
实施例5
使用与基于相关性的方法相比的基于互信息的方法检测复杂微生物群落的关系的能力
在乳脂降低发作期间通过插管从三头泌乳中期的荷斯坦奶牛采集一系列瘤胃样品。在第0天、第7天、第10天、第16天和第28天在早上4点采集瘤胃样品。根据从瘤胃内容物纯化的dna制备测序文库并且进行测序。
使用原始测序读段识别存在于样品池中的所有微生物株系—在样品池中识别了4,729个独特的株系。然后计算每种微生物株系的相对丰度并且用于随后的分析。
表8a
在每个时间点由各动物产生的乳脂的测得镑数在表8a中给出。通过取乳脂值并减去1来创建用于该分析的模拟株系,以便确保模拟株系和乳脂值随时间的变化趋势相同,即,模拟株系与乳脂值之间存在已知的线性趋势/关系。然后将该模拟株系添加到先前在群落中识别的所有株系的矩阵中。针对各种条件(下文描述)同时计算所产生的乳脂的磅数与矩阵内的所有株系之间的mic值和皮尔逊系数,以建立作为关系预测因子的这些测量值的灵敏度和稳健性。
为了测试所公开的方法相对于传统方法检测关系的能力,逐一去除模拟株系的数据点(相对丰度设为0)。在去除每个数据点之后,重新计算mic和皮尔逊系数,并且记录模拟株系的排名(表8b)。可以看出,mic是远比皮尔逊系数更稳健的测量值。当没有去除点时,两种方法均将模拟株系识别为与所产生的乳脂的镑数相关的第一位株系。然而,当去除一个点时,相关性方法将模拟株系的排名下降至55,当去除另外一个点时下降至2142。mic仍然将模拟株系预测为最高排名的株系,直至去除6个点。
表8b
通过去除某些点来测试灵敏度的一个基本原理是当查看一组目标(例如,动物)的微生物时,存在所有这些目标所共有的特定株系,该株系可被称为核心微生物组。该组可表示特定目标(例如,特定动物)的微生物群体的少数部分,并且可存在仅存在于目标/动物的子集/小部分中的整个单独的株系群体。在一些实施方案中,更独特的株系(即,并非在所有动物中均存在的那些株系)可以是具有特定相关性的株系。开发了所公开方法的一些实施方案来解决数据集中的此类“缺口”,并因此靶向尤其相关的微生物和株系。
实施例6
提高肉鸡的饲料效率的活性微生物株系群集的选择
将96只雄性cobb500饲养21天。针对各只鸡确定重量和进食量,并在处死之后采集盲肠刮样。使用本公开的方法处理盲肠样品以识别当施用到生产环境中的肉鸡时将提高饲料效率的微生物群集。
实验设计
基于饮食处理将120只cobb500鸡分开放到围栏中。按照从第0-14天的处理将鸡放在地面围栏中。将测试设施分成2个围栏的1个区和每个区各2个笼子的48个区。使用完全随机区组设计将处理分配到围栏/笼子;在整个研究期间,围栏/笼子保持其处理。通过数字代码标识各处理。将鸡随机分配到笼子/围栏中。特定处理组如表9所示。
表9
圈养:
使用计算机程序将各处理分配到笼子/围栏。计算机生成的分配如下:
将鸡圈养在由固体塑料(4'高)构造的具有干净褥草的大混凝土地面围栏(4'x8')中的环境受控设施中。在第14天,将96只鸡移到在相同的环境受控设施内的笼子中。每个笼子的尺寸为24"x18"x24"。
通过白炽灯照明,并使用商业照明程序。每24小时时间段的连续灯光时间如表10中所示。
表10
对于所有处理组而言,鸡的环境条件(即0.53平方英尺、鸡密度、温度、照明、饲槽和储水空间)均相似。
为了防止鸡迁移,检查每个围栏以确保对于围栏之间的约14英寸的高度,不存在大于1英寸的开口。
疫苗接种:
在孵化场为鸡接种马立克氏病(mareks)疫苗。在收到鸡时(研究第0天),通过喷雾施用为鸡接种新城病(newcastle)和传染性支气管炎疫苗。疫苗制造商、批号和失效期的文档与最终报告一起提供。
水:
在整个研究过程中随意提供水。地面围栏水通过自动钟式饮水器提供。层架式鸡笼水通过一个乳头式饮水嘴提供。饮水器按需要每天检查两次并清理以保证在所有时间对鸡的清洁水供应。
饲料:
在整个研究过程中随意提供饲料。地面围栏饲料通过悬挂的~17英寸直径的管式饲槽提供。层架式鸡笼饲料通过一个9"x4"的饲槽提供。在大约前4天,将鸡饲槽托盘放在每个地面围栏中。
每日观察:
每天至少观察两次测试设施、围栏和鸡以了解一般鸡群状况、照明、水、饲料、通风和意外事件。每天记录一次测试设施的最低-最高温度。
死亡和淘汰:
从研究第0天开始,对发现已死亡或被去除并处死的任何鸡进行尸检。将不能取得饲料或水的淘汰鸡处死并尸检。在围栏死亡记录上记录可能的死因以及尸检发现。
体重和进食量:
每天单个地对~96只鸡称重。从第14-21天每天对每个笼子中剩余的饲料进行称重和记录。每天确定每个笼子对应的进食量。
增重和饲料转化率:
从第14-21天确定按照笼子的体重增加以及按照处理的平均体重增加。使用笼子的总饲料消耗量除以鸡重量来计算每天的和第14-21天时间段总体的饲料转化率。通过对来自处理内的每个笼子的各饲料转化率进行平均化来确定第14-21天时间段的平均处理饲料转化率。
兽医护理、干预和安乐死:
使发展显著并发病、受到伤害及其状态可能影响研究结果的动物从研究中去除并在做出此确定时实施安乐死。在挑战后六天,将笼子中的所有鸡取出并进行病变评分。
采集的数据:
从第14-21天每天单独得到的鸡重量和饲料转化率。
从第0天至研究结束向每个地面围栏和笼子添加的和从中去除的饲料量。
死亡:第0天至研究结束的可能的死因。
去除的鸡:第0天至研究结束的淘汰原因。
设施和鸡的每日观察、每日设施温度。
第21天来自每只鸡的盲肠内容物。
结果
使用以上所公开的方法(例如,如参考图1a、图1b和图2以及在整个说明书中所讨论的)分析结果。针对每只鸡的盲肠内容物确定株系水平上的微生物量和活性。在所有96个肉鸡盲肠样品中检测出总计22,461种独特的株系。根据活性阈值过滤每种株系的绝对细胞计数,以创建活性微生物株系及其相应绝对细胞计数的列表。平均而言,在处死时,每只肉鸡中仅48.3%的株系被认为是有活性的。在过滤之后,将每只鸡中的活性微生物的图谱与各种鸡元数据(包括饲料效率、最终体重以及饮食中沙利霉素的存在/不存在)整合,以便选择改善所有这些性状的表现的群集。
使用本公开的互信息方法对所有96只鸡的活性株系的绝对细胞计数与表现测量值之间的关系以及两种不同活性株系之间的关系进行评分。在应用阈值之后,4039个元数据-株系关系被认为是显著的,并且8842个株系-株系关系被认为是显著的。然后将通过mic得分加权的这些链接用作边缘(以元数据和株系作为节点)来创建网络以用于随后的群落检测分析。将louvain方法群落检测算法应用于该网络以将节点分类到子组中。
louvain方法通过首先将节点从其当前的子组去除并放置到相邻的子组中来优化网络模块性。如果节点的相邻节点的模块性得到改善,则将该节点重新分配到新的子组中。如果多个组具有改善的模块性,则选择具有最正向变化的子组。针对网络中的每个节点重复此步骤,直至没有进行新的分配为止。下一步骤涉及创建新的粗粒度网络,即所发现的子组变成新的节点。节点之间的边缘通过每个子组内的所有较低水平的节点的总和限定。从这里开始,重复第一步骤和第二步骤,直至不能进行更多的模块性优化变化为止。可研究局部最大值(即,在迭代步骤中形成的组)和全局最大值(即,最终分组)以解析总微生物群落内出现的子组,以及识别可能存在的潜在层次结构。
模块性:
其中a是元数据-株系和株系-株系关系的矩阵;ki=∑jaij是附接到节点i的总链接权重;并且m=1/2σijaij。当将节点i和j分配到相同的群落时,克罗内克符号(kroneckerdelta)δ(ci,cj)是1,否则是0。
计算移动节点时的模块性变化:
δq是子组c中模块性的增益。σin是c中链接的权重的总和,σtot是c中节点易发生的链接的权重的总和,ki是节点i易发生的链接的权重的总和,ki,in是c中从i到节点的链接的权重的总和,并且m是网络中所有链接的权重的总和。
使用louvain群落检测法在鸡微生物群落中检测到五个不同的子组。虽然自然界中存在大量的微生物多样性,但功能多样性要少得多。代谢能力的相似性和重叠性造成了冗余。对应于相同环境刺激或营养物的微生物株系可能趋势相似—这通过本公开的方法捕捉,并且这些微生物将最终被分组在一起。所得的分类和层次结构基于群落检测分析后株系所落入的组来展示对所述株系的功能性的预测。
在完成株系分类之后,从样品培养微生物株系。由于与分离和生长来自异质微生物群落的纯性培养物相关联的技术困难,通过本公开的方法的活性和关系阈值两者的株系只有少部分能在实验室环境中纯性繁殖。在完成培养后,基于纯性培养物是否存在以及株系所归类到的子组来选择微生物株系的群集。创建群集以包含尽可能多的功能性多样性—即,选择株系使得各种各样的子组呈现在所述群集中。然后在效力和现场研究中测试这些群集以确定株系群集作为产品的有效性,并且如果株系群集证明对生产有贡献,则该株系群集可作为产品生产和分销。
实施例7
使用小样品大小来识别活性微生物株系
如下所详述,少至两个样品即可有效识别活性微生物株系。具体地讲,以下实验表明本公开的方法针对所有比较(包括2个样品的比较)均正确地将产气荚膜梭菌识别为活性微生物株系以及肠道病变和坏死性肠炎的病原体。
实验设计
将鸡圈养在混凝土地板围栏中的环境受控设施内(~4'×4'减去2.25平方英尺的饲槽空间),其提供[~0.55平方英尺/鸡(第0天),~0.69平方英尺/鸡(病变评分后第21天)]的地面空间和鸡密度,所有测试组的温度、湿度、照明、饲槽和储水空间都将是相似的。将鸡放入铺有适当厚度干净木刨花的干净围栏中,为鸡提供舒适的环境。为了保持鸡的舒适环境,还向围栏添加了额外的木刨花。通过白炽灯照明,并如下使用商业照明程序。
表11
对于所有处理组而言,鸡的环境条件(即鸡密度、温度、照明、饲槽和储水空间)均相似。为了防止鸡迁移和细菌在围栏间扩散,每个围栏都有一个在围栏之间的高度约为24英寸的固体(塑料)分隔器。
疫苗接种和治疗药物:
在孵化场为鸡接种马立克氏病(mareks)疫苗。在收到鸡时(研究第0天),通过喷雾施用为鸡接种新城病(newcastle)和传染性支气管炎疫苗。疫苗制造商、批号和失效期的文档与最终报告一起提供。
水:
在整个研究期间,通过每个围栏一个plasson饮水器随意提供水。饮水器按需要每天检查两次并清理以保证对鸡的清洁且恒定的水供应。
饲料:
在整个研究期间,每个围栏通过一根悬挂的直径为~17英寸的管式饲槽随意提供饲料。在大约前4天,将鸡饲槽托盘放在每个围栏中。根据实验设计,在收到鸡时(第0天)使鸡接受它们相应的处理饮食。称重并记录从第0天到研究结束向围栏中添加和从中去除的饲料。
每日观察:
每天至少观察两次测试设施、围栏和鸡以了解一般鸡群状况、照明、水、饲料、通风和意外事件。如果在每日两次观察中的任何一次观察到异常情况或异常行为,则对其进行记载,并将该记载包括在研究记录中。每天记录一次测试设施的最低-最高温度。
围栏卡片:
每个围栏附有2张卡片。一张卡片标识围栏编号,第二张卡片表示处理编号。
动物处理:
将动物保持在理想的宜居条件下。以减少伤害和不必要压力的方式处理动物。严格执行人道措施。
兽医护理、干预和安乐死:
根据现场sop,可将发展与测试程序不相关的临床上显著的并发疾病的鸡按照研究者或指定人员的判断从研究中去除并实施安乐死。另外,在现场兽医或合格技术人员授权后,也可对濒死的或受伤的鸡实施安乐死。记载退出的原因。如果动物死亡,或因人道原因而去除并实施安乐死,则将其记录在围栏的死亡表上,并实施尸检且记录以记载去除的原因。
如果研究者认为安乐死是必要的,则通过颈脱位法对动物实施安乐死。
死亡和淘汰:
从研究第0天开始,对发现已死亡或被去除并处死的任何鸡进行称重和尸检。将不能取得饲料或水的淘汰鸡处死、称重和记载。在围栏死亡记录上记录重量和可能的死因以及尸检发现。
产气荚膜梭菌挑战
施用方法:
通过饲料施用本研究中的产气荚膜梭菌(cl-15,α型,α和β2毒素)培养物。使用来自每个围栏的饲槽的饲料与培养物混合。在将培养物放置在围栏中之前,将处理饲料从鸡中去除,保持约4-8小时。对于每个围栏的鸡,将浓度为约2.0–9.0x108cfu/ml的基于研究设计的固定量的肉汤培养物与饲槽托盘中的固定量的饲料(~25g/鸡)混合,并且所有挑战的围栏均以相同的方式处理。大多数培养物-饲料在1-2小时内被消耗。为了使所有处理中的鸡均以类似的方式处理,未挑战的组也在与挑战组相同的时间段内去除饲料。
梭菌挑战:
将产气荚膜梭菌培养物(cl-15)在~37℃下在含有淀粉的液体硫乙醇酸盐培养基中生长~5小时。cl-15是来自科罗拉多州的肉鸡爆发的产气荚膜梭菌的野外株系(fieldstrain)。每天制备并使用新鲜肉汤培养物。对于每个围栏的鸡,将固定量的过夜肉汤培养物与饲槽托盘中的固定量的处理饲料混合。将饲料的量、培养接种物的体积和定量以及给药天数记载在最终报告中,并且所有围栏将以相同的方式处理。鸡将接受产气荚膜梭菌培养物一天(研究第17天)。
采集的数据
用于使用本申请的方法进行分析的肠内容物
在大约第14天和第21天按照围栏和个体得到的鸡重量,以及按围栏得到的饲料效率。
从第0天至研究结束向每个围栏添加的和从中去除的饲料量。
死亡:第0天至研究结束的性别、重量和可能的死因。
去除的鸡:第0天至研究结束的淘汰原因、性别和重量。
设施和鸡的每日观察、每日设施温度。
大约第21天的5只鸡/围栏的病变得分
从48只进行病变评分的鸡采集的样品
病变评分:
在上一次产气荚膜梭菌培养物施用后四天,从每个围栏随机选择五只鸡(抓到哪只是哪只)、处死并且针对坏死性肠炎对肠病变进行评分。病变评分如下:
0=正常:没有ne病变,小肠具有正常弹性(打开后卷回正常位置)
1=轻度:小肠壁薄而松弛(打开时保持平坦并且打开后不卷回正常位置);过量粘液覆盖粘膜
2=中度:可观察到的小肠壁发红和肿胀;肠膜的轻微溃疡和坏死;过量粘液
3=重度:小肠膜的大面积坏死和溃疡;大量出血;粘膜上存在纤维蛋白层和坏死碎片(土耳其式毛巾外观)
4=死亡或濒死:可能在24小时内死亡并且具有2或更大ne病变得分的鸡
结果
使用本申请的方法分析结果。测定了所有48只鸡的小肠内容物的株系水平上的微生物绝对细胞计数和活性。本申请的方法整合了每个样品的活性信息以及绝对细胞计数信息。
本申请的互信息方法用于对10只随机选择的肉鸡的活性株系的绝对细胞计数与个体病变得分之间的关系进行评分。从数据集中随机去除一个样品,并重复分析。重复该过程,直到仅比较两个肉鸡样品。
通过针对从样品池识别的生物列表进行的全局比对搜索来确认致病株系产气荚膜梭菌。表12列出了针对所有分析株系的排名(排名第1位的是与病变得分最为相关的株系):
表12说明了使用所公开的方法针对所有比较(包括2个样品的比较)将产气荚膜梭菌正确地识别为活性微生物株系和病变得分的病原体。随着样品数量的减少,从7个样品的比较开始,假阳性的数量(即其他株系也被识别为病原体)增加,其中两种株系(包括产气荚膜梭菌)并列排第1位。这种趋势继续至减少到2个样品的比较,其中31种株系(包括产气荚膜梭菌)并列排第1位。
通常,虽然使用另外的样品可以减少噪音/假阳性的数量,但是可以使用对所得株系的进一步分析和处理来将产气荚膜梭菌识别为致病株系,包括来自总共31种识别的株系。根据实施方案、配置和应用,本公开的方法可以用少量样品来实践,并且所用样品的数量可以根据样品来源、样品类型、元数据、目标微生物组的复杂性等而变化。
另外的示例实施方案
实施方案a1是一种方法,所述方法包括:获得共有至少一个共同特征并具有至少一个不同特征的至少两个样品;针对每个样品,检测每个样品中的一种或多种微生物类型的存在;确定每个样品中的所述一种或多种微生物类型中的每种检出微生物类型的数量;测量每个样品中独特的第一标记物的数量及其量,每个独特的第一标记物是微生物株系的标记物;整合每种微生物类型的所述数量和所述第一标记物的所述数量,以得到存在于每个样品中的每种微生物株系的绝对细胞计数;基于指定阈值测量每种微生物株系的至少一个独特的第二标记物,以确定每个样品中所述微生物株系的活性水平;根据所述确定的活性来过滤所述绝对细胞计数,以提供所述至少两个样品中的每一个的活性微生物株系及其相应绝对细胞计数的列表;对所述至少两个样品中的每一个的活性微生物株系的所述经过滤的绝对细胞计数与所述至少两个样品中的每一个的至少一个测得元数据或另外的活性微生物株系进行比较,并且基于预测的功能和/或化学性质将所述活性微生物株系分类成至少两组;从所述至少两组中选择至少一种微生物株系;以及组合来自所述至少两组的所述选择的至少一种微生物株系以形成微生物的群集,所述群集被配置为改变与所述至少一个元数据相对应的性质。
实施方案a2是根据实施方案a1所述的方法,其中测量独特的第一标记物的所述数量包括测量每个样品中独特的基因组dna标记物的数量。实施方案a3是根据实施方案a1所述的方法,其中测量独特的第一标记物的所述数量包括测量每个样品中独特的rna标记物的数量。实施方案a4是根据实施方案a1所述的方法,其中测量独特的第一标记物的所述数量包括测量每个样品中独特的蛋白质标记物的数量。实施方案a5是根据实施方案a1所述的方法,其中测量独特的第一标记物的所述数量包括测量每个样品中独特的代谢物标记物的数量。实施方案a6是根据实施方案a5所述的方法,其中测量独特的代谢物标记物的所述数量包括测量每个样品中独特的脂质标记物的数量。实施方案a7是根据实施方案a5所述的方法,其中测量独特的代谢物标记物的所述数量包括测量每个样品中独特的碳水化合物标记物的数量。实施方案a8是根据实施方案a1所述的方法,其中测量独特的第一标记物的所述数量及其量包括对来自每个样品的基因组dna进行高通量测序反应。实施方案a9是根据实施方案a1所述的方法,其中测量独特的第一标记物的所述数量及其量包括对来自每个样品的基因组dna进行元基因组测序。实施方案a10是根据实施方案a1所述的方法,其中所述独特的第一标记物包括mrna标记物、sirna标记物和/或核糖体rna标记物中的至少一种。实施方案a11是根据实施方案a1所述的方法,其中所述独特的第一标记物包括σ因子、转录因子、核苷相关蛋白和/或代谢酶中的至少一种。
实施方案a12是根据实施方案a1至a11中任一个所述的方法,其中测量所述至少一个独特的第二标记物包括测量每个样品中所述至少一个独特的第二标记物的表达水平。实施方案a13是根据实施方案a12所述的方法,其中测量所述至少一个独特的第二标记物的所述表达水平包括对所述样品中的mrna进行基因表达分析。实施方案a14是根据实施方案a13所述的方法,其中所述基因表达分析包括测序反应。实施方案a15是根据实施方案a13所述的方法,其中所述基因表达分析包括定量聚合酶链式反应(qpcr)、元转录组测序和/或转录组测序。实施方案a16是根据实施方案a12所述的方法,其中测量所述至少一个独特的第二标记物的所述表达水平包括对每个样品或其部分进行质谱分析。实施方案a17是根据实施方案a12所述的方法,其中测量所述至少一个独特的第二标记物的所述表达水平包括对每个样品或其部分进行元核糖体图谱分析或核糖体图谱分析。
实施方案a18是根据实施方案a1至a17中任一个所述的方法,其中所述一种或多种微生物类型包括细菌、古细菌、真菌、原生动物、植物、其他真核生物、病毒、类病毒或它们的组合。实施方案a19是根据实施方案a1至a18中任一个所述的方法,其中所述一种或多种微生物株系是一种或多种细菌株系、古细菌株系、真菌株系、原生动物株系、植物株系、其他真核生物株系、病毒株系、类病毒株系或它们的组合。实施方案a20是根据实施方案a19所述的方法,其中所述一种或多种微生物株系是一种或多种真菌种或亚种;和/或其中所述一种或多种微生物株系是一种或多种细菌种或亚种。
实施方案a21是根据实施方案a1至a20中任一个所述的方法,其中确定每个样品中所述一种或多种微生物类型中每一种的数量包括对每个样品或其部分进行测序、离心、光学显微镜检查、荧光显微镜检查、染色、质谱分析、微流体分析、定量聚合酶链式反应(qpcr)、凝胶电泳和/或流式细胞术。
实施方案a22是根据实施方案a1所述的方法,其中所述独特的第一标记物包括系统发育标记物,其包含5s核糖体亚基基因、16s核糖体亚基基因、23s核糖体亚基基因、5.8s核糖体亚基基因、18s核糖体亚基基因、28s核糖体亚基基因、细胞色素c氧化酶亚基基因、β-微管蛋白基因、延伸因子基因、rna聚合酶亚基基因、内部转录间隔区(its)或它们的组合。
实施方案a22a是根据实施方案a1所述的方法,其中所述独特的第一标记物不包括系统发育标记物。实施方案a22b是根据实施方案a1所述的方法,其中所述独特的第一标记物不包括包含5s核糖体亚基基因的系统发育标记物。实施方案a22c是根据实施方案a1所述的方法,其中所述独特的第一标记物不包括包含16s核糖体亚基基因的系统发育标记物。实施方案a22d是根据实施方案a1所述的方法,其中所述独特的第一标记物不包括包含23s核糖体亚基基因的系统发育标记物。实施方案a22e是根据实施方案a1所述的方法,其中所述独特的第一标记物不包括包含5.8s核糖体亚基基因的系统发育标记物。实施方案a22f是根据实施方案a1所述的方法,其中所述独特的第一标记物不包括包含18s核糖体亚基基因的系统发育标记物。实施方案a22g是根据实施方案a1所述的方法,其中所述独特的第一标记物不包括包含28s核糖体亚基基因的系统发育标记物。实施方案a22h是根据实施方案a1所述的方法,其中所述独特的第一标记物不包括包含细胞色素c氧化酶亚基基因的系统发育标记物。实施方案a22i是根据实施方案a1所述的方法,其中所述独特的第一标记物不包括包含β-微管蛋白基因的系统发育标记物。实施方案a22j是根据实施方案a1所述的方法,其中所述独特的第一标记物不包括包含延伸因子基因的系统发育标记物。实施方案a22k是根据实施方案a1所述的方法,其中所述独特的第一标记物不包括包含rna聚合酶亚基基因的系统发育标记物。实施方案a22l是根据实施方案a1所述的方法,其中所述独特的第一标记物不包括包含内部转录间隔区(its)的系统发育标记物。
实施方案a23是根据实施方案a22所述的方法,其中测量独特的标记物的所述数量及其量包括对来自每个样品的基因组dna进行高通量测序反应。实施方案a24是根据实施方案a22所述的方法,其中测量独特的标记物的所述数量及其量包括对基因组dna进行基因组测序。实施方案a25是根据实施方案a22所述的方法,其中测量独特的标记物的所述数量及其量包括对基因组dna进行扩增子测序。
实施方案a26是根据实施方案a1至a25中任一个所述的方法,其中所述至少一个不同特征包括采集时间,在所述采集时间采集所述至少两个样品中的每一个,使得第一样品的所述采集时间不同于第二样品的所述采集时间。
实施方案a27是根据实施方案a1至a25中任一个所述的方法,其中所述至少一个不同特征包括采集位置,在所述采集位置处采集所述至少两个样品中的每一个,使得第一样品的所述采集位置不同于第二样品的所述采集位置。
实施方案a28是根据实施方案a1至a27中任一个所述的方法,其中所述至少一个共同特征包括样品来源类型,使得第一样品的所述样品来源类型与第二样品的所述样品来源类型相同。实施方案a29是根据实施方案a28所述的方法,其中所述样品来源类型是动物类型、器官类型、土壤类型、水类型、沉积物类型、油类型、植物类型、农产品类型、根外土壤类型、根际土壤类型或植物部分类型中的一种。
实施方案a30是根据实施方案a1至a27中任一个所述的方法,其中所述至少一个共同特征包括所述至少两个样品中的每一个是胃肠样品。
实施方案a31是根据实施方案a1至a27中任一个所述的方法,其中所述至少一个共同特征包括动物样品来源类型,每个样品具有另外的共同特征,使得每个样品是组织样品、血液样品、牙齿样品、汗液样品、指甲样品、皮肤样品、毛发样品、粪便样品、尿液样品、精液样品、粘液样品、唾液样品、肌肉样品、脑样品或器官样品。
实施方案a32是根据实施方案a1至a31中任一个所述的方法,还包括:基于所述至少一个测得元数据从目标获得至少一个另外的样品,其中来自所述目标的所述至少一个另外的样品与所述至少两个样品共有至少一个共同特征;以及对于来自所述目标的所述至少一个另外的样品,检测一种或多种微生物类型的存在,确定所述一种或多种微生物类型中的每种检出微生物类型的数量,测量独特的第一标记物的数量及其量,整合每种微生物类型的所述数量和所述第一标记物的所述数量以得到存在的每种微生物株系的所述绝对细胞计数,测量每种微生物株系的至少一个独特的第二标记物以确定所述微生物株系的活性水平,根据所述确定的活性来过滤所述绝对细胞计数以提供来自所述目标的所述至少一个另外的样品的活性微生物株系及其相应绝对细胞计数的列表;其中从所述至少两组的每一组中选择所述至少一种微生物株系是基于来自所述目标的所述至少一个另外的样品的活性微生物株系及其相应绝对细胞计数的所述列表,使得所述形成的群集被配置为改变与所述至少一个元数据相对应的所述目标的性质。
实施方案a33是根据实施方案a1至a32中任一个所述的方法,其中对所述至少两个样品中的每一个的活性微生物株系的所述经过滤的绝对细胞计数与所述至少两个样品中的每一个的至少一个测得元数据或另外的活性微生物株系进行比较包括确定每个样品中所述一种或多种活性微生物株系与所述至少一个测得元数据或另外的活性微生物株系的共现。实施方案a34是根据实施方案a33所述的方法,其中所述至少一个测得元数据包括一个或多个参数,其中所述一个或多个参数是样品ph、样品温度、脂肪丰度、蛋白质丰度、碳水化合物丰度、矿物质丰度、维生素丰度、天然产物丰度、指定化合物丰度、所述样品来源的体重、所述样品来源的进食量、所述样品来源的重量增加、所述样品来源的饲料效率、一种或多种病原体的存在或不存在、所述样品来源的物理特征或测量结果、所述样品来源的生产特征中的至少一个或它们的组合。实施方案a35是根据实施方案a34所述的方法,其中所述一个或多个参数是奶中的乳清蛋白丰度、酪蛋白丰度和/或脂肪丰度中的至少一个。
实施方案a36是根据实施方案a33至a35中任一个所述的方法,其中确定每个样品中所述一种或多种活性微生物株系和所述至少一个测得元数据的共现包括创建使用表示元数据和微生物株系关联的链接、所述一种或多种活性微生物株系的所述绝对细胞计数以及所述一种或多种独特的第二标记物的所述测量结果填充的矩阵,以代表一个或多个异质微生物群落的一个或多个网络。实施方案a37是根据实施方案a36所述的方法,其中所述至少一个测得元数据包括第二微生物株系的存在、活性和/或量。
实施方案a38是根据实施方案a33至a37中任一个所述的方法,其中确定所述一种或多种活性微生物株系和所述至少一个测得元数据的共现并对所述活性微生物株系进行分类包括网络分析和/或聚类分析,以测量网络内每种微生物株系的连通性,其中所述网络表示共有共同特征、测得元数据和/或相关环境参数的所述至少两个样品的集合体。实施方案a39是根据实施方案a38所述的方法,其中所述至少一个测得元数据包括第二微生物株系的存在、活性和/或量。实施方案a40是根据实施方案a38或a39所述的方法,其中所述网络分析和/或聚类分析包括链接分析、模块性分析、稳健性测量、中介性测量、连通性测量、传递性测量、中心性测量或其组合。实施方案a41是根据实施方案a38至a40中任一个所述的方法,其中所述聚类分析包括建立连通性模型、子空间模型、分布模型、密度模型或质心模型。
实施方案a42是根据实施方案a38或实施方案a39所述的方法,其中所述网络分析包括通过链接挖掘和预测、集体分类、基于链接的聚类、关系相似性或其组合的网络预测建模。实施方案a43是根据实施方案a38或实施方案3a9所述的方法,其中所述网络分析包括基于微分方程的群体建模。实施方案a44是根据实施方案a43所述的方法,其中所述网络分析包括lotka-volterra建模。实施方案a45是根据实施方案a38或实施方案a39所述的方法,其中所述聚类分析是启发式方法。实施方案a46是根据实施方案a45所述的方法,其中所述启发式方法是louvain方法。
实施方案a47是根据实施方案a38或实施方案a39所述的方法,其中所述网络分析包括用于建立变量之间的连通性的非参数方法。实施方案a48是根据实施方案a38或实施方案a39所述的方法,其中所述网络分析包括用于建立连通性的变量之间的互信息和/或最大信息系数计算。
实施方案a49是一种基于两个或更多个样品集合形成活性微生物株系的群集的方法,所述群集被配置为改变环境中的性质或特征,所述两个或更多个样品集合在其之间共有至少一个共同的或相关的环境参数并且在其之间具有至少一个不同的环境参数,每个样品集合包含至少一个含有异质微生物群落的样品,其中所述一个或多个微生物株系是一种或多种生物类型的亚分类单元,所述方法包括:检测每个样品中多种微生物类型的存在;确定每个样品中每种检出微生物类型的绝对细胞数量;测量每个样品中独特的第一标记物的数量及其量,其中独特的第一标记物是微生物株系的标记物;在蛋白质或rna水平上,测量一种或多种独特的第二标记物的表达水平,其中独特的第二标记物是微生物株系活性的标记物;基于超过指定阈值的所述一种或多种独特的第二标记物的所述表达水平,确定每个样品的所述检出微生物株系的活性;基于所述一种或多种第一标记物的所述量和所述一种或多种微生物株系是其中的亚分类单元的微生物类型的绝对细胞数量计算每个样品中每种检出活性微生物株系的绝对细胞计数,其中所述一种或多种活性微生物株系表达高于所述指定阈值的所述第二独特的标记物;基于最大信息系数网络分析确定所述样品中的所述活性微生物株系与至少一个环境参数或另外的活性微生物株系的共现,以测量网络内每种微生物株系的连通性,其中所述网络是所述至少两个或更多个样品集合与至少一个共同或相关环境参数的集合体;基于所述网络分析从所述一种或多种活性微生物株系中选择多种活性微生物株系;以及从所述选择的多种活性微生物株系形成活性微生物株系的群集,所述活性微生物株系的群集被配置为当被引入到某环境中时选择性地改变所述环境的性质或特征。
实施方案a50是根据实施方案a49所述的方法,其中所述至少一个环境参数包括第二微生物株系的存在、活性和/或量。实施方案a51是根据实施方案a49或实施方案a50所述的方法,其中第一样品集合的至少一个共同或相关环境因素的至少一个测得指标不同于第二样品集合的至少一个共同或相关环境因素的测得指标。
实施方案a52是根据实施方案a49或实施方案a50所述的方法,其中每个样品集合包含多个样品,并且样品集合内每个样品的至少一个共同或相关环境因素的测得指标基本相似,并且一个样品集合的平均测得指标不同于另一个样品集合的平均测得指标。实施方案a53是根据实施方案a49或实施方案a50所述的方法,其中每个样品集合包含多个样品,并且从第一群体采集第一样品集合,从第二群体采集第二样品集合。实施方案a54是根据实施方案a49或a50所述的方法,其中每个样品集合包含多个样品,并且第一样品集合在第一时间从第一群体采集,第二样品集合在与所述第一时间不同的第二时间从所述第一群体采集。实施方案a55是根据实施方案a49至a54中任一个所述的方法,其中至少一个共同或相关环境因素包括营养物信息。
实施方案a56是根据实施方案a49至a54中任一个所述的方法,其中至少一个共同或相关环境因素包括饮食信息。实施方案a57是根据实施方案a49至a54中任一个所述的方法,其中至少一个共同或相关环境因素包括动物特征。实施方案a58是根据实施方案a49至a54中任一个所述的方法,其中至少一个共同或相关环境因素包括感染信息或健康状态。
实施方案a59是根据实施方案a51所述的方法,其中至少一个测得指标是样品ph、样品温度、脂肪丰度、蛋白质丰度、碳水化合物丰度、矿物质丰度、维生素丰度、天然产物丰度、指定化合物丰度、所述样品来源的体重、所述样品来源的进食量、所述样品来源的重量增加、所述样品来源的饲料效率、一种或多种病原体的存在或不存在、所述样品来源的物理特征或测量结果、所述样品来源的生产特征或它们的组合。
实施方案a60是根据实施方案a49或实施方案a50所述的方法,其中所述至少一个参数是奶中的乳清蛋白丰度、酪蛋白丰度和/或脂肪丰度中的至少一个。实施方案a61是根据实施方案a49至a60中任一个所述的方法,其中测量每个样品中独特的第一标记物的所述数量包括测量独特的基因组dna标记物的数量。实施方案a62是根据实施方案a49至a60中任一个所述的方法,其中测量所述样品中独特的第一标记物的所述数量包括测量独特的rna标记物的数量。实施方案a63是根据实施方案a49至a60中任一个所述的方法,其中测量所述样品中独特的第一标记物的所述数量包括测量独特的蛋白质标记物的数量。
实施方案a64是根据实施方案a49至a63中任一个所述的方法,其中所述多种微生物类型包括一种或多种细菌、古细菌、真菌、原生动物、植物、其他真核生物、病毒、类病毒或它们的组合。实施方案a65是根据实施方案a49至a64中任一个所述的方法,其中确定每个样品中每种所述微生物类型的所述绝对细胞数量包括对所述样品或其部分进行测序、离心、光学显微镜检查、荧光显微镜检查、染色、质谱分析、微流体分析、定量聚合酶链式反应(qpcr)、凝胶电泳和/或流式细胞术。实施方案a66是根据实施方案a49至a65中任一个所述的方法,其中一种或多种活性微生物株系是选自一种或多种细菌、古细菌、真菌、原生动物、植物、其他真核生物、病毒、类病毒或它们的组合的一种或多种微生物类型的亚分类单元。
实施方案a67是根据实施方案a49至a65中任一个所述的方法,其中一种或多种活性微生物株系是一种或多种细菌株系、古细菌株系、真菌株系、原生动物株系、植物株系、其他真核生物株系、病毒株系、类病毒株系或它们的组合。实施方案a68是根据实施方案a49至a67中任一个所述的方法,其中一种或多种活性微生物株系是一种或多种真菌种、真菌亚种、细菌种和/或细菌亚种。实施方案a69是根据实施方案a49至a68中任一个所述的方法,其中至少一个独特的第一标记物包括系统发育标记物,其包含5s核糖体亚基基因、16s核糖体亚基基因、23s核糖体亚基基因、5.8s核糖体亚基基因、18s核糖体亚基基因、28s核糖体亚基基因、细胞色素c氧化酶亚基基因、β-微管蛋白基因、延伸因子基因、rna聚合酶亚基基因、内部转录间隔区(its)或它们的组合。
实施方案a70是根据实施方案a49或实施方案a50所述的方法,其中测量独特的第一标记物的所述数量及其量包括对来自每个样品的基因组dna进行高通量测序反应。实施方案a71是根据实施方案a49或a50所述的方法,其中测量独特的第一标记物的所述数量及其量包括对来自每个样品的基因组dna进行元基因组测序。实施方案a72是根据实施方案a49或a50所述的方法,其中独特的第一标记物包括mrna标记物、sirna标记物或核糖体rna标记物。实施方案a73是根据实施方案a49或实施方案a50所述的方法,其中独特的第一标记物包括σ因子、转录因子、核苷相关蛋白、代谢酶或它们的组合。
实施方案a74是根据实施方案a49至a73中任一个所述的方法,其中测量一种或多种独特的第二标记物的所述表达水平包括对所述样品中的mrna进行基因表达分析。实施方案a75是根据实施方案a74所述的方法,其中所述基因表达分析包括测序反应。实施方案a76是根据实施方案a74所述的方法,其中所述基因表达分析包括定量聚合酶链式反应(qpcr)、元转录组测序和/或转录组测序。
实施方案a77是根据实施方案a49至a68和实施方案a74至a76中任一个所述的方法,其中测量一种或多种独特的第二标记物的所述表达水平包括对每个样品或其部分进行质谱分析。实施方案a78是根据实施方案a49至a68和实施方案a74至a76中任一个所述的方法,其中测量一种或多种独特的第二标记物的所述表达水平包括对所述样品或其部分进行元核糖体图谱分析和/或核糖体图谱分析。
实施方案a79是根据实施方案a49至a78中任一个所述的方法,其中所述样品的所述来源类型是动物、土壤、空气、盐水、淡水、污水污泥、沉积物、油、植物、农产品、根外土壤、根际土壤、植物部分、蔬菜、极端环境中的一种或它们的组合。
实施方案a80是根据实施方案a49至a78中任一个所述的方法,其中每个样品是胃肠样品。实施方案a81是根据实施方案a49至a78中任一个所述的方法,其中每个样品是组织样品、血液样品、牙齿样品、汗液样品、指甲样品、皮肤样品、毛发样品、粪便样品、尿液样品、精液样品、粘液样品、唾液样品、肌肉样品、脑样品或器官样品中的一种。
实施方案a82是一种处理器实施的方法,所述方法包括:接收来自共有至少一个共同特征并具有至少一个不同特征的至少两个样品的样品数据;针对每个样品,确定每个样品中的一种或多种微生物类型的存在;确定每个样品中的所述一种或多种微生物类型中的每种检出微生物类型的数量;确定每个样品中独特的第一标记物的数量及其量,每个独特的第一标记物是微生物株系的标记物;通过处理器整合每种微生物类型的所述数量和所述第一标记物的所述数量,以得到存在于每个样品中的每种微生物株系的绝对细胞计数;基于超过指定阈值的每种微生物株系的至少一个独特的第二标记物的测量值,确定每个样品中每种微生物株系的活性水平,如果微生物株系的至少一个独特的第二标记物的测量值超过对应阈值,则该株系被识别为有活性的;根据确定的活性来过滤每种微生物株系的所述绝对细胞计数,以提供所述至少两个样品中的每一个的活性微生物株系及其相应绝对细胞计数的列表;通过至少一个处理器对所述至少两个样品中的每一个的活性微生物株系的所述经过滤的绝对细胞计数与所述至少两个样品中的每一个的至少一个测得元数据或另外的活性微生物株系进行网络分析,所述网络分析包括确定每种活性微生物株系与每种其他活性微生物株系之间的最大信息系数得分以及确定每种活性微生物株系与相应的至少一个测得元数据或另外的活性微生物株系之间的最大信息系数得分;根据预测的功能和/或化学性质对所述活性微生物株系进行分类;基于所述分类识别多种活性微生物株系;以及输出所述识别的多种活性微生物株系。
实施方案a83是如实施方案a82所述的处理器实施的方法,所述方法还包括:聚集活性微生物群集,所述活性微生物群集被配置为当被应用于目标时改变与所述至少一个测得元数据相对应的性质。实施方案a84是如实施方案a82所述的处理器实施的方法,其中所述输出的多种活性微生物株系用于聚集活性微生物群集,所述活性微生物群集被配置为当被应用于目标时改变与所述至少一个测得元数据相对应的性质。实施方案a85是如实施方案a82所述的处理器实施的方法,所述方法还包括:基于所述输出的多种识别的活性微生物株系识别至少一种病原体。实施方案a86是如实施方案a82至a85中任一个所述的处理器实施的方法,其中所述输出的多种活性微生物株系进一步用于聚集活性微生物群集,所述活性微生物群集被配置为当被应用于目标时靶向所述至少一种识别的病原体,并治疗和/或预防与所述至少一种识别的病原体相关的症状。
实施方案a87是一种形成活性微生物株系的活性微生物生物群集的方法,所述活性微生物生物群集被配置为改变目标生物环境中的性质,所述方法包括:获得共有至少一个共同特征并具有至少一个不同特征的至少两个样品;针对每个样品,检测每个样品中的一种或多种微生物类型的存在;确定每个样品中的所述一种或多种微生物类型中的每种检出微生物类型的数量;测量每个样品中独特的第一标记物的数量及其量,每个独特的第一标记物是微生物株系的标记物;整合每种微生物类型的所述数量和所述第一标记物的所述数量,以得到存在于每个样品中的每种微生物株系的绝对细胞计数;基于指定阈值测量每种微生物株系的至少一个独特的第二标记物,以确定每个样品中所述微生物株系的活性水平;根据所述确定的活性来过滤所述绝对细胞计数,以提供所述至少两个样品中的每一个的活性微生物株系及其相应绝对细胞计数的列表;对所述至少两个样品中的每一个的活性微生物株系的所述经过滤的绝对细胞计数与所述至少两个样品中的每一个的至少一个测得元数据进行比较,所述比较包括确定每个样品中的所述活性微生物株系与所述至少一个测得元数据的共现,确定每个样品中所述活性微生物株系和所述至少一个测得元数据的共现包括创建使用表示元数据和微生物株系关系的链接、所述活性微生物株系的所述绝对细胞计数以及所述独特的第二标记物的测量结果的矩阵,以代表一个或多个异质微生物群落网络;根据预测的功能和/或化学性质,基于非参数网络分析和聚类分析中的至少一种,将所述活性微生物株系分组成至少两组,所述非参数网络分析和聚类分析识别活性异质微生物群落网络内的每种活性微生物株系和测得元数据的连通性;从所述至少两组的每一组中选择至少一种微生物株系;以及将所述选择的微生物株系与载体培养基组合以形成活性微生物的生物群集,所述生物群集被配置为当被引入到目标生物环境中时改变与所述目标生物环境的所述至少一个元数据相对应的性质。
实施方案a88是根据实施方案a87所述的方法,所述方法还包括:基于所述至少一个测得元数据获得至少一个另外的样品,其中所述至少一个另外的样品与所述至少两个样品共有至少一个特征;以及对于所述至少一个另外的样品,检测一种或多种微生物类型的存在,确定所述一种或多种微生物类型中的每种检出微生物类型的数量,测量独特的第一标记物的数量及其量,整合每种微生物类型的所述数量和所述第一标记物的所述数量以得到存在的每种微生物株系的绝对细胞计数,测量每种微生物株系的至少一个独特的第二标记物以确定所述微生物株系的活性水平,根据所述确定的活性来过滤所述绝对细胞计数以提供所述至少一个另外的样品的活性微生物株系及其相应绝对细胞计数的列表;其中对活性微生物株系的所述经过滤的绝对细胞计数进行比较包括对所述至少两个样品中的每一个和所述至少一个另外的样品的活性微生物株系的所述经过滤的绝对细胞计数与所述至少一个测得元数据进行比较,使得选择所述活性微生物株系至少部分地基于所述至少一个另外的样品的活性微生物株系及其相应绝对细胞计数的所述列表。
实施方案a89是一种基于各自具有多个环境参数的两个或更多个样品集合形成活性微生物株系的合成群集的方法,所述合成群集被配置为改变生物环境中的性质,所述多个环境参数中的至少一个参数是在所述两个或更多个样品集合之间相似的共同环境参数,并且至少一个环境参数是在所述两个或更多个样品集合中的每一个之间不同的不同环境参数,每个样品集合包括至少一个包含获自生物样品来源的异质微生物群落的样品,所述活性微生物株系中的至少一种是一种或多种生物类型的亚分类单元,所述方法包括:检测每个样品中多种微生物类型的存在;确定每个样品中每种检出微生物类型的绝对细胞数量;测量每个样品中独特的第一标记物的数量及其量,独特的第一标记物是微生物株系的标记物;测量一种或多种独特的rna标记物的表达水平,其中独特的rna标记物是微生物株系活性的标记物;基于超过指定阈值的所述一种或多种独特的rna标记物的所述表达水平,确定每个样品的每种所述检测微生物株系的活性;基于所述一种或多种第一标记物的所述量和所述一种或多种微生物株系是其中的亚分类单元的微生物类型的绝对细胞数量计算每个样品中每种检出活性微生物株系的绝对细胞计数,所述一种或多种活性微生物株系表达高于所述指定阈值的一种或多种独特的rna标记物;分析所述两个或更多个样品集合的所述活性微生物株系,所述分析包括对所述两个或更多个样品集合中的每一个的每种所述活性微生物株系、所述至少一个共同环境参数以及所述至少一个不同环境参数进行非参数网络分析,所述非参数网络分析包括(1)确定每种活性微生物株系与每种其他活性微生物株系之间的最大信息系数得分以及(2)确定每种活性微生物株系与所述至少一个不同环境参数之间的最大信息系数得分;基于所述非参数网络分析从所述一种或多种活性微生物株系中选择多种活性微生物株系;以及形成包含选择的多种活性微生物株系和微生物载体培养基的活性微生物株系的合成群集,所述活性微生物株系的合成群集被配置为当被引入到生物环境中时选择性地改变该生物环境的性质。
实施方案a90是一种形成活性微生物生物群集的方法,所述活性微生物生物群集被配置为改变目标生物环境中的性质,所述方法包括:获得共有至少一个共同环境参数并具有至少一个不同环境参数的至少两个样品;针对每个样品,检测每个样品中的一种或多种微生物类型的存在;确定每个样品中的所述一种或多种微生物类型中的每种检出微生物类型的数量;测量每个样品中独特的第一标记物的数量及其量,每个独特的第一标记物是检出微生物类型的微生物株系的标记物;基于每种检出微生物类型的所述数量和所述微生物类型的对应或相关的独特的第一标记物的比例/相对数量,确定存在于每个样品中的每种微生物株系的绝对细胞计数;基于指定阈值测量每种微生物株系的至少一个独特的第二标记物,以确定每个样品中所述微生物株系的活性水平;根据确定的活性来过滤每种微生物株系的所述绝对细胞计数,以提供所述至少两个样品中的每一个的活性微生物株系及其相应绝对细胞计数的列表;对所述至少两个样品中的每一个的活性微生物株系的所述经过滤的绝对细胞计数与所述至少两个样品中的每一个的至少一个测得元数据进行比较,所述比较包括确定每个样品中的所述活性微生物株系与所述至少一个测得元数据的共现,确定每个样品中所述活性微生物株系和所述至少一个测得元数据的共现包括创建使用表示元数据和微生物株系关系的链接、所述活性微生物株系的所述绝对细胞计数以及所述独特的第二标记物的测量结果的矩阵,以代表一个或多个异质微生物群落网络;根据预测的功能和/或化学性质,基于非参数网络分析和聚类分析中的至少一种,将所述活性微生物株系分组成至少两组,所述非参数网络分析和聚类分析识别活性异质微生物群落网络内的每种活性微生物株系和测得元数据的连通性;从所述至少两组的每一组中选择至少一种微生物株系;以及将所述选择的微生物株系与载体培养基组合以形成合成的活性微生物生物群集,所述生物群集被配置为当被引入到目标生物环境中时改变与所述目标生物环境的所述至少一个元数据相对应的性质。
*********
虽然已参考具体实施方案传达了本公开,但是本领域技术人员应当理解,在不脱离本公开的真实精神和范围的情况下,可以进行各种改变并且可以替换等同物。另外,在所描述的实施方案和公开的客观精神和范围内,可以进行许多修改以采用特定情况、材料、物质组成、过程、一个或多个过程步骤。所有这些修改都旨在处于本公开的范围内。本文引用的专利、专利申请、专利申请公布、期刊文章和方案全文出于所有目的以引用方式并入,包括以下pct申请公布:wo/2016/210251、wo/2017/120495和wo/2017/181203。
虽然本文已描述和说明了各种实施方案,但是本领域技术人员将容易想到用于执行功能和/或获得本文描述的结果和/或一个或多个优点的各种其他方式和/或结构,并且每个这样的变型和/或修改均被认为是在本公开的范围内。更一般地,本领域技术人员将容易理解,本文描述的参数、尺寸、材料和配置是作为说明性实例提供的,并且实际的参数、尺寸、材料和/或配置将取决于所公开的教导内容将用到的具体应用或实现方式。本领域技术人员将认识到或只使用常规实验便能够确定本文所述的具体实施方案的等同物。因此,应该理解,前述实施方案仅以实例的方式呈现,并且应该理解,在所附权利要求书及其等同物的范围内,除了具体描述和要求保护的实施方案之外,还可以实施其他实施方案。本公开的实施方案涉及本文描述的每个单独的特征、系统、制品、材料、试剂盒和/或方法。此外,如果这样的特征、系统、制品、材料、试剂盒和/或方法不相互矛盾,则两个或更多个这样的特征、系统、制品、材料、试剂盒和/或方法的任何组合也包括在本公开的范围内。
上述实施方案可以按多种方式中的任一种来实现。例如,可以使用硬件、软件或其组合来实现这些实施方案。当以软件实现时,软件代码可以在任何合适的处理器或处理器集合体上执行,无论其是提供在单台计算机中还是分布在多台计算机之间。
此外,应当理解,所公开的方法可以与计算机结合使用,该计算机可以多种形式中的任一种来体现,例如机架式计算机(rack-mountedcomputer)、台式计算机、膝上型计算机或平板电脑。另外,计算机可以嵌入通常不被视为计算机但具有合适处理能力的设备中,包括平板电脑、个人数字助理(pda)、智能电话或任何其他合适的便携式或固定电子设备。
此外,计算机可以具有一个或多个输入和输出设备,包括一个或多个显示器。这些设备尤其可用于呈现用户界面。可用于提供用户界面的输出设备的实例包括用于输出视觉呈现的打印机或显示屏以及用于输出听觉呈现的扬声器或其他声音生成设备。可用于用户界面的输入设备的实例包括键盘和指示设备,诸如鼠标、触摸板和数字面板。又如,计算机可以通过语音识别或以其他听觉格式接收输入信息。
此类计算机可以通过一个或多个网络以任何合适的形式互连,包括局域网或广域网(例如企业网络)以及智能网络(in)或因特网。此类网络可以基于任何合适的技术,并且可以根据任何合适的协议操作且可包括无线网络、有线网络或光纤网络。
本文(和/或其一部分)概述的各种方法和过程可以被编码为可在采用各种操作系统或平台中的任一种的一个或多个处理器上执行的软件。另外,这种软件可以使用多种合适的编程语言和/或编程工具或脚本工具中的任一种来编写,并且还可以编译为在框架或虚拟机上执行的可执行机器语言代码或中间代码。
在这方面,各种公开的概念可以体现为用一个或多个程序编码的计算机可读存储介质(或多个计算机可读存储介质)(例如,计算机存储器、一张或多张软盘、光盘、磁带、闪存存储器、现场可编程门阵列或其他半导体器件中的电路配置、或其他非暂态介质或有形计算机存储介质),所述一个或多个程序在一个或多个计算机或其他处理器上执行时实施实现上述公开内容的各种实施方案的方法。所述一种或多种计算机可读介质可以是可移动的,使得存储在其上的一个或多个程序可以被加载到一个或多个不同的计算机或其他处理器上,以实现如上所述的本公开的各个方面。
术语“程序”或“软件”在本文中以一般含义使用,以指代任何类型的计算机代码或计算机可执行指令集,其可用于对计算机或其他处理器进行编程以实现如上所讨论的实施方案的各个方面。另外,应当理解,根据一个方面,在执行时实施本公开的方法的一个或多个计算机程序不需要驻留在单个计算机或处理器上,而是可以以模块化方式分布在多个不同的计算机或处理器之间,以实现本公开的各个方面。
计算机可执行指令可以呈由一台或多台计算机或其他设备执行的多种形式(例如程序模块)。一般地,程序模块包括执行特定任务或实现特定抽象数据类型的例程、程序、对象、部件、数据结构等。通常,在各种实施方案中,可以根据需要组合或分布程序模块的功能。
另外,数据结构可以按任何合适的形式存储在计算机可读介质中。为了简化说明,数据结构可以示出为具有在数据结构中通过位置关联的字段。此类关系同样可以通过以下方式实现:为在计算机可读介质中具有传达字段之间的关系的位置的字段分配存储区。然而,可以使用任何合适的机制来建立数据结构的字段中的信息之间的关系,包括通过使用指针、标签或建立数据元素之间的关系的其他机制。
另外,各种公开的概念可以体现为一种或多种方法,已经提供了所述方法的实例。作为方法的一部分执行的动作可以按任何合适的方式排序。因此,可以构造这样的实施方案,其中以不同于所示顺序的顺序执行动作,这可以包括同时执行一些动作,即使它们在说明性实施方案中示为顺序动作。
如本文定义和使用的所有定义应理解为优先于字典定义、以引用方式并入的文献中的定义和/或所限定术语的普通含义。
本文使用流程图。使用流程图并不意味着对所执行的操作的顺序进行限制。本文描述的主题有时示出不同的部件,其包含在不同的其他部件内或与不同的其他部件连接。应当理解,这样描绘的架构仅是示例性的,并且事实上可以实施实现相同功能性的许多其他架构。在概念意义上,实现相同功能的任何部件布置均有效地“关联”,使得实现期望的功能。因此,本文加以组合以实现特定功能的任何两个部件可以被视为彼此“关联”,使得实现期望的功能,而不论架构或中间部件如何。同样,如此关联的任何两个部件也可以被视为彼此“可操作地连接”或“可操作地耦合”以实现期望的功能,并且能够如此关联的任何两个部件也可以被视为彼此“能可操作地耦合”以实现期望的功能。能可操作地耦合的具体实例包括但不限于能以物理方式配对的和/或以物理方式相互作用的部件和/或能以无线方式相互作用的和/或以无线方式相互作用的部件和/或以逻辑方式相互作用的和/或能以逻辑方式相互作用的部件。
除非有明确的相反指示,否则本说明书和权利要求书中使用的不定冠词“一个/种”应理解为表示“至少一个/种”。
如本说明书和权利要求书中使用的短语“和/或”应理解为表示如此结合的“一个或两个”要素,即在某些情况下联合存在而在其他情况下分离存在的要素。用“和/或”列出的多个要素应以相同的方式解释,即,如此结合的“一个或多个”要素。除了由“和/或”分句具体标识的要素之外,可以任选地存在其他要素,无论与具体标识的那些要素相关还是不相关。因此,作为非限制性实例,当与开放式语言(诸如“包括”)结合使用时,对“a和/或b”的引用在一个实施方案中可以仅指代a(任选地包括除b之外的要素);在另一个实施方案中,可以仅指代b(任选地包括除a之外的要素);在又一个实施方案中,可以指代a和b两者(任选地包括其他要素);等等。
如本说明书和权利要求书中所用,“或”应理解为具有与如上所定义的“和/或”相同的含义。例如,当分隔列表中的项目时,“或”或“和/或”应被解释为包含性的,即包含至少一个,但也包含多于一个、多个或一系列要素,以及任选地另外的未列出的项目。只有明确表示相反含义的术语,诸如“只有一个”或“恰好一个”或在权利要求书中使用时的“由……组成”,才指代恰好包含多个要素或一系列要素中的一个要素。一般而言,如本文所用的术语“或”当前面是排他性术语(诸如“任一个”、“……中的一个”、“……中的仅一个”或“……中的恰好一个”)时,仅应被解释为表示排他性的替代方案(即,“一个或另一个,但不是两个”)。当在权利要求书中使用时,“基本上由……组成”应具有其在专利法领域中使用的普通含义。
如本说明书和权利要求书中所用,关于一个或多个要素的列表,短语“至少一个”应理解为表示选自该要素列表中的任何一个或多个要素的至少一个要素,但不一定包括在该要素列表中具体列出的每一个要素中的至少一个要素,并且不排除该要素列表中的要素的任何组合。该定义还允许可任选地存在除了在短语“至少一个”所指的要素列表内具体标识的要素之外的要素,无论与具体标识的那些要素相关还是不相关。因此,作为非限制性实例,“a和b中的至少一个”(或等效地,“a或b中的至少一个”,或等效地,“a和/或b中的至少一个”)在一个实施方案中可以指代至少一个a,任选地包括多于一个a,而不存在b(并且任选地包括除b之外的要素);在另一个实施方案中,可以指代至少一个b,任选地包括多于一个b,而不存在a(并且任选地包括除a之外的要素);在又一个实施方案中,可以指代至少一个a,任选地包括多于一个a,以及至少一个b,任选地包括多于一个b(以及任选地包括其他要素);等等。
在权利要求书以及上面的说明书中,所有过渡型短语诸如“包含”、“包括”、“携带”、“具有”、“含有”、“涉及”、“持有”、“由……组成”等应理解为开放式的,即,意指包括但不限于。如在美国专利局专利审查程序手册第2111.03节中所述,只有过渡型短语“由……组成”和“基本上由……组成”才应分别是封闭式或半封闭式过渡型短语。
- 还没有人留言评论。精彩留言会获得点赞!